

Abstract
Transcriptional co-activator with PDZ-binding motif (TAZ) and Yes-associated protein (YAP) are critical transcriptional co-activators downstream of the Hippo pathway involved in the regulation of organ size, tissue regeneration, proliferation, and apoptosis. Recent studies suggested common and distinct functions of TAZ and YAP and their diverse impact under several pathological conditions. Here we report differential regulation of TAZ and YAP in response to oxidative stress. H2O2 exposure leads to increased stability and activation of TAZ but not of YAP. H2O2 induces reversible S-glutathionylation at conserved cysteine residues within TAZ. We further demonstrate that TAZ S-glutathionylation is critical for reactive oxygen species (ROS)-mediated, TAZ-dependent TEA domain transcription factor (TEAD) trans-activation. Lysophosphatidic acid, a physiological activator of YAP and TAZ, induces ROS elevation and, subsequently, TAZ S-glutathionylation, which promotes TAZ-mediated target gene expression. TAZ expression is essential for renal homeostasis in mice, and we identify basal TAZ S-glutathionylation in murine kidney lysates, which is elevated during ischemia/reperfusion injury in vivo. This induced nuclear localization of TAZ and increased expression of connective tissue growth factor. These results describe a novel mechanism by which ROS sustains total cellular levels of TAZ. This preferential regulation suggests TAZ to be a redox sensor of the Hippo pathway.
Keywords: connective tissue growth factor (CTGF), Hippo pathway, oxidative stress, reactive oxygen species (ROS), Yes-associated protein (YAP), S-glutathionylation, transcriptional co-activator with PDZ-binding motif (TAZ)
Introduction
The Hippo signaling pathway has been under intense scrutiny because of its conserved ability to regulate organ size and cell proliferation (1). The canonical Hippo pathway revolves around the kinase cascade of Mst1/2, Lats1/2, and their coactivators Sav1 and Mob, which leads to phosphorylation of transcriptional co-activator with a PDZ-binding motif (TAZ)3 and Yes Associated protein (YAP). Both TAZ and YAP are transcriptional co-activators and downstream effectors of the Hippo pathway. The stability of these proteins depends on Lats1/2 phosphorylation of TAZ at Ser-89 (Ser-127 in YAP), leading to 14-3-3 binding and cytoplasmic retention. Furthermore, phosphorylation of Ser-314 of TAZ and Ser-381 of YAP primes them for subsequent casein kinase 1-mediated ubiquitination and proteasomal degradation (2). In the absence of Lats1/2 activity, both TAZ and YAP function as transcription co-activators and initiate gene expression, promoting proliferation and cell survival by binding to transcription factors such as TEAD or RUNX2 (3). Thereby, TAZ and YAP modulate the expression of a variety of target genes (2, 4). Recently, it has been shown that G protein-coupled receptor (GPCR) ligands, lysophosphatidic acid (LPA), and sphingosine 1-phosphate can lead to dephosphorylation and promote nuclear localization of YAP and TAZ (5). Both TAZ and YAP have been shown to be up-regulated in various cancers and involved in epithelial-mesenchymal transition, leading to metastasis (6, 7).
Despite overlapping functions of both TAZ and YAP, recent evidence suggests that they have distinct physiological roles. Knockout of Yap in mice leads to embryonic lethality (8), whereas Taz knockout mice are viable but develop cystic kidney disease and lung emphysema (9, 10). On the protein level, TAZ and YAP display ∼50% sequence similarity, including the conserved phosphorylation residues. They have many common but also few distinct protein-protein interaction partners that are essential for their stability and function (11). Interestingly, three conserved cysteine residues are only present in TAZ, highlighting its potential redox regulation.
The human proteome contains ∼200,000 cysteine residues, making cysteine one of the least commonly used amino acids (12). This implies the evolutionary importance of cysteine content, which correlates with the degree of biological complexity of an organism (13). Homeostasis between cellular oxidation and reduction reactions plays a critical role in signal transduction. Reactive oxygen species (ROS) are in general regarded as cytotoxic, mutagenic, and inducers of oxidative stress. However, several studies implicate ROS in the stimulation or inhibition of cell proliferation, apoptosis, and cell senescence (14). Posttranslational modifications of cysteines can directly influence these processes. S-glutathionylation is a well characterized protein modification. An oxidized glutathione (GSSG) forms a disulfide linkage with the reactive thiol group of cysteine, resulting in a protein-glutathione complex. Mild oxidative stress upon elevated superoxide (O2˙̄) or hydrogen peroxide (H2O2) can induce S-glutathionylation at cysteine residues, which can lead to alteration of protein function (15–17).
In this study, we demonstrate that TAZ is S-glutathionylated at residues Cys-261, Cys-315, and Cys-358 in vitro. Circumstantial S-glutathionylation of TAZ may lead to activation of its transcriptional activity and could function as a redox sensor of the Hippo pathway. We propose a previously unidentified signaling pathway whereby ROS can directly modulate TAZ activity.
Experimental Procedures
Cell Culture
HEK 293T cells were cultured in Dulbecco's minimum essential medium (Gibco) supplemented with 10% FCS. Transient transfection was performed using Lipofectamine (Invitrogen) or the calcium phosphate method. Cells were treated with the indicated concentrations of hydrogen peroxide (Sigma-Aldrich) and lysophosphatidic acid (Tocris) for 1 or 6 h and harvested for protein or RNA preparations. Cells were incubated with antioxidant-reduced glutathione (Sigma-Aldrich), N-acetylcysteine (Sigma-Aldrich), or the NOX inhibitors apocynin (Sigma-Aldrich) and diphenyleneiodonium (Sigma-Aldrich) as indicated. NIH3T3 TAZ flp-in stable cell lines were cultured in Dulbecco's minimum essential medium (Gibco) supplemented with 10% FCS in the presence of 200 μg/ml hygromycin (Invitrogen).
Plasmids and Antibodies
Murine TAZ (NP_598545.2) and YAP (NP_033560.1) cDNA was provided by M. Yaffe (Massachusetts Institute of Technology, Boston, MA). TAZ cysteine mutants were generated using site-directed mutagenesis to mutate the cysteine residues to alanine in positions 261, 315, and 358 in murine FLAG.TAZ pcDNA6 (Invitrogen). The mutant constructs were verified by sequencing and restriction digestion. The FLAG.EPS plasmid was used as a control protein. Antibodies were purchased from Sigma-Aldrich (anti-FLAG/M2, catalog no. F3165), Cell Signaling Technology (anti-YAP/TAZ, catalog no. 8418; anti-TAZ, catalog no. 4883; anti-YAP catalog no. 4912; anti-phospho-YAP/TAZ, catalog no. 13008; anti-β-tubulin, catalog no. 2128; and anti-β-actin, catalog no. 3700), Virogen (anti-GSH, catalog no.101-A-100), and Millipore (anti-V5, catalog no. AB3792).
Reverse Transcription and Quantitative Real-time PCR (qPCR) Analysis
One microgram of total RNA from HEK 293T cells was used to generate cDNA templates for reverse transcriptase PCR. The first-strand cDNA synthesis was performed using a hexamer (Applied Biosystems) and Superscript II reverse transcriptase (Applied Biosystems). The first-strand cDNA products were further diluted 1:1 and used as qPCR templates. The SYBR Green-based qPCR analysis was carried out in a real-time PCR system (7900HT, Applied Biosystems). Duplicate reactions were carried out for each sample. All samples were normalized by the expression level of the housekeeping gene β-actin. The primer sequences used in the qPCR assays were as follows: hCYR61, 5′-AGCCTCGCATCCTATACAACC-3′ (forward) and 5′-TTCTTTCACAAGGCGGCACTC-3′ (reverse); β-actin, 5′-GGACTTCGAGCAAGAGATGG-3′ (forward) and 5′-AGCACTGTGTTGGCG TACAG3-′ (reverse); mTAZ, 5′-CCGGGTGGGAGATGACCTT-3′ (forward) and 5′-AGGTTCACATGATTCAGAGGCT-3′ (reverse); and mYAP, 5′-GTCCTCCTTTGAGATCCCTGA-3′ (forward) and 5′-TGTTGTTGTCTGATCGTTGTGAT-3′ (reverse). TaqMan assays (Applied Biosystems) were used to evaluate CTGF (Hs00170014_m1) and β-actin (4326315E) levels.
Immunoprecipitation
HEK 293T cells were transiently transfected using the calcium phosphate method, and the total amount of DNA was always adjusted with empty pcDNA6 (Invitrogen). For TAZ and NOX4 expression studies, plasmids were co-transfected in a ratio of 5:1. The following day, cells were harvested with ice-cold PBS. The harvested cells were lysed in a 1% Triton X-100 buffer (1% Triton X-100, 20 mm Tris-HCl (pH 7.5), 50 mMNaCl, 50 mMNaF, 15 mm Na4P2O7, 2 mm Na3VO4, 25 mm N-ethylmaleimide, and complete protease inhibitors (PIM complete, Roche)) for 15 min on ice. After centrifugation at 15,000 × g for 15 min at 4 °C and ultracentrifugation at 100,000 g for 30 min at 4 °C, the supernatant was incubated at 4 °C for 1 h with the anti-FLAG (M2) covalently coupled to agarose beads (Sigma-Aldrich) or with 1 μg of the appropriate first antibody and 30 μl protein G-Sepharose beads (GE Healthcare). Before the addition of antibodies, a small aliquot of each supernatant was preserved and diluted with 2× SDS-PAGE sample buffer for later Western blot analysis (lysate). The beads were washed extensively with lysis buffer, and bound proteins were resolved by SDS-PAGE, blotted on to polyvinylidene fluoride membranes. Visualization was performed by infra-red detection using donkey anti mouse IRDye 680 or donkey anti rabbit IRDye 800 in the Odyssey Infrared Imaging System (LI-COR Biosciences). Quantification was performed with the LI-COR Image StudioTM analysis software provided in a linear dynamic range.
Luciferase Assays
The luciferase reporter plasmid (pGBD-Hyg-Luc) was transfected together with an activator plasmid (pGal4-TEAD), pGL4.74 (Promega), for normalization and the indicated expression plasmids (TAZ, YAP, NOX4, and control empty pcDNA6) into HEK 293T cells in a 96-well format using Lipofectamine 2000 (Invitrogen) as a transfection reagent. The total amount of DNA was always adjusted with empty pcDNA6. Renilla luciferase and firefly luciferase activities were measured by using a reporter assay system (Dual-Luciferase, Promega) in a luminometer (Mithras LB 940, Berthold) at 6 or 24 h after treatment. Transfections and measurements were performed in triplicates for each single experiment, and each experiment was repeated at least three times. The remaining lysates were pooled in 2× Laemmli, and TAZ expression was determined by immunoblotting. The error bars shown in the figures represent standard error. p values were calculated using two-tailed unpaired Student's t test.
Measurement of Intracellular ROS
2 × 104 HEK 293T cells were seeded in optical-bottomed 96-well plates (Nunc) in complete DMEM. 16 h later, cells were challenged with H2O2 or LPA for 1 h. Cells were loaded either with 10 μm 2′,7′-dichlorodihydrofluorescein diacetate (excitation, 488 nm; emission, 520 nm; Sigma-Aldrich) or 10 μm DHE (excitation, 510 nm; emission, 595 nm; Sigma-Aldrich) for 30 min at 37 °C. Cells were gently rinsed in Hanks' balanced salt solution once, and fluorescence intensity was immediately quantified in a multimode plate reader (PerkinElmer Life Sciences).
Ischemia/Reperfusion Injury
We used a rodent warm ischemia/reperfusion model. The surgical procedure was approved by the Landesamt für Natur, Umwelt, und Verbraucherschutz NRW (LANUV 84–02.04.2013.A158). Male 10- to 12-week-old C57BL/6J mice were anesthetized with a Ketavet/xylazine injection intraperitoneally. The following steps were conducted on a heat pad to preserve the physiological body temperature of the experimental animals. After cessation of reflexes, a median laparotomy was carried out. The right kidney pedicle was mobilized and clamped for 30 min with an atraumatic microvascular clamp. Successful ischemia was confirmed by discoloration of the kidney. During ischemia, the abdomen was covered with gauze soaked with normal saline. At the end of ischemia, the clamp was released, and the kidney was controlled for visual signs of reperfusion. 5 μm Hoechst 33342 and dihydroethidium (DHE) was injected via retro-orbital injection. Five minutes after reperfusion, two-photon imaging of the intact kidney was performed (Leica TCS SP8 MP-OPO) with a ×20 water immersion objective as described previously (18). Animals were sacrificed by cervical dislocation, and the right ischemic and left unharmed kidney was harvested in PBS for protein extraction. 500 μg of total protein extract was used for immunoprecipitation experiments.
Immunohistochemistry
Tissue sections were deparaffinized by two changes of xylene for 5 min each. Slides were then rehydrated in two changes of 100% ethanol for 3 min, followed by 95% and 70% ethanol for 1 min, and rinsed in TBS. Sections were boiled in sodium citrate buffer (10 mm sodium citrate (pH 6.0)) for 20 min. Slides were allowed to cool, and endogenous peroxidase activity was quenched using 3% H2O2 in methanol. Sections were pretreated with unconjugated avidin followed by biotin treatment (Vector Laboratories, SP2001). Slides were then incubated with primary anti-YAP (1:500) or anti-TAZ (V386) (1:500) at 4 °C overnight. Slides were washed three times with TBS, followed by incubation with biotinylated donkey anti-rabbit IgG diluted secondary antibody (1:500) for 1 h. After final washing, slides were incubated with ABC reagent (Vector Laboratories) for 30 min. Finally, DAB reagent (Jackson ImmunoResearch Laboratories, Inc.) was added to the sections for 5 min and counterstained with hematoxylin. Sections were dehydrated and mounted with Histomount. The slides were then scanned with an automated slide scanner (Leica), and images were quantified using the positive pixel count feature in Imagescope (Leica).
Confocal Imaging
NIH-3T3 TAZ or YAP GFP flp-in cells were grown on μ-Dish 35-mm, high glass bottom dishes (Ibidi). Cells were then exposed to 250 μm H2O2 or vehicle control (PBS) for 1 h at 37 °C. Cells were washed once in complete medium, and fluorescent images were captured at 488-nm excitation using the Zeiss LSM710 META confocal imaging system with a ×40/1.3 numerical aperture oil objective at room temperature. Images were analyzed and quantified using ImageJ (National Institutes of Health).
Statistics
All statistical comparisons between groups were analyzed using two-tailed Student's t test. Differences in means among multiple groups were analyzed using one-way ANOVA with Bonferroni post test unless otherwise indicated. p < 0.05 was considered significant in all analyses. The data were computed using GraphPad Prism version 5.0.
Results
H2O2 Elevates Total Protein Levels of TAZ and Increases TAZ/TEAD Activity
A previous study observed increased levels of TAZ but not YAP upon stimulation with the GPCR agonist LPA, suggesting that TAZ might be differently regulated than YAP (5). It has been well characterized that LPA induces ROS via the phospholipase C/PKC/NOX pathway (19, 20). Hence, we hypothesized that ROS could specifically augment total TAZ levels. To assess whether ROS modulates TAZ/YAP expression, we first investigated the role of exogenous H2O2 in HEK 293 cells transiently expressing either wild-type YAP or TAZ. HEK 293T cells exposed to increasing concentrations of H2O2 (0, 50 100, or 200 μm) for 1 h showed a dose-dependent increase in intracellular ROS levels, as determined by dichlorodihydrofluorescein staining in live cells, which was inhibited by preincubating cells with antioxidant-reduced GSH (Fig. 1A). In this model, we observed an increase in total TAZ (Fig. 1B), whereas YAP was significantly degraded (Fig. 1C). To validate whether H2O2 differentially alters TAZ and YAP stability, we pretreated cells expressing TAZ and YAP with cycloheximide, followed by exposure H2O2 for 1 h. We observed that total YAP levels were decreased, whereas TAZ levels remained stable under these conditions (Fig. 1D). YAP and TAZ are essential co-activators of the TEAD family of transcription factors. To determine whether H2O2 influences the trans-activation of YAP/TAZ, we performed a TEAD reporter gene assay as described previously (21). Interestingly, TAZ/TEAD4 trans-activation principally remained unaltered following 6-h incubation, with a slight but significant increase at 50 μm H2O2 (Fig. 1E), whereas there was a significant decrease of YAP/TEAD4 trans-activation when exposed to H2O2 (Fig. 1F). NAD(P)H oxidase 4 (NOX4) serves as the endogenous source of H2O2 in cellular systems (22) and has been implicated in several murine renal disease models (23). Hence, we investigated whether NOX4-derived H2O2 could alter TAZ/YAP/TEAD activity in a similar manner. NOX4 co-expression significantly up-regulates TAZ/TEAD activity, whereas YAP/TEAD activity was diminished, consistent with our findings using exogenous H2O2 (Fig. 1G). In addition, increasing amounts of exogenously expressed NOX4 decreased YAP expression without affecting TAZ (Fig. 1H), which is again congruent with H2O2 treatment (Fig. 1, B and C). Similar results were obtained using NOX1, NOX3, and p22-phox overexpression constructs (data not shown).
FIGURE 1.

H2O2 elevates total protein levels of TAZ and increases TAZ/TEAD activity. A, HEK 293T cells were challenged with 50, 100, and 200 μm H2O2 for 1 h. After treatment, cells were loaded with a cellular ROS indicator (H2DCFDA), and changes in fluorescence arbitrary units (f.a.u.) were quantified using a microplate reader. B and C, HEK 293T cells expressing FLAG.TAZ (F.TAZ) or FLAG.YAP (F.YAP) were challenged with 50, 100, and 200 μm H2O2 for 1 h. Western blotting and densitometry analysis reveals elevated protein levels of TAZ but not YAP. D, HEK 293T cells expressing FLAG.TAZ or FLAG.YAP were pretreated with cycloheximide (CHX, 40 μg/ml) for 1 h. Cells were then challenged with 50, 100, and 200 μm H2O2 for an additional 1 h. Western blotting and densitometry analysis reveals stable protein levels of TAZ but not YAP. E and F, HEK 293T cells were treated with 50, 100, and 200 μm H2O2 for 6 h. H2O2 enhances the activity of the transcriptional coactivator TAZ (E) but not YAP (F) in TEAD reporter assays, as described under “Experimental Procedures.” G, HEK 293T cells expressing FLAG.TAZ or FLAG.YAP were co-transfected with NOX4. NOX4 enhances the activity of the transcriptional coactivator TAZ but not YAP in TEAD reporter assays. H, immunoblotting of whole cell lysates reveals that increasing amounts of NOX4 degrades YAP in a dose-dependent manner but not TAZ. I and J, HEK 293T cells expressing FLAG.TAZ and V5.TEAD4 or FLAG.YAP and V5.TEAD4 were treated with increasing concentrations of H2O2 for 1 h. Cells expressing FLAG.EPS and V5.TEAD4 served as negative control. Co-immunoprecipitation reveals that H2O2 does not significantly alter the TAZ/TEAD4 interaction but that the YAP/TEAD4 interaction is perturbed. K and L, CTGF expression levels analyzed by qPCR reveal that TAZ induces CTGF expression following 6-h H2O2 treatment, which is reversed by antioxidant pretreatment. In contrast, YAP-induced CTGF expression is not significantly altered by H2O2. M, NIH-3T3 TAZ or YAP GFP flp-in stable cells were exposed to 250 μm H2O2 for 1 h. Shown are representative confocal images and quantification of GFP fluorescence. Scale bars = 50 μm. Data are mean ± S.E. from three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant).
H2O2 did not significantly alter the TAZ/TEAD interaction, but the YAP/TEAD4 interaction was considerably diminished (Fig. 1, I and J). To substantiate the role of H2O2-induced TAZ/YAP target gene expression, we quantified the mRNA expression of CTGF, which is a bona fide target gene for both TAZ and YAP (2). H2O2 exposure increased CTGF expression in a dose-dependent manner in cells expressing TAZ/TEAD4 constructs, which was attenuated by pretreating cells with antioxidant-reduced GSH (Fig. 1K). However, YAP failed to induce CTGF expression above the basal levels despite H2O2 exposure (Fig. 1L). Next we generated stable cell lines expressing wild-type GFP-tagged TAZ and YAP at low expression levels in NIH-3T3 Flp-in cells as described previously (11). NIH-3T3 Flp-in cells have the advantage of single-copy genomic integration. TAZ GFP wild-type showed increased accumulation and nuclear localization upon 1-h H2O2 exposure compared with YAP GFP wild-type (Fig. 1M), confirming that ROS could differentially regulate TAZ and YAP.
H2O2 Induces S-Glutathionylation in TAZ at Three Cysteine Residues
TAZ and YAP are transcriptional co-activators downstream of the Hippo pathway and have been well characterized in several pathophysiological models (24). Comparing the amino acid sequences of mouse proteins reveals that TAZ has three evolutionarily conserved cysteine residues (Cys-261, Cys-315, and Cys-358), whereas YAP has none. Of the three cysteine residues in TAZ, Cys-315 is evolutionarily conserved from lower organisms to higher mammals (Fig. 2A). We speculated that the cysteine residues in TAZ could undergo H2O2 induced S-glutathionylation and that this could be responsible for the H2O2 effect on TAZ expression and activation. Oxidative stress can directly modify cysteine residues, leading to formation of intramolecular disulfide bonds. Exposing recombinant His TAZ protein with 100 μm H2O2 for 30 min did not alter protein mobility under non-reducing conditions, suggesting that oxidant stress did not directly modify TAZ (Fig. 2B). HEK 293T cells were co-transfected with wild-type TAZ and YAP. 100 μm H2O2 induced S-glutathionylation in TAZ but not YAP, as detected in precipitates with an antibody directed against GSH. TAZ S-glutathionylation was partially reversed by addition of 1 mm DTT to cells 30 min after H2O2 exposure (Fig. 2C). H2O2 induced TAZ S-glutathionylation in a dose-dependent manner, which was inhibited by pretreating cells with antioxidants such as reduced GSH or N-acetylcysteine. Interestingly, there was no detectable phosphorylated TAZ (Ser-89) upon S-glutathionylation (Fig. 2D). Protein S-glutathionylation can occur at single or multiple cysteine residues in a context-dependent manner (25). Analysis of different Cys-Ala mutants revealed that only the triple mutant lacking all three cysteine residues but not the single or double mutants was negative for S-glutathionylation. These observations suggest that TAZ can undergo S-glutathionylation at all three residues, with higher specificity toward the evolutionarily conserved residue Cys-315 (Fig. 2, E–I).
FIGURE 2.

H2O2 induces S-glutathionylation of TAZ at three cysteine residues. A, sequence alignment of mouse TAZ (NP_598545.2) reveals conserved cysteine residues at position Cys-261, Cys-315, and Cys-358. B, Coomassie staining of recombinant His.TAZ protein following 100 μm H2O2 exposure for 30 min in vitro. C, HEK 293T cells transiently co-transfected with FLAG.TAZ and FLAG.YAP were treated with 100 μm H2O2 for 1 h. 1 mm DTT was added to cells 30 min after H2O2 exposure. After immunoprecipitation (IP) with anti-GSH antibody, Western blotting analysis revealed that wild-type FLAG.TAZ (55 kDa) undergoes S-glutathionylation but not YAP (72 kDa). D, HEK 293T cells transiently transfected with FLAG.TAZ were treated with 50, 100, and 200 μm H2O2 for 1 h. After IP with anti-GSH antibody, Western blotting (WB) analyses of lysates (LYS) and precipitates (IP) revealed that wild-type FLAG.TAZ undergoes S-glutathionylation in a dose-dependent manner, which was inhibited by the antioxidants GSH and N-acetylcysteine. E–I, HEK 293T cells transiently transfected with FLAG.TAZ cysteine mutants as indicated were treated with 50, 100, and 200 μm H2O2 for 1 h. After IP with anti-GSH antibody, Western blotting analysis revealed that TAZ undergoes S-glutathionylation in all mutants analyzed (C261A (E), C315A (F), C358A (G), C261A/C315A (H), and C261A/C315A/C358A (I)), indicating the presence of multiple S-glutathionylation sites within TAZ. Mouse IgG (isotype) is shown as a control. Shown are representative data from three independent experiments.
H2O2-mediated Activation of TAZ Depends on S-Glutathionylation at Three Cysteine Residues
Because all three cysteines undergo S-glutathionylation, we then investigated the sensitivity of the TAZ cysteine triple mutant toward H2O2-mediated activation using a TEAD reporter assay. Strikingly, the TAZ triple mutant did not show any significant activation following 6 h exposure of H2O2 compared with the wild type (Fig. 3A). We then characterized the TAZ cysteine mutants in TAZ/TEAD reporter assays. The TAZ C315A mutant evoked a significantly elevated transactivation compared with the wild type, whereas both the C261A and C358A mutants did not show any significant change (Fig. 3B). Quantitative PCR analysis confirmed elevated expression of the TAZ target genes CTGF and CYR61 in HEK 293 cells expressing the TAZ C315A mutant, supporting the idea that Cys-315 may enhance TAZ stability and increase the expression of target genes (Fig. 3, C and D). In contrast, co-immunoprecipitation experiments demonstrated that affinity toward TEAD4 is not significantly altered in the Cys-Ala mutants (Fig. 3E). Interestingly, despite enhanced trans-activation of the C315A mutant, total protein levels were reduced in these cell lysates, which might be due to nuclear translocation because expression levels in whole-cell-lysates were found to be equal (Fig. 3B). Phosphorylation of Ser-89 was increased in the TAZ C261A and C261/315/358A mutants (Fig. 3F). In summary, these findings indicate that all three cysteine residues play an essential role in ROS-mediated activation of TAZ.
FIGURE 3.

H2O2-mediated activation of TAZ depends on S-glutathionylation at three cysteine residues. A, TEAD reporter assay showing increased activity of TAZ wild-type, whereas the TAZ triple mutant fails to elicit the H2O2-induced response. B, basal TEAD reporter activity of TAZ cysteine mutants. Immunoblotting of whole cell extracts from the luciferase assay shows basal TAZ protein levels. C and D, qPCR analysis reveals increased basal CTGF and CYR61 expression in the TAZ C315A mutant compared with the wild type in HEK 293 cells. E, HEK293T cells expressing FLAG.TAZ (F.TAZ) and V5.TEAD4 were immunoprecipitated using M2 beads; F.EPS served as negative control. Co-immunoprecipitation reveals that TAZ cysteine mutants do not alter the TAZ/TEAD4 interaction. F, basal total and phosphorylated (Ser-89) levels of TAZ cysteine mutants in HEK 293 cells. Data are presented as mean ± S.E. from three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant).
The GPCR Agonist LPA Induces S-glutathionylation of TAZ
LPA is a potent phospholipid-derived GPCR agonist. LPA has been demonstrated previously to induce elevation of superoxide (O2˙̄) via the phospholipase C/PKC/NOX pathway in cancer cell lines (19, 20). To corroborate LPA-induced ROS elevation, we treated HEK 293T cells with increasing concentrations of LPA for 1 h and quantified superoxide generation using DHE. LPA induces a significant elevation of O2˙̄, which was mitigated by supplementing with N-acetylcysteine or the pan NOX inhibitor apocynin (Fig. 4A). To elucidate the physiological relevance of TAZ S-glutathionylation, we hypothesized that LPA-derived ROS could induce TAZ S-glutathionylation in HEK 293T cells. LPA stimulation of HEK 293T cells expressing TAZ wild-type resulted in TAZ S-glutathionylation in a dose-dependent manner, which was inhibited by preincubation with antioxidants or the NOX inhibitor apocynin (Fig. 4B). Because LPA is a physiological agonist of both TAZ and YAP, we performed a TEAD reporter assay with increasing concentrations of LPA. We observed an increase in reporter activity for both TAZ and YAP at 1 μm LPA. However, YAP/TEAD activity was significantly reduced at 5 μm LPA, whereas TAZ/TEAD activity was slightly altered at this concentration, suggesting that LPA-induced TAZ S-glutathionylation could sustain TAZ/TEAD activity. This is further supported by the finding that the TAZ cysteine triple mutant is insensitive to LPA treatment (Fig. 4C). This enhanced trans-activation of TAZ/TEAD at higher concentrations of LPA might correlate to the increased levels of intracellular ROS. Moreover, LPA induced TAZ target gene expression, as demonstrated by qPCR analyses for CTGF and CYR61 even at the high concentration of 5 μm, which was reduced by ROS suppression using NOX inhibitors (apocynin) or antioxidants (N-acetylcysteine) (Fig. 4, E and F). Taken together, our results indicate that LPA-mediated NOX-derived ROS promotes S-glutathionylation of TAZ and thus increases TAZ target gene expression predominantly through TAZ.
FIGURE 4.
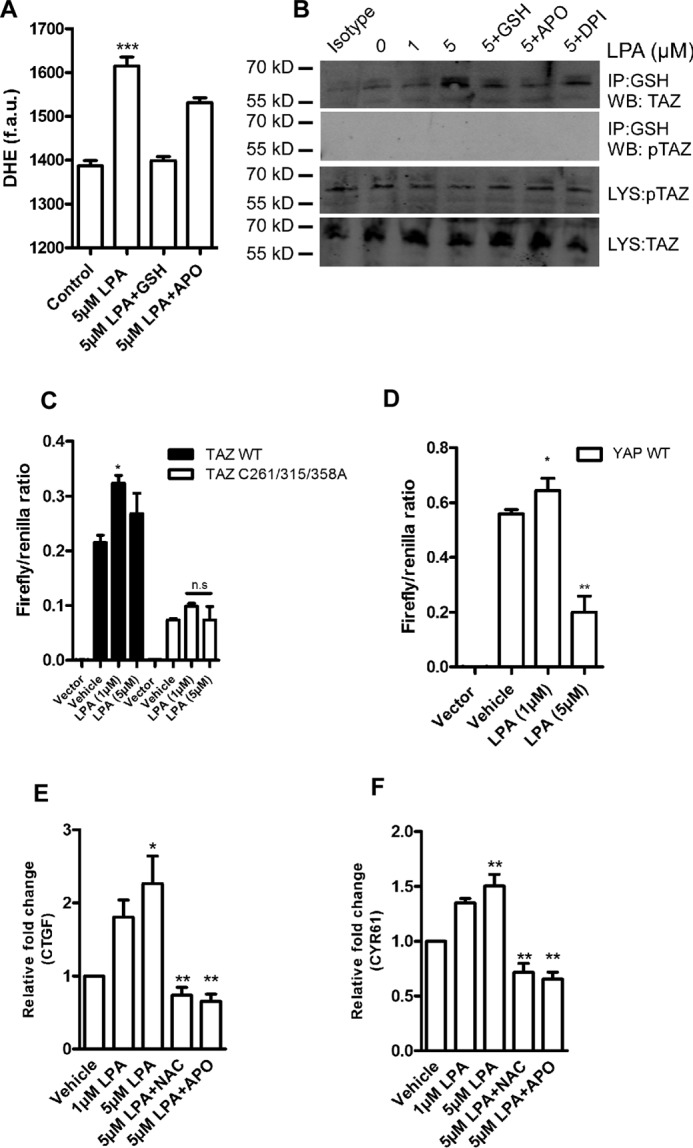
LPA induces TAZ S-glutathionylation. A, HEK 293T cells were treated with 5 μm LPA for 1 h. After treatment, cells were loaded with DHE, and superoxide elevation was quantified as fluorescence arbitrary units (f.a.u.) in a fluorescence plate reader. APO, apocynin. B, HEK 293T transiently expressing FLAG.TAZ were challenged with 1 and 5 μm LPA for 1 h. After IP with anti-GSH antibody, Western blotting (WB) analyses revealed that wild-type TAZ undergoes S-glutathionylation in a dose-dependent manner, which was inhibited with pretreatment of antioxidants and the pan NOX inhibitor apocynin or diphenyliodonium (DPI). Mouse IgG IP (isotype) is shown as a control. C, TEAD reporter activity of TAZ wild-type and the cysteine triple mutant 6 h following LPA treatment. D, TEAD reporter activity of YAP wild-type 6 h following LPA exposure. E and F, HEK 293T cells transiently expressing FLAG.TAZ were challenged with 1 or 5 μm LPA for 6 h. TAZ target gene CTGF and CYR61 levels were quantified by qPCR. LPA induces TAZ-mediated CTGF and CYR61 expression, which is attenuated with pretreatment with 2 mm N-acetylcysteine (NAC) and 10 μm apocynin. Data are mean ± S.E. from three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant).
TAZ S-Glutathionylation in Vivo
Both TAZ and YAP play crucial roles in renal development. YAP knockout mice are embryonic lethal (8), whereas TAZ knockout mice develop a severe cystic kidney disease (9, 10). Conditional knockout models underline the importance of TAZ and YAP in kidney development (26). In our study, immunohistochemical staining reveals higher expression of TAZ in the medullary region in the adult murine kidney compared with YAP, indicating a prominent role of TAZ in renal homeostasis (Fig. 5, A and B). To understand whether TAZ S-glutathionylation is a relevant physiological mechanism, we first investigated basal TAZ S-glutathionylation in murine kidney tissue homogenates. Strikingly, co-immunoprecipitation experiments identified basal TAZ S-glutathionylation in renal homogenates (Fig. 5C). Renal ischemia/reperfusion (I/R) injury has been well documented to induce oxidative stress by elevation of superoxide and H2O2 (27). We propose that oxidative stress could also lead to TAZ S-glutathionylation in kidneys. We induced I/R injury in the left kidney (the right kidney served as a control) by clamping the kidney pedicle for 40 min, followed by 5-min reperfusion in mice preinjected with the DNA marker Hoechst 3342 and the superoxide sensitive dye DHE. DHE undergoes oxidation by superoxide, which then enhances its DNA binding and fluorescence. Dual-photon imaging shows a significant increase in superoxide levels, as seen by the nuclear DHE signal when compared with the control right kidney (Fig. 5D). Total tissue homogenates from the control and I/R kidneys were precipitated using the total YAP/TAZ antibody and probed with anti-GSH antibody under non-reducing conditions. The results indicate a significant increase in TAZ S-glutathionylation upon renal I/R injury (Fig. 5E). Immunohistochemical staining reveals total and nuclear TAZ levels to be increased in the tubular compartments in I/R kidneys, whereas YAP levels remain unchanged. Furthermore, CTGF levels were also found to be elevated following I/R injury, indicating the significance of the redox activation of TAZ (Fig. 5F). These results indicate that TAZ S-glutathionylation is an important pathophysiological response upon oxidative stress signaling that could lead to enhanced stability and activation, leading to tissue repair.
FIGURE 5.

TAZ S-glutathionylation is induced by ischemia/reperfusion in the kidney. A, immunohistochemical staining of wild-type mouse kidneys with total TAZ and YAP antibodies. Neg, negative. B, positive pixel count analysis of sections in A, showing expression levels of TAZ and YAP (n = 3). C, TAZ/YAP was immunoprecipitated using total anti-YAP/TAZ antibody from whole kidney lysates of wild-type mice. S-Glutathionylated TAZ (TAZ-SSG) was detected using anti-GSH antibody under non-reducing conditions. WB, Western blot. D, in vivo elevation of superoxide (nuclear DHE) in tubular epithelial cells upon renal I/R injury by two-photon microscopy (n = 3). E, whole kidney lysates from healthy and I/R kidney reveal increased TAZ glutathionylation following I/R injury (n = 3). Rabbit IgG IP (Isotype) is shown as a control. F, immunohistochemical staining of wild-type mouse kidneys following I/R injury showing increased cytoplasmic and nuclear TAZ and CTGF levels in I/R kidneys compared with the control (n = 3). G, proposed model in which ROS derived from physiological or oxidative stress pathways induce TAZ glutathionylation, leading to activation and stability. Data are mean ± S.E. from three independent experiments (*, p < 0.05; ***, p < 0.001). Scale bars = 200 μm.
Discussion
Regulation of cellular protein levels, phosphorylation, and localization of YAP and TAZ determines the functional output of the Hippo pathway. Increased YAP/TAZ protein levels have been well documented during physiological responses such as cell proliferation and wound healing (1) as well as in cystic kidney diseases (28). Previous studies of the regulation of YAP and TAZ relied on phosphorylation events in the conserved C-terminal and N-terminal phosphodegron (6, 29). However, several studies also indicate that biological responses mediated by these proteins are quite different, with tissue and organ specificities (1, 30). Despite the similarities, outstanding questions such as context-dependent activation and factors that uniquely regulate nuclear and cytoplasmic localization of TAZ and YAP still remain. It has been shown previously that Hippo signaling induces YAP inactivation upon I/R injury in cardiomyocytes (31), but the role of TAZ during oxidative stress has not been investigated.
Because of its greater stability in comparison with all other oxidant species, H2O2 is the major source of cellular ROS. Under basal conditions, cellular compartments are maintained in a reduced state. Under conditions of metabolic stress, ROS accumulate in cells and directly interfere with the intracellular pathway or directly damage cellular components via oxidation. However, receptor-mediated signaling can lead to a subtle elevation of ROS that participate in signal transduction. Our data indicate accumulation of TAZ upon short-term exposure to H2O2 or NOX4. This also led to an increased activation of TAZ but not YAP. TEAD reporter assays and target gene expression assert a striking difference between the two proteins in response to ROS in vitro. As a defense against ROS-mediated oxidation, cells use antioxidants to revert to a reduced state. Furthermore, localization studies in stable cell lines expressing TAZ GFP or YAP GFP reveal increased nuclear localization of TAZ but not YAP upon H2O2 exposure. This complementary response authenticates the differential response of TAZ and YAP toward H2O2-induced oxidative stress. We hypothesized that the noticeable TAZ response to H2O2 could be due to redox regulation of conserved cysteine residues in TAZ.
Here we demonstrate for the first time that TAZ, but not YAP, undergoes S-glutathionylation under basal conditions and upon mild oxidative stress in HEK 293T cells. The physiological relevance of S-glutathionylation is further substantiated by the reversibility of the process. TAZ S-glutathionylation is reversed by restoring cells to a reduced state either by supplementing with excess antioxidant (reduced GSH) or the reducing agent DTT. By mutational analysis, we identify that TAZ undergoes S-glutathionylation at all three cysteine residues (Cys-261, Cys-315, and Cys-358). Interestingly, neither exposure to exogenous H2O2 nor mutation of these cysteine residues altered the TAZ/TEAD4 interaction. S-Glutathionylation can lead to a gain or loss and, in some cases, to no noticeable alterations in protein function (25). In our study, H2O2- or LPA-induced TAZ S-glutathionylation enhanced the transcriptional activation of TEAD as well as increased target gene expression compared with YAP. This is consistent with the fact that H2O2 or LPA did not lead to TEAD activation in the TAZ cysteine triple mutant, which fails to undergo S-glutathionylation. Phosphorylation of TAZ at Ser-89 is crucial for 14-3-3 binding and cytoplasmic localization (32). Results from immunoprecipitation experiments reveal that glutathionylated TAZ is not phosphorylated at Ser-89 and, hence, could participate in activation of TEAD upon ROS exposure. The evolutionarily conserved Cys-315 residue lies in the TAZ C-terminal phosphodegron hot spot. TAZ Cys-315 could also be the favorable site for S-glutathionylation because the incorporated GSH moiety might interact with surrounding amino acid bases, bringing about a structural change in the protein and thereby modulating function.
Previous studies have demonstrated that sequential phosphorylation at TAZ Ser-311 and Ser-314 by large tumor suppressor kinase 1 and CK1-ϵ is essential for SCFβ-TrCP-mediated TAZ ubiquitylation and degradation. We speculate that TAZ C315A behaves similar to the TAZ S314A mutant, which could also lead to inhibition of SCFβ-TrCP-mediated TAZ ubiquitylation and degradation, leading to nuclear translocation and enhanced target gene expression (33). The stability and cytoplasmic retention of TAZ depend on the physical interaction with 14-3-3 (32) and angiomoitin (34). We speculate that the conformational change caused by the C315A mutation could prevent interaction between TAZ and 14-3-3 or angiomoitin, leading to enhanced trans-activation. However, further mechanistic studies are warranted to validate the importance of the Cys-315 residue in redox activation of TAZ.
It has been demonstrated previously that phosphorylation of Mst1/2 as well as Mob and Lats1 can be induced by H2O2 exposure (35). H2O2-induced phosphorylation of MST1 leads to neuronal cell death (36). However, the role of the classical Hippo cascade and its downstream effectors YAP and TAZ remained unclear in this study. H2O2-induced activation of the canonical Hippo cascade could promote phosphorylation and nuclear localization of the downstream effectors, which has been demonstrated recently for YAP (37). This inactivation of YAP would promote cell death, and consistently it has been shown that both decreased Lats1 activity (31) as well as increased YAP activity could protect cardiomyocytes against H2O2-induced cell death (38). However, the role of TAZ is not as clear. TAZ could also undergo similar regulation because of its conserved phosphorylation sites with YAP at the 14-3-3 binding motif. However, our data indicate that cysteine S-glutathionylated TAZ fails to undergo Lats1/2-induced phosphorylation at the Ser-89 residue despite potentially high Mst1/2 activity (Fig. 2D). Therefore, we assume that TAZ S-glutathionylation could counteract Hippo-dependent phosphorylation and deactivation and thereby partially circumvent Mst1/2-dependent H2O2-induced apoptosis.
GPCR agonists play an important role in cell signaling to mediate cell growth, migration, and gene expression via elevation of physiological ROS (38–40). These different signaling pathways converge at downstream targets such as PKC that, upon activation, triggers PI3K and subsequent activation of the small GTPase Rac, which is required for the assembly of a functional NADPH oxidase (NOX) complex (14). Our data show that LPA-induced ROS promote TAZ S-glutathionylation and target gene expression in HEK 293T cells. These observations highlight an exclusive regulatory mechanism of TAZ in a physiological setting.
TAZ is highly expressed in the renal cortex, and TAZ knockout mice develop polycystic kidneys, implying a fundamental role in renal development (9, 10). Furthermore, our study indicates lower YAP levels in murine kidneys, suggestive of a critical role of TAZ in renal homeostasis. NAD(P)H oxidase is a multimeric enzyme complex composed of the rac1, gp91phpox, p67phcox, p47phox, and p22phox subunits. Several NOX isoforms have been documented with tissue-specific expression and activation (41). Our data indicate that renal I/R injury leads to TAZ S-glutathionylation and nuclear localization in vivo. This validates our hypothesis that TAZ and YAP could undergo differential regulation in response to I/R-induced oxidative stress.
We demonstrate that NOX4-derived H2O2 modulates TAZ activity in vitro, suggestive of a novel redox regulation of the Hippo pathway. Furthermore, it has to be noted that NOX4 is the major isoform expressed in the kidney and constitutively generates H2O2 (23). LPA receptors (LPA1, LPA2, and LPA3) are expressed in both renal cortical and medullary regions (42). At lower concentrations, LPA initiates proliferative pathways. However, excess LPA precursors have been observed upon I/R injury, leading to proinflammatory signaling and tissue damage (39). Basal TAZ S-glutathionylation observed in the kidney could be modulated by NOX4-derived ROS under physiological conditions. Increased TAZ S-glutathionylation following I/R injury could indicate that oxidative stress stabilizes TAZ from degradation, leading to enhanced CTGF expression, and may contribute to tissue repair or renal fibrosis.
In summary, we have demonstrated a unique regulatory mechanism by which TAZ undergoes S-glutathionylation, leading to enhanced stability and function. Because ROS are important signal transducers in both physiological and pathological mechanisms, TAZ could function as a redox sensor of the Hippo pathway (Fig. 5H). However, further studies are needed to determine the redox activation of TAZ under various pathological conditions of renal fibrosis, inflammation, and cancer for effective therapeutic strategies.
Author Contributions
R. K. G., B. S., and T. B. conceived the idea for the project, analyzed the results, and wrote most of the paper. R. K. G., B. W., M. Jain, M. P. B., M. H. A., and M. M. R. conducted most of the experiments. M. Johnsen conducted the in vivo I/R experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Astrid Schauss and Dr. Christian Jungst for technical assistance with two-photon imaging. We thank Dr. Olek Krut and Dr. Raja Ganesan for the NOX expression plasmids.
The authors declare that they have no conflicts of interest with the contents of this article.
- TAZ
- transcriptional co-activator with PDZ-binding motif
- YAP
- Yes-associated protein
- TEAD
- TEA domain transcription factor
- GPCR
- G protein-coupled receptor
- LPA
- lysophosphatidic acid
- ROS
- reactive oxygen species
- NOX
- NAD(P)H oxidase
- qPCR
- quantitative PCR
- CTGF
- connective tissue growth factor
- DHE
- dihydroethidine
- I/R
- ischemia/reperfusion
- IP
- immunoprecipitation.
References
- 1. Piccolo S., Dupont S., and Cordenonsi M. (2014) The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 94, 1287–1312 [DOI] [PubMed] [Google Scholar]
- 2. Zhang H., Liu C. Y., Zha Z. Y., Zhao B., Yao J., Zhao S., Xiong Y., Lei Q. Y., and Guan K. L. (2009) TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 284, 13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu S., Liu Y., Zheng Y., Dong J., and Pan D. (2008) The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell 14, 388–398 [DOI] [PubMed] [Google Scholar]
- 4. Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., Lai Z. C., and Guan K. L. (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu F. X., Zhao B., Panupinthu N., Jewell J. L., Lian I., Wang L. H., Zhao J., Yuan H., Tumaneng K., Li H., Fu X. D., Mills G. B., and Guan K. L. (2012) Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lei Q. Y., Zhang H., Zhao B., Zha Z. Y., Bai F., Pei X. H., Zhao S., Xiong Y., and Guan K. L. (2008) TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the Hippo pathway. Mol. Cell. Biol. 28, 2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Su D., Gaffrey M. J., Guo J., Hatchell K. E., Chu R. K., Clauss T. R., Aldrich J. T., Wu S., Purvine S., Camp D. G., Smith R. D., Thrall B. D., and Qian W. J. (2014) Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic. Biol. Med. 67, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morin-Kensicki E. M., Boone B. N., Howell M., Stonebraker J. R., Teed J., Alb J. G., Magnuson T. R., O'Neal W., and Milgram S. L. (2006) Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell. Biol. 26, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hossain Z., Ali S. M., Ko H. L., Xu J., Ng C. P., Guo K., Qi Z., Ponniah S., Hong W., and Hunziker W. (2007) Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc. Natl. Acad. Sci. U.S.A. 104, 1631–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tian Y., Kolb R., Hong J. H., Carroll J., Li D., You J., Bronson R., Yaffe M. B., Zhou J., and Benjamin T. (2007) TAZ promotes PC2 degradation through a SCFβ-Trcp E3 ligase complex. Mol. Cell. Biol. 27, 6383–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohli P., Bartram M. P., Habbig S., Pahmeyer C., Lamkemeyer T., Benzing T., Schermer B., and Rinschen M. M. (2014) Label-free quantitative proteomic analysis of the YAP/TAZ interactome. Am. J. Physiol. Cell Physiol. 306, C805–C818 [DOI] [PubMed] [Google Scholar]
- 12. Jones D. P. (2008) Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 295, C849–C868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miseta A., and Csutora P. (2000) Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evolution 17, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 14. Bae Y. S., Oh H., Rhee S. G., and Yoo Y. D. (2011) Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32, 491–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hawkins B. J., Irrinki K. M., Mallilankaraman K., Lien Y. C., Wang Y., Bhanumathy C. D., Subbiah R., Ritchie M. F., Soboloff J., Baba Y., Kurosaki T., Joseph S. K., Gill D. L., and Madesh M. (2010) S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 190, 391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reynaert N. L., van der Vliet A., Guala A. S., McGovern T., Hristova M., Pantano C., Heintz N. H., Heim J., Ho Y. S., Matthews D. E., Wouters E. F., and Janssen-Heininger Y. M. (2006) Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. U.S.A. 103, 13086–13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Velu C. S., Niture S. K., Doneanu C. E., Pattabiraman N., and Srivenugopal K. S. (2007) Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 46, 7765–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hall A. M., Crawford C., Unwin R. J., Duchen M. R., and Peppiatt-Wildman C. M. (2011) Multiphoton imaging of the functioning kidney. J. Am. Soc. Nephrol. 22, 1297–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saunders J. A., Rogers L. C., Klomsiri C., Poole L. B., and Daniel L. W. (2010) Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic. Biol. Med. 49, 2058–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin C. C., Lin C. E., Lin Y. C., Ju T. K., Huang Y. L., Lee M. S., Chen J. H., and Lee H. (2013) Lysophosphatidic acid induces reactive oxygen species generation by activating protein kinase C in PC-3 human prostate cancer cells. Biochem. Biophys. Res. Commun. 440, 564–569 [DOI] [PubMed] [Google Scholar]
- 21. Habbig S., Bartram M. P., Müller R. U., Schwarz R., Andriopoulos N., Chen S., Sägmüller J. G., Hoehne M., Burst V., Liebau M. C., Reinhardt H. C., Benzing T., and Schermer B. (2011) NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 193, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang F., Liu G. S., Dusting G. J., and Chan E. C. (2014) NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Babelova A., Avaniadi D., Jung O., Fork C., Beckmann J., Kosowski J., Weissmann N., Anilkumar N., Shah A. M., Schaefer L., Schroder K., and Brandes R. P. (2012) Role of Nox4 in murine models of kidney disease. Free Radic. Biol. Med. 53, 842–853 [DOI] [PubMed] [Google Scholar]
- 24. Johnson R., and Halder G. (2014) The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Disc. 13, 63–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grek C. L., Zhang J., Manevich Y., Townsend D. M., and Tew K. D. (2013) Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 288, 26497–26504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reginensi A., Scott R. P., Gregorieff A., Bagherie-Lachidan M., Chung C., Lim D. S., Pawson T., Wrana J., and McNeill H. (2013) Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 9, e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Granger D. N., and Kvietys P. R. (2015) Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 6, 524–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Happé H., de Heer E., and Peters D. J. (2011) Polycystic kidney disease: the complexity of planar cell polarity and signaling during tissue regeneration and cyst formation. Biochim. Biophys. Acta 1812, 1249–1255 [DOI] [PubMed] [Google Scholar]
- 29. Huang W., Lv X., Liu C., Zha Z., Zhang H., Jiang Y., Xiong Y., Lei Q. Y., and Guan K. L. (2012) The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J. Biol. Chem. 287, 26245–26253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Plouffe S. W., Hong A. W., and Guan K. L. (2015) Disease implications of the Hippo/YAP pathway. Trends Mol. Med. 21, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shao D., Zhai P., Del Re D. P., Sciarretta S., Yabuta N., Nojima H., Lim D. S., Pan D., and Sadoshima J. (2014) A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 5, 3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanai F., Marignani P. A., Sarbassova D., Yagi R., Hall R. A., Donowitz M., Hisaminato A., Fujiwara T., Ito Y., Cantley L. C., and Yaffe M. B. (2000) TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hall A. M., Rhodes G. J., Sandoval R. M., Corridon P. R., and Molitoris B. A. (2013) In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 83, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan S. W., Lim C. J., Chong Y. F., Pobbati A. V., Huang C., and Hong W. (2011) Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J. Biol. Chem. 286, 7018–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Praskova M., Xia F., and Avruch J. (2008) MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lehtinen M. K., Yuan Z., Boag P. R., Yang Y., Villén J., Becker E. B., DiBacco S., de la Iglesia N., Gygi S., Blackwell T. K., and Bonni A. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125, 987–1001 [DOI] [PubMed] [Google Scholar]
- 37. Bao Y., Nakagawa K., Yang Z., Ikeda M., Withanage K., Ishigami-Yuasa M., Okuno Y., Hata S., Nishina H., and Hata Y. (2011) A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J. Biochem. 150, 199–208 [DOI] [PubMed] [Google Scholar]
- 38. Dorsam R. T., and Gutkind J. S. (2007) G-protein-coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94 [DOI] [PubMed] [Google Scholar]
- 39. Goetzl E. J., Graeler M., Huang M. C., and Shankar G. (2002) Lysophospholipid growth factors and their G protein-coupled receptors in immunity, coronary artery disease, and cancer. The Scientific World Journal 2, 324–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang M., Zhong W. W., Srivastava N., Slavin A., Yang J., Hoey T., and An S. (2005) G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the β-catenin pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 42. Okusa M. D., Ye H., Huang L., Sigismund L., Macdonald T., and Lynch K. R. (2003) Selective blockade of lysophosphatidic acid LPA3 receptors reduces murine renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 285, F565–F574 [DOI] [PubMed] [Google Scholar]