

Abstract
The fatty aldehyde dehydrogenase (FALDH) ALDH3A2 is the causative gene of Sjögren Larsson syndrome (SLS). To date, the molecular mechanism underlying the symptoms characterizing SLS has been poorly understood. Using Aldh3a2−/− mice, we found here that Aldh3a2 was the major FALDH active in undifferentiated keratinocytes. Long-chain base metabolism was greatly impaired in Aldh3a2−/− keratinocytes. Phenotypically, the intercellular spaces were widened in the basal layer of the Aldh3a2−/− epidermis due to hyperproliferation of keratinocytes. Furthermore, oxidative stress-induced genes were up-regulated in Aldh3a2−/− keratinocytes. Upon keratinocyte differentiation, the activity of another FALDH, Aldh3b2, surpassed that of Aldh3a2. As a result, Aldh3a2−/− mice were indistinguishable from wild-type mice in terms of their whole epidermis FALDH activity, and their skin barrier function was uncompromised under normal conditions. However, perturbation of the stratum corneum caused increased transepidermal water loss and delayed barrier recovery in Aldh3a2−/− mice. In conclusion, Aldh3a2−/− mice replicated some aspects of SLS symptoms, especially at the basal layer of the epidermis. Our results suggest that hyperproliferation of keratinocytes via oxidative stress responses may partly contribute to the ichthyosis symptoms of SLS.
Keywords: ceramide, epidermis, gene knockout, genetic disease, keratinocyte, lipid, lipid ether, lipid metabolism, oxidative stress, sphingolipid
Introduction
Sjögren-Larsson syndrome (SLS)2 is a hereditary neurocutaneous disorder caused by mutations in the fatty aldehyde dehydrogenase (FALDH) gene, ALDH3A2. The major symptoms of SLS are mental retardation, spastic di- or tetraplegia, and ichthyosis, with crystalline macular dystrophy sometimes comorbid (1). ALDH3A2 catalyzes the conversion of fatty aldehydes with medium-chain (MC) to very-long-chain fatty acids (FA) (MC, C5–C10; long-chain (LC), C11–C20; and very-long-chain, ≥C21), with the most preferred substrates being C16 and C18 aldehydes (2, 3). To date, more than 70 mutations have been found in the ALDH3A2 gene of SLS patients, and most of them cause >90% reduction in enzyme activity (4). Because aldehyde molecules are reactive and toxic in general, it is considered that accumulated fatty aldehydes cause the SLS pathology by reacting with certain important proteins in the nervous system and epidermis and compromising their functions. Furthermore, the literature lacks a detailed description of the characteristics of Aldh3a2 knock-out mice.
The skin symptom ichthyosis is characterized by dry, thickened, and scaly skin, often likened to fish scales, and is caused by a skin permeability barrier defect related to multilayered lipids (lipid lamellae) in the epidermis. The epidermis is composed of four cell layers, the stratum basale, stratum spinosum, stratum granulosum, and the stratum corneum, the last of which is the site of these lipid lamellae, and accordingly has the most important role in skin barrier formation (5, 6). Keratinocytes proliferate in the stratum basale and migrate outward, differentiating into cell layer-specific cell types. The major lipid components of lipid lamellae are ceramides, cholesterol, and FAs, with ceramides being the most abundant (5, 6). A variety of ceramide species exist in the epidermis. Among them, acylceramides are a class of epidermis-specific ceramides that play an essential role in skin barrier formation (7, 8). Ceramides are normally composed of a long-chain base (LCB) and a FA (9). However, acylceramides contain an additional hydrophobic chain (linoleic acid), which is esterified with the hydroxylated ω-carbon of the FA. In the epidermis of SLS patients, some ceramide species, including the acylceramide EOS (a combination of an esterified ω-hydroxy FA and the LCB sphingosine), are greatly reduced (10).
Several metabolic pathways generate the substrates of ALDH3A2, fatty aldehydes. These include metabolic pathways of leukotriene B4, diet-derived phytol, plasmalogens, and fatty alcohols (11–15). Furthermore, we recently revealed that metabolism of LCBs also generates fatty aldehydes, i.e. hexadecanal (C16:0 aldehyde) from dihydrosphingosine (DHS) and trans-2-hexadecenal (C16:1 aldehyde) from sphingosine (16–18). However, it is still unclear which fatty aldehyde or which metabolic pathway mainly contributes to the pathogenesis of SLS.
ALDH3A2 belongs to the aldehyde dehydrogenase (ALDH) family, which is well represented in mammals (19 genes in human and 21 in mouse) (19, 20). The mammalian ALDH family is divided into 11 subfamilies (ALDH1–9, -16, and -18). Mouse Aldh3 in particular includes five subfamily members (20), Aldh3a2, Aldh3b1, Aldh3b2, and Aldh3b3, which exhibit high activities toward LC aldehydes, and Aldh3a1, which shows only weak activity toward them but high activity toward MC aldehydes (2). Human ALDH3, on the other hand, contains four ALDH3 subfamily members (ALDH3A1, ALDH3A2, ALDH3B1, and ALDH3B2, with no Aldh3b3 homolog). As their mouse homologs do, ALDH3A1, ALDH3A2, and ALDH3B1 exhibit high activity toward MC or LC fatty aldehydes (21–23). However, human ALDH3B2 lacks 94 N-terminal amino acids present in the corresponding mouse Aldh3b2 and has no enzyme activity, suggesting that human ALDH3B2 is a pseudogene (19).
In the present study, to gain insight into the molecular mechanism behind SLS pathology, especially as it relates to ichthyosis symptoms, we analyzed Aldh3a2 KO mice. We revealed the contribution of Aldh3a2 to total FALDH activities as well as mRNA expression profiles of Aldh3 subfamily members in various tissues. Although Aldh3a2 is the major FALDH in undifferentiated keratinocytes, Aldh3b2 mRNA levels began to surpass Aldh3a2 mRNA levels upon differentiation. Accordingly, gene disruption of Aldh3a2 had little effect on skin barrier formation and ceramide composition. However, Aldh3a2 KO mice did exhibit broadened intercellular spaces in the stratum basale and delayed skin barrier recovery. Furthermore, proliferation and oxidative stress responses were enhanced in Aldh3a2 keratinocytes. Thus, our findings give a clue to understanding the molecular mechanism behind the early-stage pathogenesis of the SLS symptom ichthyosis.
Experimental Procedures
Generation of Aldh3a2−/− Mice
Aldh3a2 gene trap mice (C57BL/6N background), in which exon 4 of the Aldh3a2 gene is flanked by two loxP sequences, were obtained from the European Mouse Mutant Archive. Aldh3a2 heterozygous KO mice (Aldh3a2+/−) were generated by crossing Aldh3a2 gene trap mice with CAG-Cre transgenic mice (24) obtained from the Riken BioResource Center (BRC) through the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). The Aldh3a2+/− mice were maintained by repeated back-crossing with C57BL/6J mice. Aldh3a2 homozygous KO mice (Aldh3a2−/−) were generated by intercrossing Aldh3a2+/− mice born from the second or higher generations of back-crossing. Genotyping was conducted by PCR using genomic DNAs and primers (p1 and p2 for detection of wild-type alleles; p3 and p2 for detection of KO alleles (Table 1)). Mice were kept at 23 ± 1 °C in a 12-h light/dark cycle with a standard chow diet (PicoLab Rodent Diet 20, LabDiet, St. Louis, MO; or CRF-1, Oriental Yeast Co., Ltd., Tokyo, Japan) and with water available ad libitum. The food intake of mice was measured, from the ages of 16 to 23 weeks, every 24 h for 7 days. Statistical analysis was performed by one-way analysis of variance using StatView (SAS Institute, Cary, NC). The animal experiments performed in this study were approved by the institutional animal care and use committees of Hokkaido University and Fujita Health University.
TABLE 1.
Primers used in this study
| Primera | Sequence |
|---|---|
| p1 | 5′-TGGAAGCTGCTGCCAAGCATCTGAC-3′ |
| p2 | 5′-GTGAACACAATGCCCTTCCAACTGAG-3′ |
| p3 | 5′-CACTGCATTCTAGTTGTGGTTTGTC-3′ |
| Aldh3a1-F | 5′-CGGTGATGCCCATTGTGTGTGTTCG-3′ |
| Aldh3a1-R | 5′-TTCTTCATTCCGCAGAGACCTCACC-3′ |
| Aldh3a2-F | 5′-TTCTCGTAACAATAAGCTCATCAAACG-3′ |
| Aldh3a2-R | 5′-CAGCATCCCCAGCCTTCCTTTGTTG-3′ |
| Aldh3b1-F | 5′-GCTGTATGCCTTCTCCAAGAGAAGC-3′ |
| Aldh3b1-R | 5′-GCAGCTGCAGCACCTCTCCTCCATGG-3′ |
| Aldh3b2-F | 5′-TGAGTTCATCAACCGGCGGGAGAAGC-3′ |
| Aldh3b2-R | 5′-GTTGTTGGTTCCAGGGACCATAAGG-3′ |
| Aldh3b3-F | 5′-CTTTATGCCTATTCCAACAACGCAG-3′ |
| Aldh3b3-R | 5′-GGGTGCAGCTCTCAGAGCCGATAGC-3′ |
| Ki67-F | 5′-CCTCAGCTCCTGCCTGTTTGGAAGG-3′ |
| Ki67-R | 5′-GTTCCTTTCCAAGGGACTTTCCTGG-3′ |
| Hmox1-F | 5′-ATGACACCTGAGGTCAAGCACAGGG-3′ |
| Hmox1-R | 5′-TTACATGGCATAAATTCCCACTGCC-3′ |
| Sod1-F | 5′-TTAACTGAAGGCCAGCATGGGTTCC-3′ |
| Sod1-R | 5′-TCTTTGTACTTTCTTCATTTCCACC-3′ |
| Gclc-F | 5′-GAGAACATGAAAGTGGCCCAGAAGC-3′ |
| Gclc-R | 5′-GTCCACATCAACTTCCATGTTCTCC-3′ |
| Gclm-F | 5′-AGTTGGAGCAGCTGTATCAGTGGGC-3′ |
| Gclm-R | 5′-ACGATGACCGAGTACCTCAGCAGCC-3′ |
| ALDH3A1-F | 5′-CCTGGCCCTCTACATGTTCTCCAGC-3′ |
| ALDH3A1-R | 5′-TGCTGGGTCATCTTGGCCGGGCTCG-3′ |
| ALDH3A2-F | 5′-AATGACGTCATTATGCACTTCACGC-3′ |
| ALDH3A2-R | 5′-AAGCACAGCGGCTACAATACCCAGG-3′ |
| ALDH3B1-F | 5′-GGTCAAGCGGGTGCTGACCCAGACC-3′ |
| ALDH3B1-R | 5′-TGTGCAGCTGCAGCCTTGGGCCTCC-3′ |
| Gapdh/GAPDH-F | 5′-GAACGGGAAGCTCACTGGCATGGCC-3′ |
| Gapdh/GAPDH-R | 5′-TGTCATACCAGGAAATGAGCTTGAC-3′ |
a F, forward; R, reverse.
Cell Culture
Mouse primary keratinocytes were isolated as described elsewhere (25). Briefly, skins prepared from P0 pups were treated with 3 ml of 5 mg/ml dispase (Thermo Fisher Scientific, Waltham, MA) for 24 h at 4 °C. Epidermis was then separated from dermis and washed with CnT-Prime epithelial culture medium (CELLnTEC Advanced Cell Systems AG, Bern, Switzerland). Keratinocytes were isolated by incubation with 500 μl of 1× TrypLETM Select (Thermo Fisher Scientific) for 15 min at room temperature followed by rubbing the basal side of the epidermis. Keratinocytes were then washed with CnT-Prime epithelial culture medium, collected by centrifugation, suspended in the medium, and seeded at a cell density of 5 × 104 cells/cm2. Differentiation was induced by replacing the medium with CnT-Prime 3D barrier culture medium (CELLnTEC) at subconfluency. The medium was freshly replaced every 3 days.
RT-PCR
Total RNAs were isolated from the various tissues and keratinocytes of mice using the NucleoSpin RNA II kit (Machery-Nagel, Dueren, Germany), according to the manufacturer's instructions. RT-PCR was performed using the One-Step PrimeScript RT-PCR Kit II (Takara Bio, Shiga, Japan) and the following primers: for Aldh3a2, Aldh3a2-F and Aldh3a2-R; for Gapdh, Gapdh/GAPDH-F and Gapdh/GAPDH-R) (Table 1).
Real-time quantitative PCR was performed using the One-Step SYBR PrimeScript RT-PCR Kit II (Takara Bio) on a CFX96 Touch real-time PCR detection system (Bio-Rad) according to the manufacturer's manual. Forward (F) and reverse (R) primers for the respective genes were used (Table 1). The gene amplification efficiencies of the primers used were all >90%. The mRNA levels were normalized with those of Gapdh. The reaction was conducted by incubating the samples at 42 °C for 5 min and at 95 °C for 10 s followed by 40 cycles of 95 °C for 5 s, 63 °C for 30 s, and 72 °C for 30 s.
Skin Permeability Assays
Transepidermal water loss (TEWL) was measured as described previously (26) using an AS-VT100RS evaporimeter (Asahi Biomed, Yokohama, Japan). Dye exclusion assays were performed using toluidine blue essentially as described elsewhere (26, 27). Newborn pups at P0 were incubated with methanol for 5 min and washed in PBS, after which they were incubated with 0.1% toluidine blue for 24 h. Before and after staining, pups were washed with PBS and photographed by a digital camera.
For acetone treatment, back of the mice at P0 was wiped with absorbent cotton swabs containing acetone or saline. The fluid was removed with absorbent cotton swabs and the skin dried for 1 min. TEWL was measured at 0, 1, and 3 h after drying.
In Vitro FALDH Assay
Total cell lysates were prepared from various tissues of mice by homogenizing the tissues using a homogenizer in buffer A (50 mm HEPES/NaOH (pH 7.4), 150 mm NaCl, 10% glycerol, 1× protease inhibitor mixture (Complete, EDTA-free, Roche Diagnostics), 1 mm PMSF, and 1 mm DTT) followed by removal of cell debris by centrifugation (200 × g for 3 min at 4 °C). In vitro FALDH assays were conducted by incubating total cell lysates (1–30 μg) with 500 μm NAD+ and 100 μm hexadecanal (C16:0) (28), trans-2-hexadecenal (C16:1; Avanti Polar Lipids, Alabaster, AL), or octanal (C8:0; Wako Pure Chemical Industries) in reaction buffer (50 mm Tris/HCl (pH 8.5), 150 mm NaCl, 10% glycerol, and 0.1% Triton X-100) at 37 °C for 15–30 min. The reaction was monitored by measuring the fluorescence of the reaction product NADH (excitation wavelength, 356 nm; emission wavelength, 460 nm) using an Infinite M200 monochromator (Tecan, Männedorf, Switzerland).
H&E Staining
After P0 pups were subjected to perfusion fixation with 3.7% formaldehyde in saline, and skins were isolated and fixed with 3.7% formaldehyde in PBS at 4 °C for >48 h. Fixed skins were then subjected to dehydration, paraffin embedding, preparation of sections, deparaffinization, and staining with hematoxylin and then eosin using an automatic staining system (Tissue-Tek DRS 2000, Sakura, Torrance, CA) as described previously (26). Bright field images were captured using a Leika DM5000B microscope equipped with a DFC295 digital color camera (Leica Microsystems, Wetzlar, Germany).
Electron Microscopy
Skins isolated from P0 pups were fixed with 5% glutaraldehyde in 0.1 m sodium cacodylate buffer (pH 7.4) for >72 h at 4 °C and postfixed with 1% OsO4 in 0.05 m sodium cacodylate buffer (pH 7.4) for 1 h and in 0.5% RuO4 (Electron Microscopy Sciences, Hatfield, PA) for 30 min followed by overnight immersion in 0.1 m sodium cacodylate buffer (pH 7.4). The fixed skins were then dehydrated in graded ethanol and propylene oxide and subsequently embedded in Epon 812 resin (TAAB Laboratories, Berkshire, United Kingdom) followed by incubation for >4 days at 70 °C. Ultra-thin sections (70 nm) were prepared using a Leica EM UC7 Ultramicrotome (Leica Microsystems). The skin sections obtained were then stained with uranyl acetate and lead citrate followed by observation and photography of images using a CCD camera affixed to a JEM-1400 transmission electron microscope (Jeol, Tokyo, Japan).
Lipid Labeling Assay
[3H]DHS labeling assays were performed as described previously (16). For deuterium (d7)-sphingosine labeling assays, primary keratinocytes were treated with 1 μm d7-sphingosine (Avanti Polar Lipids) at 37 °C for 4 h. After washing with PBS, cells were suspended in PBS, collected by scraping, and cell numbers were counted using a hemocytometer. Samples consisting of equal cell numbers were then subjected to lipid extractions and LC/MS/MS analysis.
LC/MS/MS Analysis
To measure epidermal ceramide/acylceramide levels, skins isolated from P0 pups were treated with 5 mg/ml dispase (Thermo Fisher Scientific) in PBS at 4 °C for 24 h, and epidermis and dermis were separated by manipulation under a stereomicroscope. The resulting epidermis (20–30 mg) was suspended in 600 μl of buffer A and homogenized using a homogenizer. After the addition of 1 ml of buffer A and 6 ml of CHCl3/MeOH (1:2, v/v), samples were mixed vigorously for 3 min, incubated at 50 °C for 10 min, sonicated for 1 min using a bath sonicator, mixed vigorously again for 3 min, and treated with 2 ml of CHCl3 and 2 ml of 1% KCl. After centrifugation (2600 × g at room temperature for 3 min), the organic phase was recovered. The aqueous phase was treated with 4 ml of CHCl3, mixed, and centrifuged. The resulting organic phase was pooled with the previous one, dried, and stored at −30 °C. Lipids were dissolved in 50 μl of CHCl3/MeOH (1:2, v/v) for LC/MS/MS analysis.
Fatty aldehydes were derivatized for detection as described previously (29) with minor modifications. Lipids prepared from epidermis and liver were dissolved in methanol. In amounts corresponding to 8 mg of epidermis or 4 mg of liver, lipids in 46 μl of methanol were treated with 2 μl of 1.69 mm 2-diphenylacetyl-1,3-indandione-1-hydrazone (Sigma) in acetonitrile and 2 μl of 250 mm N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Sigma) in ethanolic 3% pyridine, incubated at 60 °C for 30 min, and subjected to LC/MS/MS analysis.
To measure the levels of ceramide, phosphatidylcholine (PC), phosphatidylethanolamine (PE), plasmanyl/plasmenylcholine (PlsC), and plasmanyl/plasmenylethanolamine (PlsE) species in keratinocytes, the cells were suspended in 200 μl of PBS, and lipids were extracted by the successive addition and mixing of 750 μl of CHCl3/MeOH/HCl (100:200:1, v/v), 250 μl of CHCl3, and 250 μl of 1% KCl. After centrifugation (2600 × g at room temperature for 3 min), the organic phase was collected, dried, and suspended in 150 μl of CHCl3/MeOH (1:2, v/v).
LC/MS/MS analyses were performed as described previously (8). Lipids were resolved by ultra-performance LC on a reverse-phase column (ACQUITY UPLC BEH C18 column, length 150 mm; Waters, Milford, MA) coupled with electrospray ionization tandem triple quadrupole MS (Xevo TQ-S, Waters) and detected by multiple reaction monitoring by selecting the specific m/z at quadrupole mass filters Q1 and Q3 (Tables 2–4). Data were analyzed and quantified using MassLynx software.
TABLE 2.
Selected m/z values for ceramide species in LC/MS/MS analysis
| FA | Precursor ions (Q1) |
Product ion (Q3) | |
|---|---|---|---|
| [M-H2O+H]+ | [M+H]+ | ||
| C16:1 | 518.2 | 536.2 | 264.2 |
| C16:0 | 520.2 | 538.2 | 264.2 |
| C18:1 | 546.2 | 564.2 | 264.2 |
| C18:0 | 548.2 | 566.2 | 264.2 |
| C20:1 | 574.2 | 592.2 | 264.2 |
| C20:0 | 576.2 | 594.2 | 264.2 |
| C22:1 | 602.2 | 620.2 | 264.2 |
| C22:0 | 604.2 | 622.2 | 264.2 |
| C24:1 | 630.2 | 648.2 | 264.2 |
| C24:0 | 632.2 | 650.2 | 264.2 |
| C26:1 | 658.2 | 676.2 | 264.2 |
| C26:0 | 660.2 | 678.2 | 264.2 |
| C30:0a | 716.2 | 734.2 | 264.2 |
| d7-C16:1 | 525.2 | 543.2 | 271.2 |
| d7-C16:0 | 527.2 | 545.2 | 271.2 |
| d7-C18:1 | 553.2 | 571.2 | 271.2 |
| d7-C18:0 | 555.2 | 573.2 | 271.2 |
| d7-C20:1 | 581.2 | 599.2 | 271.2 |
| d7-C20:0 | 583.2 | 601.2 | 271.2 |
| d7-C22:1 | 609.2 | 627.2 | 271.2 |
| d7-C22:0 | 611.2 | 629.2 | 271.2 |
| d7-C24:1 | 637.2 | 655.2 | 271.2 |
| d7-C24:0 | 639.2 | 657.2 | 271.2 |
| d7-C26:1 | 665.2 | 683.2 | 271.2 |
| d7-C26:0 | 667.2 | 685.2 | 271.2 |
| C26:1 EOS | 936.9 | 954.9 | 264.2 |
| C26:0 EOS | 938.9 | 956.9 | 264.2 |
| C28:1 EOS | 964.9 | 982.9 | 264.2 |
| C28:0 EOS | 966.9 | 984.9 | 264.2 |
| C30:1 EOS | 992.9 | 1010.9 | 264.2 |
| C30:0 EOSb | 994.9 | 1012.9 | 264.2 |
| C32:1 EOS | 1020.9 | 1038.9 | 264.2 |
| C32:0 EOS | 1022.9 | 1040.9 | 264.2 |
| C34:1 EOS | 1048.9 | 1066.9 | 264.2 |
| C34:0 EOS | 1050.9 | 1068.9 | 264.2 |
| C36:1 EOS | 1076.9 | 1094.9 | 264.2 |
| C36:0 EOS | 1078.9 | 1096.9 | 264.2 |
| C38:1 EOS | 1104.9 | 1122.9 | 264.2 |
| C38:0 EOS | 1106.9 | 1124.9 | 264.2 |
a Ceramide (C30:0, Matreya, State College, PA) was used as an external standard.
b EOS (C30:0, Matreya) was used as an external standard.
TABLE 3.
Selected m/z values for PC, PE, and fatty aldehyde species in LC/MS/MS analysis
| Lipid species | Precursor ion (Q1)a | Product ion (Q3) |
|---|---|---|
| C16:1/C16:1 PC | 774.4 | 253.0 |
| C16:0/C16:1 PC | 776.4 | 255.0 |
| C16:0/C16:0 PC | 778.4 | 255.0 |
| C16:1/C18:1 PC | 802.4 | 253.0 |
| C16:1/C18:0 PC | 804.4 | 253.0 |
| C16:0/C18:1 PCb | 804.4 | 255.0 |
| C16:0/C18:0 PC | 806.4 | 255.0 |
| C18:1/C18:1 PC | 830.4 | 281.0 |
| C18:1/C18:0 PC | 832.4 | 283.0 |
| C18:0/C18:0 PC | 834.4 | 283.0 |
| d7-C16:0/C16:1 PC | 783.4 | 262.0 |
| d7-C16:0/C16:0 PC | 785.4 | 262.0 |
| d7-C16:0/C18:2 PC | 809.4 | 262.0 |
| d7-C16:0/C18:1 PC | 811.4 | 262.0 |
| d7-C16:0/C18:0 PC | 813.4 | 262.0 |
| d7-C16:0/C20:5 PC | 831.4 | 262.0 |
| d7-C16:0/C20:4 PC | 833.4 | 262.0 |
| d7-C16:0/C22:6 PC | 857.4 | 262.0 |
| C16:1/C16:1 PE | 686.5 | 253.0 |
| C16:0/C16:1 PE | 688.5 | 255.0 |
| C16:0/C16:0 PE | 690.5 | 255.0 |
| C16:1/C18:1 PE | 714.5 | 253.0 |
| C16:1/C18:0 PE | 716.5 | 253.0 |
| C16:0/C18:1 PE | 716.5 | 255.0 |
| C16:0/C18:0 PE | 718.5 | 255.0 |
| C18:1/C18:1 PEc | 742.5 | 281.0 |
| C18:1/C18:0 PE | 744.5 | 283.0 |
| C18:0/C18:0 PE | 746.5 | 283.0 |
| d7-C16:0/C16:1 PE | 695.5 | 262.0 |
| d7-C16:0/C16:0 PE | 697.5 | 262.0 |
| d7-C16:0/C18:2 PE | 721.5 | 262.0 |
| d7-C16:0/C18:1 PE | 723.5 | 262.0 |
| d7-C16:0/C18:0 PE | 725.5 | 262.0 |
| d7-C16:0/C20:5 PE | 743.5 | 262.0 |
| d7-C16:0/C20:4 PE | 745.5 | 262.0 |
| d7-C16:0/C22:6 PE | 769.5 | 262.0 |
| Trans-2-hexadecenald,e, | 573.3 | 249.0 |
| Hexadecanald,e | 575.4 | 167.1 |
a Precursor ion is [M+HCOOH-H]− (PC) or [M-H]− (PE and fatty aldehydes).
b PC (C16:0/C18:1, Avanti Polar Lipids) was used as an external standard.
c PE (C18:1/C18:1, Avanti Polar Lipids) was used as an external standard.
d 2-Diphenylacetyl-1,3-indandione-1-hydrazone derivatives.
e Trans-2-hexadecenal and hexadecanal were used as external standards.
TABLE 4.
Selected m/z values for PlsC and PlsE species in LC/MS/MS analysis
| d7-Alcohol/FA | Precursor ion (Q1)a | Product ion (Q3) |
|---|---|---|
| d7-C16:1/C16:1 PlsC | 767.4 | 253.0 |
| d7-C16:0/C16:1 PlsC | 769.4 | 253.0 |
| d7-C16:1/C16:0 PlsC | 769.4 | 255.0 |
| d7-C16:0/C16:0 PlsC | 771.4 | 255.0 |
| d7-C16:1/C18:1 PlsC | 795.4 | 281.0 |
| d7-C16:0/C18:1 PlsC | 797.4 | 281.0 |
| d7-C16:1/C18:0 PlsC | 797.4 | 283.0 |
| d7-C16:0/C18:0 PlsC | 799.4 | 283.0 |
| C18:1/C18:1 PlsCb | 816.4 | 281.0 |
| d7-C16:1/C16:1 PlsE | 679.3 | 253.0 |
| d7-C16:0/C16:1 PlsE | 681.3 | 253.0 |
| d7-C16:1/C16:0 PlsE | 681.3 | 255.0 |
| d7-C16:0/C16:0 PlsE | 683.3 | 255.0 |
| d7-C16:1/C18:1 PlsE | 707.3 | 281.0 |
| d7-C16:0/C18:1 PlsE | 709.3 | 281.0 |
| d7-C16:1/C18:0 PlsE | 709.3 | 283.0 |
| d7-C16:0/C18:0 PlsE | 711.3 | 283.0 |
| C18:1/C18:1 PlsEc | 728.3 | 281.0 |
a Precursor ion is [M+HCOOH-H]− (PlsC) or [M-H]− (PlsE).
b PlsC (C18:1/C18:1, Avanti Polar Lipids) was used as an external standard.
c PlsE (C18:1/C18:1, Avanti Polar Lipids) was used as an external standard.
[3H]Thymidine Uptake Assay
Primary keratinocytes were labeled with 0.2 mCi of [3H]thymidine (2.0 Ci/mmol; PerkinElmer Life Sciences) at 37 °C for 24 h. After the medium was collected, cells were washed with PBS and harvested with 1× SDS sample buffer (62.5 mm Tris-HCl (pH 6.8) and 2% SDS, 10% glycerol). Radioactivities associated with cells and medium were measured using a MicroBeta TriLux scintillation counter (PerkinElmer Life Sciences).
Immunoblotting
Immunoblotting was performed as described previously (2, 30) using anti-Aldh3a2 antiserum (1/5,000 dilution) or anti-calnexin antibody (H-70, 0.8 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) as a primary antibody, and HRP-conjugated anti-rabbit or anti-mouse IgG(ab′)2 fragments (each at 1:7,500 dilution; GE Healthcare Life Sciences) were used as a secondary antibody. Anti-Aldh3a2 antiserum was raised against mouse Aldh3a2 recombinant protein containing amino acid residues 1–449.
Results
Contribution of Aldh3a2 to Total FALDH Activity Differs among Tissues
To generate Aldh3a2 KO mice, Aldh3a2 gene trap mice (Aldh3a2gt/+) in which β-geo (the fusion of β-galactosidase and the neomycin phosphotransferase gene) was inserted upstream of Aldh3a2 exon 4, which was flanked by two loxP sequences (Fig. 1A), were crossed with CAG-Cre mice, which produce Cre recombinase in the whole body. The resulting Aldh3a2 heterozygous KO mice (Aldh3a2+/−) were used to generate Aldh3a2 homozygous KO mice (Aldh3a2−/−) by intercrossing. The genotypes of obtained mice were identified using genomic PCR (Fig. 1B). We also confirmed the disruption of Aldh3a2, using RT-PCR (Fig. 1C) and immunoblotting (Fig. 1D). The mice were fed with a standard chow diet throughout this study, and their food intake levels were measured. The food intake levels of wild-type mice gradually increased, probably because of habituation to their new environment (Fig. 1E). The food intake levels of Aldh3a2−/− mice were slightly higher than those of the wild-type mice. However, no significant differences in body weight were observed between the two groups (Fig. 1F).
FIGURE 1.
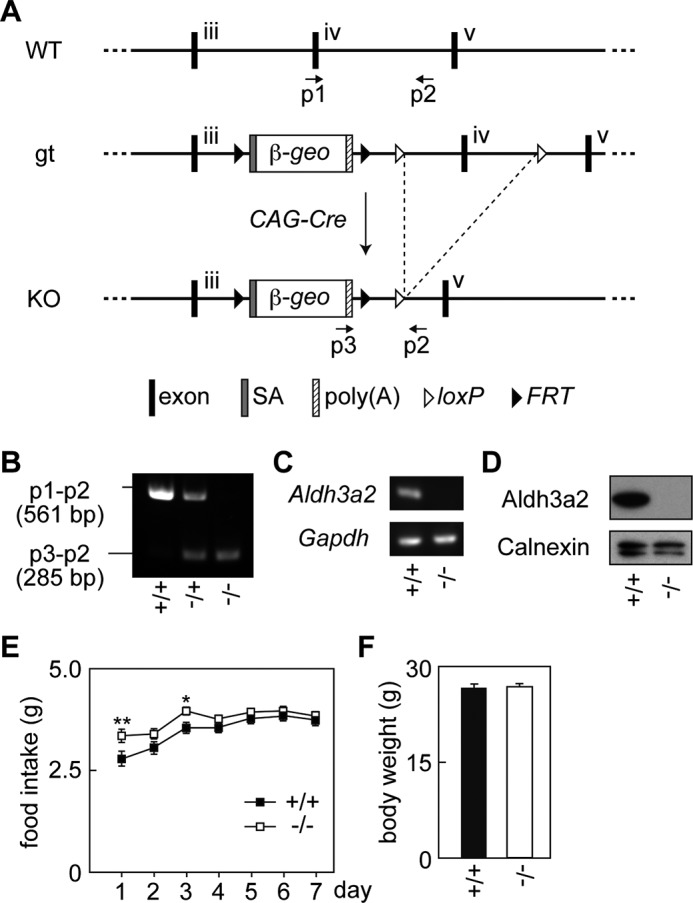
Generation of Aldh3a2 KO mice. A, schematic representation of the region surrounding exons 3 to 5 of the Aldh3a2 gene in WT, Aldh3a2 gene trap (gt), and Aldh3a2 KO mice. The positions of the primers (p1, p2, and p3) used for genomic PCR are denoted by arrows. SA, splice acceptor; poly(A), poly(A) signal; FRT, flippase recognition target. B, genomic DNAs prepared from the tails of Aldh3a2+/+, Aldh3a2+/−, and Aldh3a2−/− mice were subjected to PCR using primers p1, p2, and p3. The amplified fragments were separated by agarose gel electrophoresis followed by staining with ethidium bromide. C, total RNAs prepared from the epidermis of Aldh3a2+/+ and Aldh3a2−/− mice were subjected to RT-PCR using specific primers for Aldh3a2 and Gapdh. D, total lysates (20 μg) prepared from the livers of Aldh3a2+/+ and Aldh3a2−/− mice were separated by SDS-PAGE followed by immunoblotting using anti-Aldh3a2 antibody. Gapdh protein was detected as a loading control. E, food intake levels of wild-type and Aldh3a2 KO mice were measured every 24 h for 7 days. Values represent the means ± S.E. from 20 wild-type or 19 Aldh3a2 KO mice. Statistically significant differences are indicated (**, p < 0.01; *, p < 0.05; one-way analysis of variance). F, body weights of wild-type and Aldh3a2 KO mice were measured. Values represent the means ± S.E. from 20 wild-type or 19 Aldh3a2 KO mice.
We first measured FALDH activities toward two LC aldehydes, the DHS metabolite hexadecanal (C16:0) and the sphingosine metabolite trans-2-hexadecenal (C16:1) and the MC aldehyde octadecanal (C8:0), using various tissue lysates obtained from wild-type and Aldh3a2 KO mice. Both human ALDH3A2 and mouse Aldh3a2 proteins are active toward both LC and MC aldehydes, with higher activity toward LC aldehydes (2, 3). In wild-type mice, FALDH activity toward LC aldehydes was highest in the liver followed by the epidermis, lung, and kidney (Fig. 2A). FALDH activity was moderate in the dermis, spleen, small intestine, testis, and cornea but low in the retina and brain. Disruption of Aldh3a2 greatly affected FALDH activity in the liver, kidney, and retina; its activity toward LC aldehydes in Aldh3a2 KO mice was <10% of that in these tissues in wild-type mice (Fig. 2A). The gene disruption had a weaker effect on FALDH activity toward C8:0 aldehyde (50–66% reduction compared with wild-type activity) than that toward LC aldehydes in these tissues, consistent with the substrate specificity of the Aldh3a2 protein. The effect of Aldh3a2 gene disruption on FALDH activity toward LC aldehydes was moderate in the brain, small intestine, testis, spleen, and dermis; activities were 30–50% of that in the corresponding tissues in wild-type mice (Fig. 2A). In the cornea, lung, and epidermis, on the other hand, FALDH activity was not lower in Aldh3a2 KO mice than in wild-type mice. Thus, the contribution of Aldh3a2 to total FALDH activity varied widely among tissues.
FIGURE 2.
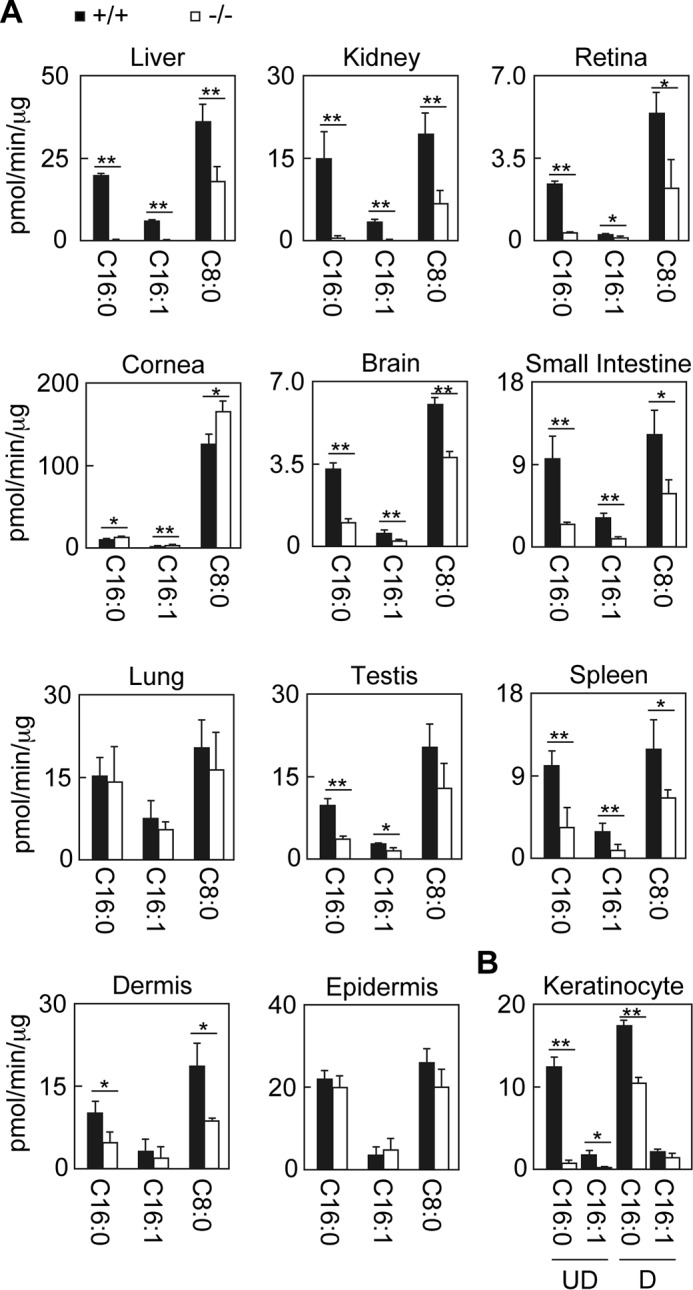
Differential contribution of Aldh3a2 to total FALDH activity depending on tissue. Total lysates were prepared from liver, kidney, retina, cornea, brain, small intestine, lung, testis, spleen, dermis, and epidermis tissue (A) and from keratinocytes kept undifferentiated or differentiated for 4 days (B) obtained from wild-type (black bars) and Aldh3a2 KO mice (white bars). Total lysates with fixed amounts of proteins (5 μg for the liver, lung, and keratinocytes; 10 μg for the kidney, cornea, small intestine, testis, spleen, epidermis, and dermis; and 15 μg for the brain and retina) were incubated with 500 μm NAD+ and 100 μm hexadecanal (C16:0), trans-2-hexadecenal (C16:1), or octanal (C8:0) for 15 min (liver, kidney, small intestine, lung, testis, and spleen) or 30 min (retina, cornea, brain, dermis, epidermis, and keratinocytes) at 37 °C. The amount of NADH product was determined by measuring the fluorescence of NADH using an Infinite M200 monochromator. Values represent the means ± S.D. of three independent experiments. Statistically significant differences are indicated (**, p < 0.01; *, p < 0.05; Student's t test). UD, undifferentiated keratinocytes; D, differentiated keratinocytes.
Because one of the primary symptoms of SLS is ichthyosis, we decided to examine FALDH activity in the skin in more detail. We prepared primary keratinocytes, which correspond to undifferentiated keratinocytes in the stratum basale, from wild-type and Aldh3a2 KO mice and measured their FALDH activity toward LC aldehydes. The primary keratinocytes from wild-type mice exhibited high activity (12.3 pmol/min/μg toward the C16:0 aldehyde), whereas activity in Aldh3a2 KO mice was extremely low (0.6 pmol/min/μg, 5.1% of wild-type activity) (Fig. 2B). When keratinocytes became differentiated, the differences became smaller, with activity toward the C16:0 aldehyde in Ald3a2 KO mice a mere 59.5% of wild-type activity. These results indicate that the contribution of Aldh3a2 to total FALDH activity in undifferentiated keratinocytes (i.e. stratum basale keratinocytes) is high, although that in keratinocytes of other parts of the epidermis is low.
Different mRNA Expression Profiles of Aldh3 Family Members Account for the Varied Contribution of Aldh3a2 to Total FALDH Activity among Tissues
We measured expression levels of the mRNAs of the five Aldh3 family members in mice in various tissues using quantitative real-time RT-PCR. The highest Aldh3a2 mRNA expression was observed in the liver followed by lung, testis, kidney, small intestine, and spleen (Fig. 3A). In these tissues, FALDH activity toward LC aldehydes was high as well (Fig. 2A), indicating a correlation between Aldh3a2 mRNA expression levels and FALDH activity toward LC aldehydes. In the liver, kidney, and retina, Aldh3a2 was almost exclusively expressed among Aldh3 family members (Fig. 3A). This expression profile of the Aldh3 family could explain the high contribution of the Aldh3a2 protein to the total FALDH activity toward LC aldehydes in these tissues (Fig. 2A). The expression of Aldh3a2 mRNA was also the highest among Aldh3 family members in the brain, small intestine, and spleen, but another Aldh3 family member was also expressed in each (Aldh3b2 in the brain and Aldh3b1 in the small intestine and spleen) (Fig. 3A). Their expression likely caused the relatively moderate contribution of Aldh3a2 to total FALDH activity toward LC aldehydes in these tissues (Fig. 2A). In the lung, the Aldh3a2 mRNA expression level was the highest, and those of Aldh3a1 and Aldh3b1 were also high (Fig. 3A). In the cornea, testis, dermis, and epidermis, the expression levels of Aldh3a2 were lower than those of other members of the Aldh3 family present there (i.e. Aldh3a1 in cornea and Aldh3b2 in testis, dermis, and epidermis). In the cornea, the expression levels of Aldh3a1 mRNA (Fig. 3A) as well as FALDH activity toward the MC aldehyde (Fig. 2A) were extremely high, matching a trend reported previously (31, 32). In the epidermis, the expression levels of Aldh3b2 were much higher than those of Aldh3a2 (Fig. 3A). Accordingly, the contributions of Aldh3a2 to total FALDH activity toward LC aldehydes were very low in the cornea and epidermis. (Fig. 2A) In summary, the expression profile of Aldh3 family members correlates well with the extent that Aldh3a2 contributes to total FALDH activity toward LC aldehydes in most tissues.
FIGURE 3.
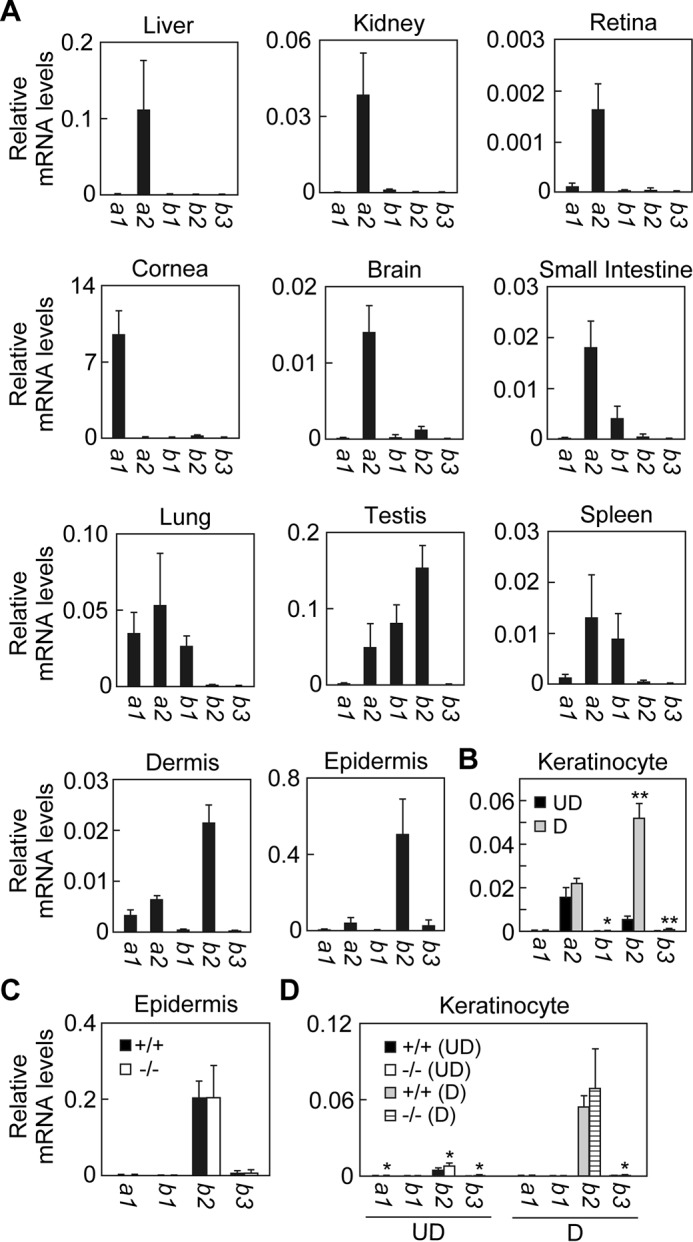
Expression profiles of Aldh3 family members in various tissues. Total RNAs prepared from liver, kidney, retina, cornea, brain, small intestine, lung, testis, spleen, dermis, and epidermis tissue (A and C) and from keratinocytes kept undifferentiated or differentiated for 4 days (B and D) obtained from wild-type (A–D) and Aldh3a2 KO mice (C and D) were subjected to SYBR Green-based real-time quantitative RT-PCR using specific primers for Aldh3a1, Aldh3a2, Aldh3b1, Aldh3b2, Aldh3b3, and Gapdh. Values are the amount of each mRNA relative to that of Gapdh, and represent the means ± S.D. for three independent experiments. A statistically significant difference is indicated (**, p < 0.01; *, p < 0.05; Student's t test). UD, undifferentiated keratinocytes; D, differentiated keratinocytes.
We next examined the expression levels of Aldh3 family members in keratinocytes. Only Aldh3a2 and Aldh3b2 mRNAs were expressed in both in undifferentiated and differentiated keratinocytes (Fig. 3B). Aldh3a2 mRNA expression was higher than that of Aldh3b2 in undifferentiated keratinocytes. Upon differentiation, Aldh3b2 mRNA greatly increased, whereas Aldh3a2 mRNA was nearly unchanged. As a result, the mRNA levels of Aldh3b2 became 2.1-fold higher than those of Aldh3a2 in differentiated keratinocytes. These results were consistent with the observed differences in the extent to which Aldh3a2 gene disruption diminishes total FALDH activity toward LC aldehydes between undifferentiated and differentiated keratinocytes (i.e. high and low effects, respectively) (Fig. 2B).
To investigate whether the expression of other Aldh3 isozymes was increased by Aldh3a2 disruption in a complementary way, we measured their expression levels in epidermis. We found no changes in the mRNA levels of other Aldh3 isozymes in Aldh3a2 KO mice with respect to wild-type mice (Fig. 3C). On the other hand, slight increases were observed in undifferentiated (Aldh3a1, 2.3-fold; Aldh3b2, 1.7-fold; and Aldh3b3, 2.6-fold) and differentiated (Aldh3b3, 2.6-fold) keratinocytes prepared from Aldh3a2 KO mice compared with corresponding levels in cells prepared from wild-type mice (Fig. 3D).
We also examined mRNA expression profiles of human ALDH3 family members in some selected tissues and keratinocytes. As described above, ALDH3B3 does not exist in humans, and ALDH3B2 is nonfunctional. Therefore, we chose to examine the mRNA expression levels of only ALDH3A1, ALDH3A2, and ALDH3B1. As in mice, ALDH3A2 mRNA was expressed highly in the liver but only moderately in the kidney and was low in the brain (Fig. 4A). ALDH3A2 was the major FALDH expressed in these three tissues, with only ALDH3B1 otherwise expressed at low levels in the kidney and brain. ALDH3A2 was almost exclusively expressed among FALDH in human keratinocytes (Fig. 4B); its expression levels increased by 6.9-fold upon differentiation.
FIGURE 4.
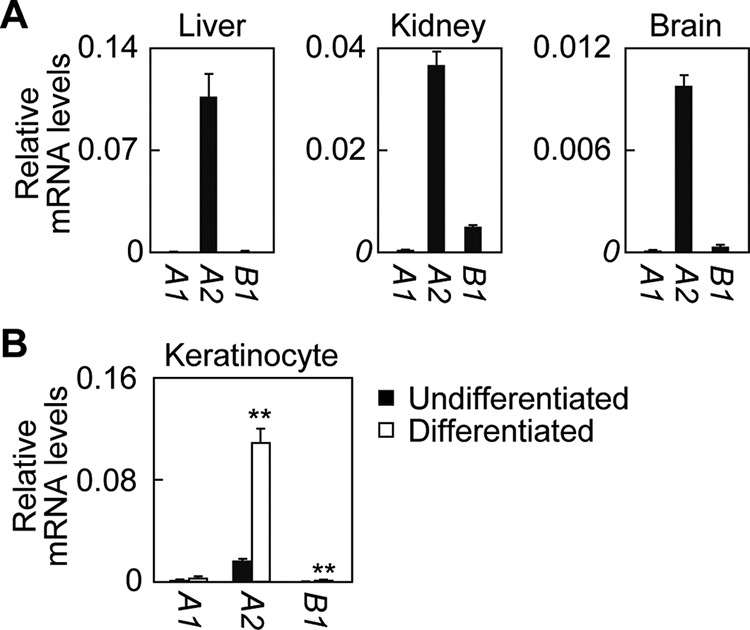
Expression profiles of ALDH3 family members in human tissues. cDNAs from human liver, kidney, and brain tissue (human MTC (multiple tissue cDNA) panels, Takara Bio) (A) or prepared from primary keratinocytes (undifferentiated or differentiated for 7 days; CELLnTEC) (B) were subjected to SYBR Green-based real-time quantitative PCR using specific primers for ALDH3A1, ALDH3A2, ALDH3B1, and GAPDH. Values represent the means ± S.D. from three independent reactions. A statistically significant difference is indicated (**, p < 0.01; Student's t test).
Impaired LCB Metabolism in Aldh3a2 KO Mice
We reported previously that ALDH3A2 is involved in the metabolism of the LCB parts of sphingolipids (16). An LCB can be metabolized to either sphingolipids or glycerolipids. In the latter pathway, the LCB is metabolized to its LCB 1-phosphate, fatty aldehyde, FA, and acyl-CoA sequentially and then is incorporated into a glycerolipid. ALDH3A2 catalyzes the conversion of the fatty aldehyde to its FA in this pathway (17). We examined the effect of Aldh3a2 disruption on the metabolism of the LCB DHS in primary keratinocytes using a [3H]DHS labeling assay. In Aldh3a2+/+ keratinocytes, DHS was metabolized to both sphingolipids (such as ceramides, sphingomyelins, and glucosylceramides) and glycerolipids (such as PE, phosphatidylserine, phosphatidylinositol, PC, and triglyceride) (Fig. 5A). In Aldh3a2−/− keratinocytes, DHS was metabolized to sphingolipids as normal; however, the metabolism of DHS to glycerolipids was greatly altered. Little DHS was converted to phosphatidylserine and phosphatidylinositol. Lipid bands were observed at the positions close to PE, PC, and triglyceride, but most products were not ester-linked PE/PC/triglyceride but rather were ether-linked glycerolipids, plasmanyl or plasmenyl (i.e. plasmalogen) types of ethanolamine/choline (PlsE/PlsC) or alkyl/alkenyl-diacylglycerol. We then performed alkaline treatment to induce hydrolysis of ester bonds in these products in order to release their constituent FAs for analysis. The amounts of FA released from glycerolipids in Aldh3a2−/− keratinocytes were much lower than those in Aldh3a2+/+ keratinocytes (Fig. 5A). Instead, alkyl/alkenyl group-containing hydrolysis products (alkyl/alkenyl-glycerol, alkyl/alkenyl-glycerophosphoethanolamine, and alkyl/alkenyl-glycerophosphocholine) were generated upon alkaline treatment. A similarly altered metabolism of DHS has been observed in Aldh3a2-null CHO-K1 cells (16). In Aldh3a2-deficient cells, a portion of the accumulated fatty aldehyde hexadecanal was reduced to the fatty alcohol hexadecanol and then incorporated into ether-linked glycerolipids.
FIGURE 5.

LCB metabolism is impaired in Aldh3a2 KO keratinocytes. A, primary keratinocytes prepared from wild-type and Aldh3a2 KO mice were labeled with [3H]DHS for 4 h at 37 °C. Lipids were extracted, treated or untreated with alkaline, separated by TLC, and detected by autoradiography. Alk-DG, alkyl/alkenyl-diacylglycerol; Alk-G, alkyl/alkenyl-glycerol; Alk-GPC, alkyl/alkenyl-glycerophosphocholine; Alk-GPE, alkyl/alkenyl-glycerophosphoethanolamine; Cer, ceramide; GlcCer, glucosylceramide; PI, phosphatidylinositol, PS, phosphatidylserine; SM, sphingomyelin; TG, triglyceride; *, unidentified breakdown products of PlsE by alkaline. B–I, primary keratinocytes prepared from wild-type and Aldh3a2 KO mice were labeled with d7-sphingosine for 4 h at 37 °C. Lipids were extracted followed by detection and quantification of d7-sphingosine metabolite-containing lipids (ceramide (B), PC (C), PE (D), PlsC (E), and PlsE (F) species) and endogenous (i.e. not containing d7-sphingosine or its metabolites) lipids (ceramide (G), PC (H), and PE (I) species) by LC/MS/MS analysis. Values are the amount of each PC/PE species containing the d7-sphingosine metabolite d7-C16:0 FA (d7-C16:0 (COOH)) and FA with the indicated chain length (C and D), that of each PlsC/PlsE species containing the sphingosine metabolite d7-C16:0/d7-C16:1 alcohol (d7-C16:0 (OH)/d7-C16:1 (OH)) and FA with the indicated chain length (E and F), that of each ceramide species containing C18:0 sphingosine and FAs with the indicated chain length (G), or that of PC/PE species containing FAs with the indicated chain length (H and I). The insets represent the total amounts of the respective lipids. Values represent the means ± S.D. from three independent reactions. Statistically significant differences are indicated (**, p < 0.01; *, p < 0.05; Student's t test). J, lipids extracted from epidermis and liver of wild-type and Aldh3a2 KO mice were subjected to LC/MS/MS analysis for detection and quantification of hexadecanal (C16:0-CHO) and trans-2-hexadecenal (C16:1-CHO). Values represent the means ± S.D. of three independent experiments. Statistically significant differences are indicated (*, p < 0.05; Student's t test).
To analyze this LCB-to-glycerolipid metabolism quantitatively, we next performed a d7-sphingosine labeling assay followed by measurement of ceramide, PC, PE, and ether-linked PlsC and PlsE levels by LC/MS/MS analysis. Sphingosine-d7-labeled ceramide levels were equivalent in Aldh3a2−/− and Aldh3a2+/+ keratinocytes (Fig. 5B). On the other hand, d7-sphingoinse was only slightly metabolized to PC (Fig. 5C) and PE (Fig. 5D) in Aldh3a2−/− keratinocytes. The PC and PE levels produced in Aldh3a2−/− keratinocytes were 17.9 and 6.9% of those in Aldh3a2+/+ keratinocytes. Instead, PlsC and PlsE levels were greatly increased (10.4- and 22.2-fold higher than those in Aldh3a2−/− keratinocytes) (Fig. 5, E and F). Both plasmanyl and plasmenyl types of PlsE were produced in Aldh3a2−/− keratinocytes, whereas the plasmanyl type was generated exclusively for PlsC.
We also measured the levels of endogenous (i.e. not containing d7-sphingosine or its metabolite FAs) ceramide, PC, and PE species. We found that endogenous ceramide levels were not affected by Aldh3a2 disruption (Fig. 5G), the same as d7-sphingosine-labeled ceramide levels (Fig. 5B). Although the levels of PC and PE containing d7-sphingosine metabolite FAs were greatly decreased in Aldh3a2−/− keratinocytes (Fig. 5, C and D), endogenous PC and PE levels were unaffected (Fig. 5, H and I). This result indicates that the contribution of the sphingosine degradation pathway to glycerolipid synthesis as a FA source is quite low, reflecting the much lower levels of sphingolipids (∼1/10) compared with glycerolipid levels in cells. In conclusion, these results indicate that the metabolism of LCBs to ester-linked glycerolipids is severely impaired in Aldh3a2−/− keratinocytes.
Although disruption of Aldh3a2 had the largest effect on FALDH activity toward LC aldehydes in liver compared with other tissues (Fig. 2A), SLS patients have not been observed to manifest symptoms in the liver. To gain insight into the reason why, we measured LC aldehyde levels in epidermis and liver. The total levels of hexadecanal (C16:0) and trans-2-hexadecanal (16:1), which is the metabolite of sphingosine, were 11.4-fold higher in the epidermis than in the liver of wild-type mice (Fig. 5J). The trans-2-hexadecenal levels were especially high in epidermis. Disruption of Aldh3a2 had no effects on the C16 aldehyde levels in epidermis, consistent with their observed lack of effects on the FALDH activity (Fig. 2A). On the other hand, the levels of C16 aldehydes were 2.5-fold higher in Aldh3a2 KO liver than in wild-type liver (Fig. 5J), reflecting the great effect of Aldh3a2 disruption on FALDH activity in this tissue (Fig. 2A). However, the accumulated levels of C16 aldehydes in the liver of Aldh3a2 KO mice were still lower than those in the epidermis of wild-type mice (Fig. 5J). These results suggest that liver has a high capacity to remove fatty aldehydes by a mechanism other than oxidation.
Aldh3a2 KO Mice Exhibit Delayed Skin Barrier Recovery
In SLS patients, some ceramide species such as acylceramide EOS are reduced (10). Therefore, we examined ceramide/acylceramide levels in the epidermis of Aldh3a2−/− mice by LC/MS/MS analysis. However, neither the levels of ceramides (containing sphingosine and non-hydroxy FA) nor those of EOS were affected by Aldh3a2 disruption (Fig. 6, A and B).
FIGURE 6.
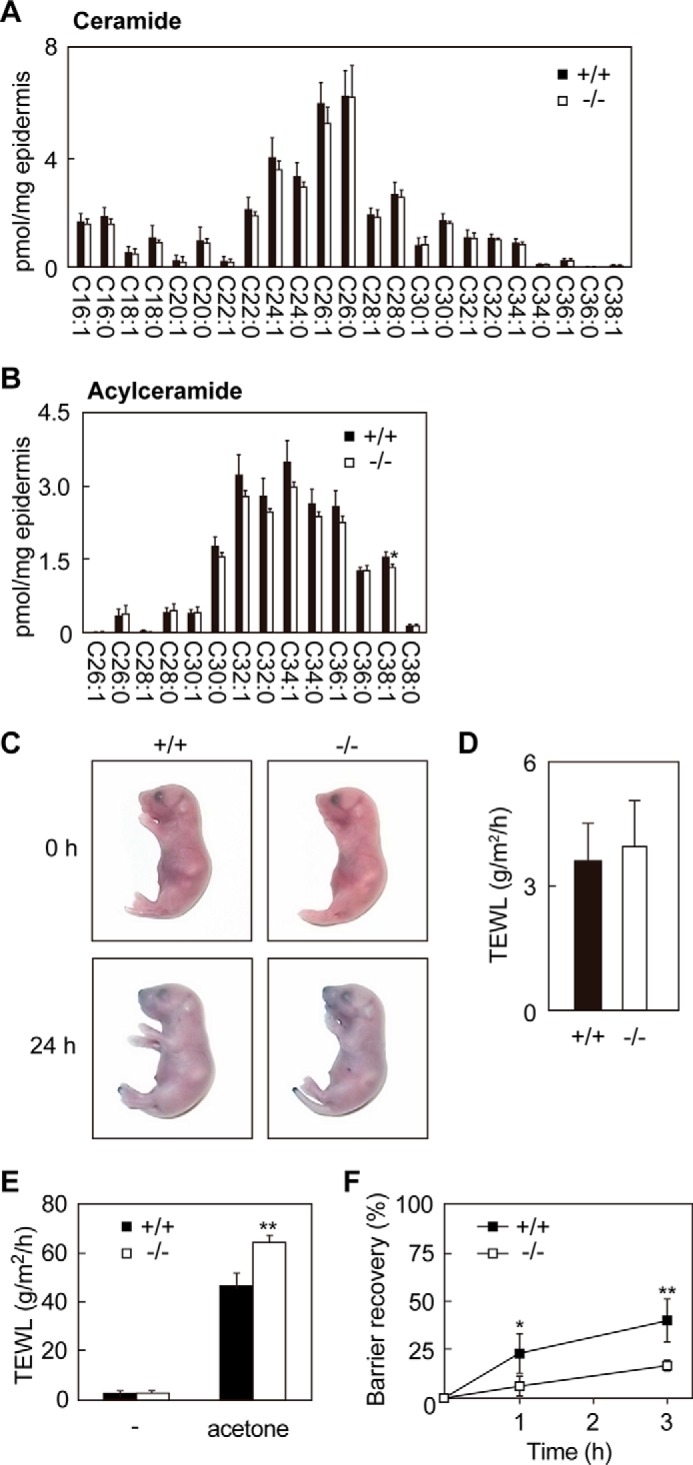
Skin barrier recovery of Aldh3a2−/− mice is delayed. A and B, lipids were extracted from the epidermis of wild-type and Aldh3a2 KO mice at P0, and ceramide (A) and acylceramide (B) species were analyzed by LC/MS/MS. Values are the amount of each ceramide species containing C18:0 sphingosine and FAs with the indicated chain length (A) or that of acylceramide species containing C18:0 sphingosine, linoleic acid, and FAs with the indicated chain length (B). They represent the means ± S.D. from four independent reactions. C, wild-type and Aldh3a2 KO mice at P0 were stained with 0.1% toluidine blue for 24 h and photographed. D, TEWL was measured for the wild-type and Aldh3a2 KO mice at P0. Values represent the means ± S.D. from eight wild-type or six Aldh3a2 KO independent mice. E and F, the skins of wild-type and Aldh3a2 KO mice at P0 were treated with acetone or saline as a control. TEWL was measured after acetone treatment (0 h, E and F; 1 h, F; 3 h, F). Values are TEWL amounts (E). In F, barrier recovery is expressed in terms of TEWL, with TEWL at 0 h after acetone set as 0% and TEWL before acetone treatment as 100%. Values represent the means ± S.D. of five wild-type or four Aldh3a2 KO mice in independent experiments. Statistically significant differences between wild-type and Aldh3a2 KO mice are indicated (**, p < 0.01; *, p < 0.05; Student's t test).
We next evaluated skin barrier function in Aldh3a2 KO mice using a dye exclusion assay. Aldh3a2 KO mice were resistant to dye penetration, the same as wild-type mice (Fig. 6C). Furthermore, there were no differences in TEWL between wild-type and Aldh3a2 KO mice (Fig. 6D). However, when the stratum corneum was perturbed by acetone, Aldh3a2 KO mice showed higher TEWL than wild-type mice (Fig. 6E). The skin barrier recovery of Aldh3a2 KO mice after the acetone treatment was also retarded (Fig. 6F). Thus, although barrier formation in the stratum corneum appeared intact in Aldh3a2 KO mice, in the lower layers of epidermis it appeared to have been compromised.
Disruption of Aldh3a2 Causes Broadened Intercellular Spaces in the Stratum Basale and Enhanced Proliferation of Keratinocytes
We next performed histological analyses. H&E staining revealed that Aldh3a2−/− mice did not show hyperkeratosis, a typical skin symptom of SLS (Fig. 7A). In terms of its overall structure, skin prepared from Aldh3a2 KO mice was very similar to that from wild-type mice. However, we observed small, intracellular vacuoles in the stratum spinosum and stratum granulosum only in Aldh3a2−/− mice (Fig. 7A, arrows). Similar vacuoles have been observed in patients suffering from autosomal recessive congenital ichthyosis caused by PNPLA1 (patatin-like phospholipase domain-containing 1) mutation (33). We then performed more detailed morphology analysis by electron microscopy, focusing especially on keratinocyte appearance in the stratum basale because the cell type in which FALDH activity and LCB metabolism were greatly impaired was undifferentiated keratinocytes. Keratinocytes formed tight cell-cell adhesion in wild-type mice (Fig. 7, B and C). However, the intercellular spaces between keratinocytes were markedly broadened in the stratum basale of Aldh3a2−/− epidermis.
FIGURE 7.
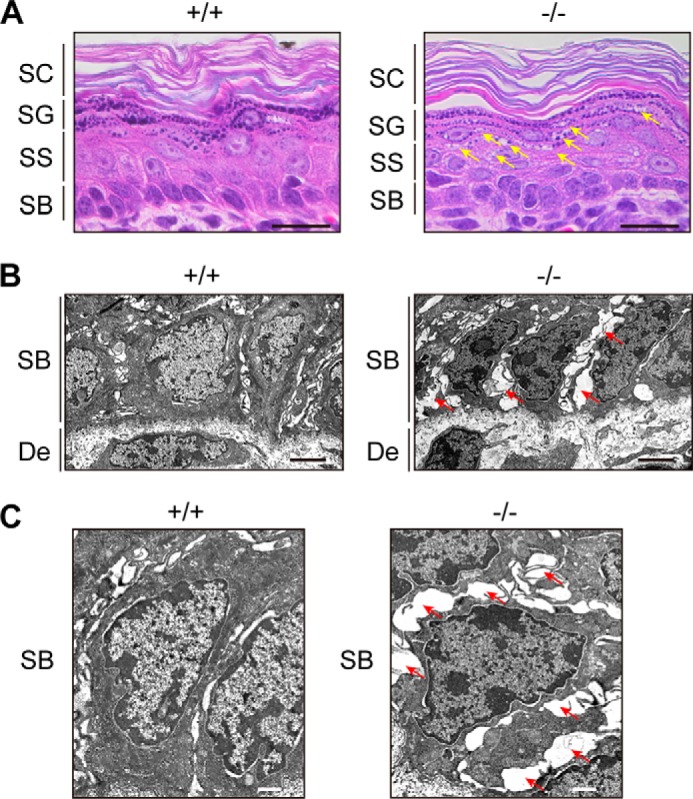
Vacuolization and broadened intercellular spaces present in Aldh3a2−/− epidermis. A, paraffin sections (4 μm) of skin prepared from wild-type and Aldh3a2 KO mice at P0 were subjected to H&E staining. Bright field images were photographed under a DM5000B light microscope. Arrows indicate vacuolization. Bar, 20 μm. B and C, skin sections of wild-type and Aldh3a2 KO mice at P0 were subjected to electron microscopy. Arrows indicate broadened intercellular spaces. Bar, 3 μm (B), 1 μm (C). SC, stratum corneum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basale; De, dermis.
Because broadened intercellular spaces are often associated with enhanced keratinocyte proliferation (34–36), we prepared primary keratinocytes from wild-type and Aldh3a2 KO mice and examined their proliferation rates by cell counting and measuring [3H]thymidine uptake (Fig. 8, A and B). We found that Aldh3a2−/− keratinocytes indeed exhibited increased proliferation compared with their wild-type counterparts. The mRNA levels of the cell proliferation marker Ki67 were also up-regulated in Aldh3a2−/− keratinocytes (Fig. 8C). Some relationships between hyperproliferation and oxidative stress-induced responses are reported for keratinocytes and cancer cells alike (37, 38). Therefore, we decided to examine the expression levels of oxidative stress-induced genes Hmox1 (heme oxygenase 1), Sod1 (superoxide dismutase 1), Gclc (glutamate-cysteine ligase, catalytic subunit), Gclm (glutamate-cysteine ligase, modifier subunit), and Gsta1 (glutathione S-transferase α1) using real-time quantitative RT-PCR (Fig. 8D). We found that expressions of these genes were increased in Aldh3a2−/− keratinocytes. These results suggest that an oxidative stress-induced response can accompany the enhanced proliferation of Aldh3a2−/− keratinocytes.
FIGURE 8.
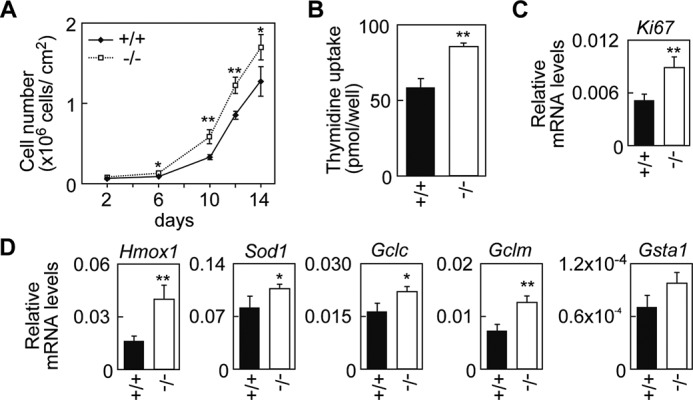
Enhanced proliferation and oxidative stress response in Aldh3a2−/− keratinocytes. A, primary keratinocytes were prepared from wild-type and Aldh3a2 KO mice and grown. Cell numbers were counted by microscopic observation. For each dish, cell numbers in three randomly chosen viewing fields were counted and summed. Values represent the means ± S.D. from four independent experiments. B, keratinocytes prepared from wild-type and Aldh3a2 KO mice were subjected to a [3H]thymidine uptake assay. Values represent the means ± S.D. from three independent experiments. C and D, total RNAs prepared from wild-type and Aldh3a2 KO keratinocytes were subjected to SYBR Green-based real-time quantitative RT-PCR using specific primers for Ki67 (C), Hmox1, Sod1, Gclc, Gclm, Gsta1, and Gapdh (D). Values are the amount of each mRNA relative to that of Gapdh and represent the means ± S.D. for three independent experiments. Statistically significant differences are indicated (**, p < 0.01; *, p < 0.05; Student's t test).
Discussion
The hereditary disease SLS is caused by mutations in ALDH3A2 gene. Aldehydes, the substrates of ALDHs including ALDH3A2, are reactive and toxic in general. This has led many to consider that the accumulation of fatty aldehydes causes SLS pathology. Although the clinical aspects of SLS have been analyzed in detail, the molecular mechanism underlying its pathology remains totally unknown. Our analysis is the first ever to analyze Aldh3a2 KO mice so as to gain insight into this molecular mechanism. We examined the effect of Aldh3a2 gene disruption on FALDH activity in various tissues and, bearing in mind the ichthyosis phenotype of SLS, expanded our investigation to include morphological and biochemical analyses of skin phenotypes. We failed to observe typical skin SLS symptoms (such as hyperkeratosis, skin barrier defects under normal conditions, and changes in ceramide composition (Figs. 6 and 7)) due to mouse-specific compensation by Aldh3b2 (Fig. 3). This Aldh3b2 compensation was detected in the upper layers of the epidermis but not in undifferentiated keratinocytes or the basal layer (i.e. stratum basale) of the epidermis (Figs. 2B and 3B). Accordingly, we observed some abnormalities in undifferentiated keratinocytes and stratum basale of Aldh3a2 KO mice, such as impaired LCB metabolism, broadened intercellular spaces, hyperproliferation of keratinocytes, increased oxidative stress response, enhanced TEWL after stratum corneum damage, and delayed skin barrier recovery (Figs. 5–8). Thus, our findings suggest that some abnormalities occur even at the basal epidermal layer in SLS.
We found that FALDH activity toward LC aldehydes was the highest in the liver followed by the epidermis, lung, and kidney in wild-type mice, among which FALDH activity in the liver and kidney was greatly affected by Aldh3a2 disruption (Fig. 2A). Despite the high contribution of Aldh3a2 in these tissues, no phenotypes corresponding to its disruption were apparent. Corroborating our results, but nonetheless puzzling, is the finding that SLS patients also do not present with any symptoms in these tissues. Therefore, it is not clear why ALDH3A2 mutations cause abnormalities only in restricted tissues such as the skin and the nervous system. Several reasons can be considered. One possibility is that most tissues (except the skin and the nervous system) have a high capacity to remove fatty aldehydes via a mechanism other than oxidation, e.g. metabolism via another metabolic pathway or excretion from cells. Indeed, we found that C16 aldehyde levels in liver were much lower than those in epidermis (Fig. 5J). One of the possible alternative metabolic pathways is reduction of fatty aldehydes to fatty alcohols. In support of this view, we found that LCBs were metabolized to fatty alcohols and incorporated into ether-linked glycerolipids in Aldh3a2 KO mice (Fig. 5). Metabolism of LCBs to ether-linked glycerolipids occurs even in ALDH3A2-active cells. This pathway is only a minor route for LCB metabolism (39, 40), but disruption of Aldh3a2 caused it to become the major pathway. Similarly, altered LCB metabolism has been observed in Aldh3a2-null CHO-K1 cells previously (16). The accumulation of fatty alcohols and their metabolism to ether-linked glycerolipids have also been reported in SLS patients (41, 42). We speculate that the generation of ether-linked glycerolipids itself is unlikely to be responsible for the SLS pathology, because ether-linked glycerolipids such as plasmalogens actually have rather cell-protective effects (43). Rather, the cause is probably that not all of the accumulated fatty aldehydes in SLS patients are converted to fatty alcohols/ether-linked glycerolipids, and some of them then react with important local proteins, leading to the SLS pathology. It is possible that the activities or expression levels of aldo-keto reductases/fatty alcohol dehydrogenases responsible for the reduction of fatty aldehydes to fatty alcohols in the skin and nervous system are lower than in other tissues.
The second possible reason for the restricted abnormalities in the skin and nervous system in SLS patients is that these tissues contain certain physiologically important proteins susceptible to damage by accumulated fatty aldehydes. Aldehydes can readily attack primary amines via Schiff base formation. ALDH3A2 is localized mainly in the endoplasmic reticulum, although a minor splicing isoform of ALDH3A2 also exists, which is localized in the peroxisome (44). Therefore, endoplasmic reticulum membrane proteins containing many lysine and arginine residues may be the primary targets of accumulated fatty aldehydes in SLS patients.
To date, it has been unclear which fatty aldehydes and which lipid metabolic pathway(s) substantively contribute to the pathogenesis of SLS. However, several lines of evidence suggest the contribution of sphingosine and its metabolic pathway. First, sphingosine metabolism occurs in the endoplasmic reticulum (17, 45, 46), the same organelle where ALDH3A2 resides (44). Second, the metabolite of sphingosine is trans-2-hexadecenal, which is classified as an α,β-unsaturated aldehyde. Normal aldehydes react with primary amines to form Schiff bases. However, α,β-unsaturated aldehydes can attack general nucleophiles including cysteine and histidine side chains through the 1,4-Michael addition (47), leading to higher toxicities of α,β-unsaturated aldehydes than normal aldehydes (48). Finally, we observed high levels of trans-2-hexadecenal in the epidermis, whereas their levels in liver were low (Fig. 5J).
SLS patients exhibit pathological symptoms of skin barrier defects under normal conditions, hyperkeratosis, and abnormal ceramide compositions; however, the Aldh3a2−/− mice exhibited none of these phenotypes (Figs. 6 and 7). These findings are likewise consistent with the results showing that epidermal FALDH activity was not affected by Aldh3a2 disruption (Fig. 2A). We found that mouse Aldh3b2 was highly expressed in the epidermis and compensated for Aldh3a2 deficiency in Aldh3a2−/− mice (Fig. 3A). On the other hand, human ALDH3B2 lacks 94 N-terminal amino acids present in the mouse homolog Aldh3b2 and, as a result, has no enzyme activity (19). The lack of functional ALDH3B2 in human may mean that just the ALDH3A2 mutation is enough to cause SLS symptoms. However, Aldh3a2 was the most expressed FALDH in undifferentiated mouse keratinocytes (Fig. 3B). Accordingly, we observed a morphological change in the stratum basale, the home of undifferentiated keratinocytes. In the stratum basale of Aldh3a2−/− mice, intercellular spaces were widened (Fig. 7, B and C), which is a typical feature of cells in a hyperproliferative state (34–36). Indeed, increased proliferation was observed in primary keratinocytes derived from Aldh3a2−/− mice (Fig. 8, A–C). Hyperproliferation of keratinocytes in SLS patients has also been observed (49).
We also found up-regulated transcription of the oxidative stress-induced genes Hmox1, Sod1, Gclc, Gclm, and Gsta1 (Fig. 8D). These genes are the targets of the transcription factor Nrf2, which is activated under oxidative stress (37, 50). Continuous activation of Nrf2 is closely related to the proliferation of cancer cells (51). Therefore, we speculated that Nrf2 is activated in Aldh3a2−/− keratinocytes, which leads to an increase in proliferation. Under normal conditions, the oxidative sensor protein Keap1 retains Nrf2 in the cytosol and acts as an adaptor for an E3 ligase to degrade Nrf2 through an ubiquitin-proteasome system (52). Oxidative stresses (such as reactive oxygen species (ROS)) and other electrophiles (including aldehydes) modify the cysteine residues in Keap1, causing Nrf2 activation, which in turn promotes the transcriptions of its target genes involved in oxidative stress resistance and cell survival (37, 53). Disruption of Keap1 and overexpression of Nrf2 in mice independently induce hyperproliferation in keratinocytes and skin barrier defect phenotypes such as increased TEWL and hyperkeratosis (54, 55). The hyperproliferation of keratinocytes observed in this study thus appears to be closely related to the pathogenesis of ichthyosis symptoms. Therefore, although Aldh3a2−/− mice are not a perfectly suitable model for typical human SLS, our results imply that increased proliferation of Aldh3a2−/− keratinocytes contributes to some aspects of the symptoms seen in this disorder.
Author Contributions
A. K. planned and organized the project. T. N., S. T., and T. Kanetake performed the experiments. T. N., S. T., T. Kanetake, and T. Kitamura analyzed the data. T. N. and T. S. prepared the Aldh3a2 KO mice. T. Kanetake, S. H., and T. M. performed and/or analyzed the food intake assay. T. N. and A. K. designed the experiments and wrote the manuscript.
Acknowledgments
Aldh3a2 gene trap mice were obtained from the European Mouse Mutant Archive. The CAG-Cre mouse was generated by Dr. Masaru Okabe (Osaka University) and provided by the Riken BioResource Center through the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We are grateful to Shotaro Suzuki (Hokkaido University) for assistance with electron microscopy analysis and helpful advice.
This work was supported by Advanced Research and Development Programs for Medical Innovation (AMED-CREST) (to A. K.) from the Japan Agency for Medical Research and Development (AMED), by Grant-in-aid for Scientific Research (A) 26251010 (to A. K.) from the Japan Society for the Promotion of Science (JSPS), and by Grant-in-aid for JSPS Fellows 25744 (to T. N.) from JSPS. The authors declare that they have no conflicts of interest with the contents of this article.
- SLS
- Sjögren-Larsson syndrome
- FALDH
- fatty aldehyde dehydrogenase
- MC
- medium-chain
- LC
- long-chain
- FA
- fatty acid
- LCB
- long-chain base
- EOS
- esterified ω-hydroxyacylsphingosine
- DHS
- dihydrosphingosine
- ALDH
- aldehyde dehydrogenase
- TEWL
- transepidermal water loss
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PlsC
- plasmanyl/plasmenylcholine
- PlsE
- plasmanyl/plasmenylethanolamine
- d7
- deuterium.
References
- 1. Rizzo W. B. (2007) Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol. Genet. Metab. 90, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kitamura T., Takagi S., Naganuma T., and Kihara A. (2015) Mouse aldehyde dehydrogenase ALDH3B2 is localized to lipid droplets via two C-terminal tryptophan residues and lipid modification. Biochem. J. 465, 79–87 [DOI] [PubMed] [Google Scholar]
- 3. Kelson T. L., Secor McVoy J. R., and Rizzo W. B. (1997) Human liver fatty aldehyde dehydrogenase: microsomal localization, purification, and biochemical characterization. Biochim. Biophys. Acta 1335, 99–110 [DOI] [PubMed] [Google Scholar]
- 4. Rizzo W. B., and Carney G. (2005) Sjögren-Larsson syndrome: diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2). Hum. Mutat. 26, 1–10 [DOI] [PubMed] [Google Scholar]
- 5. Mizutani Y., Mitsutake S., Tsuji K., Kihara A., and Igarashi Y. (2009) Ceramide biosynthesis in keratinocyte and its role in skin function. Biochimie 91, 784–790 [DOI] [PubMed] [Google Scholar]
- 6. Feingold K. R., and Elias P. M. (2014) Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim. Biophys. Acta 1841, 280–294 [DOI] [PubMed] [Google Scholar]
- 7. Uchida Y., and Holleran W. M. (2008) Omega-O-acylceramide, a lipid essential for mammalian survival. J. Dermatol. Sci. 51, 77–87 [DOI] [PubMed] [Google Scholar]
- 8. Ohno Y., Nakamichi S., Ohkuni A., Kamiyama N., Naoe A., Tsujimura H., Yokose U., Sugiura K., Ishikawa J., Akiyama M., and Kihara A. (2015) Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc. Natl. Acad. Sci. U.S.A. 112, 7707–7712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kihara A., Mitsutake S., Mizutani Y., and Igarashi Y. (2007) Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 46, 126–144 [DOI] [PubMed] [Google Scholar]
- 10. Nakajima K., Sano S., Uchida Y., Akiyama M., Morita Y., and Shimizu H. (2011) Altered lipid profiles in the stratum corneum of Sjögren-Larsson syndrome. J. Dermatol. Sci. 63, 64–66 [DOI] [PubMed] [Google Scholar]
- 11. Rizzo W. B., Heinz E., Simon M., and Craft D. A. (2000) Microsomal fatty aldehyde dehydrogenase catalyzes the oxidation of aliphatic aldehyde derived from ether glycerolipid catabolism: implications for Sjögren-Larsson syndrome. Biochim. Biophys. Acta 1535, 1–9 [DOI] [PubMed] [Google Scholar]
- 12. Willemsen M. A., Rotteveel J. J., de Jong J. G., Wanders R. J., IJlst L., Hoffmann G. F., and Mayatepek E. (2001) Defective metabolism of leukotriene B4 in the Sjögren-Larsson syndrome. J. Neurol. Sci. 183, 61–67 [DOI] [PubMed] [Google Scholar]
- 13. Verhoeven N. M., Jakobs C., Carney G., Somers M. P., Wanders R. J., and Rizzo W. B. (1998) Involvement of microsomal fatty aldehyde dehydrogenase in the α-oxidation of phytanic acid. FEBS Lett. 429, 225–228 [DOI] [PubMed] [Google Scholar]
- 14. van den Brink D. M., van Miert J. N., Dacremont G., Rontani J. F., Jansen G. A., and Wanders R. J. (2004) Identification of fatty aldehyde dehydrogenase in the breakdown of phytol to phytanic acid. Mol. Genet. Metab. 82, 33–37 [DOI] [PubMed] [Google Scholar]
- 15. Rizzo W. B. (2014) Fatty aldehyde and fatty alcohol metabolism: review and importance for epidermal structure and function. Biochim. Biophys. Acta 1841, 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakahara K., Ohkuni A., Kitamura T., Abe K., Naganuma T., Ohno Y., Zoeller R. A., and Kihara A. (2012) The Sjögren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell 46, 461–471 [DOI] [PubMed] [Google Scholar]
- 17. Kihara A. (2014) Sphingosine 1-phosphate is a key metabolite linking sphingolipids to glycerophospholipids. Biochim. Biophys. Acta 1841, 766–772 [DOI] [PubMed] [Google Scholar]
- 18. Kondo N., Ohno Y., Yamagata M., Obara T., Seki N., Kitamura T., Naganuma T., and Kihara A. (2014) Identification of the phytosphingosine metabolic pathway leading to odd-numbered fatty acids. Nat. Commun. 5, 5338. [DOI] [PubMed] [Google Scholar]
- 19. Marchitti S. A., Brocker C., Stagos D., and Vasiliou V. (2008) Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 4, 697–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson B., Brocker C., Thompson D. C., Black W., Vasiliou K., Nebert D. W., and Vasiliou V. (2011) Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genomics 5, 283–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pappa A., Estey T., Manzer R., Brown D., and Vasiliou V. (2003) Human aldehyde dehydrogenase 3A1 (ALDH3A1): biochemical characterization and immunohistochemical localization in the cornea. Biochem. J. 376, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marchitti S. A., Orlicky D. J., and Vasiliou V. (2007) Expression and initial characterization of human ALDH3B1. Biochem. Biophys. Res. Commun. 356, 792–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitamura T., Naganuma T., Abe K., Nakahara K., Ohno Y., and Kihara A. (2013) Substrate specificity, plasma membrane localization, and lipid modification of the aldehyde dehydrogenase ALDH3B1. Biochim. Biophys. Acta 1831, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 24. Matsumura H., Hasuwa H., Inoue N., Ikawa M., and Okabe M. (2004) Lineage-specific cell disruption in living mice by Cre-mediated expression of diphtheria toxin A chain. Biochem. Biophys. Res. Commun. 321, 275–279 [DOI] [PubMed] [Google Scholar]
- 25. Caldelari R., and Müller E. J. (2010) Short- and long-term cultivation of embryonic and neonatal murine keratinocytes. Methods Mol. Biol. 633, 125–138 [DOI] [PubMed] [Google Scholar]
- 26. Sassa T., Ohno Y., Suzuki S., Nomura T., Nishioka C., Kashiwagi T., Hirayama T., Akiyama M., Taguchi R., Shimizu H., Itohara S., and Kihara A. (2013) Impaired epidermal permeability barrier in mice lacking elovl1, the gene responsible for very-long-chain fatty acid production. Mol. Cell. Biol. 33, 2787–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hardman M. J., Sisi P., Banbury D. N., and Byrne C. (1998) Patterned acquisition of skin barrier function during development. Development 125, 1541–1552 [DOI] [PubMed] [Google Scholar]
- 28. Kariya Y., Kihara A., Ikeda M., Kikuchi F., Nakamura S., Hashimoto S., Choi C. H., Lee Y. M., and Igarashi Y. (2005) Products by the sphingosine kinase/sphingosine 1-phosphate (S1P) lyase pathway but not S1P stimulate mitogenesis. Genes Cells 10, 605–615 [DOI] [PubMed] [Google Scholar]
- 29. Neuber C., Schumacher F., Gulbins E., and Kleuser B. (2014) Method to simultaneously determine the sphingosine 1-phosphate breakdown product (2E)-hexadecenal and its fatty acid derivatives using isotope-dilution HPLC-electrospray ionization-quadrupole/time-of-flight mass spectrometry. Anal. Chem. 86, 9065–9073 [DOI] [PubMed] [Google Scholar]
- 30. Kihara A., Ikeda M., Kariya Y., Lee E. Y., Lee Y. M., and Igarashi Y. (2003) Sphingosine-1-phosphate lyase is involved in the differentiation of F9 embryonal carcinoma cells to primitive endoderm. J. Biol. Chem. 278, 14578–14585 [DOI] [PubMed] [Google Scholar]
- 31. Piatigorsky J. (2001) Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: the “refracton” hypothesis. Cornea 20, 853–858 [DOI] [PubMed] [Google Scholar]
- 32. Abedinia M., Pain T., Algar E. M., and Holmes R. S. (1990) Bovine corneal aldehyde dehydrogenase: the major soluble corneal protein with a possible dual protective role for the eye. Exp. Eye Res. 51, 419–426 [DOI] [PubMed] [Google Scholar]
- 33. Grall A., Guaguère E., Planchais S., Grond S., Bourrat E., Hausser I., Hitte C., Le Gallo M., Derbois C., Kim G. J., Lagoutte L., Degorce-Rubiales F., Radner F. P., Thomas A., Küry S., et al. (2012) PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat. Genet. 44, 140–147 [DOI] [PubMed] [Google Scholar]
- 34. White F. H., and Gohari K. (1984) Alterations in the volume of the intercellular space between epithelial cells of the hamster cheek-pouch: quantitative studies of normal and carcinogen-treated tissues. J. Oral Pathol. 13, 244–254 [DOI] [PubMed] [Google Scholar]
- 35. Hernández-Quintero M., Kuri-Harcuch W., González Robles A., and Castro-Muñozledo F. (2006) Interleukin-6 promotes human epidermal keratinocyte proliferation and keratin cytoskeleton reorganization in culture. Cell Tissue Res. 325, 77–90 [DOI] [PubMed] [Google Scholar]
- 36. Li D., Zhang W., Liu Y., Haneline L. S., and Shou W. (2012) Lack of plakoglobin in epidermis leads to keratoderma. J. Biol. Chem. 287, 10435–10443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taguchi K., Motohashi H., and Yamamoto M. (2011) Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140 [DOI] [PubMed] [Google Scholar]
- 38. George J., and Shukla Y. (2013) Emptying of intracellular calcium pool and oxidative stress imbalance are associated with the glyphosate-induced proliferation in human skin keratinocytes HaCaT cells. ISRN Dermatol. 2013, 825180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stoffel W., LeKim D., and Heyn G. (1970) Metabolism of sphingosine bases. XIV. Sphinganine (dihydrosphingosine), an effective donor of the alk-1′-enyl chain of plasmalogens. Hoppe Seylers Z. Physiol. Chem. 351, 875–883 [DOI] [PubMed] [Google Scholar]
- 40. Wakashima T., Abe K., and Kihara A. (2014) Dual functions of the trans-2-enoyl-CoA reductase TER in the sphingosine 1-phosphate metabolic pathway and in fatty acid elongation. J. Biol. Chem. 289, 24736–24748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rizzo W. B., and Craft D. A. (2000) Sjögren-Larsson syndrome: accumulation of free fatty alcohols in cultured fibroblasts and plasma. J. Lipid Res. 41, 1077–1081 [PubMed] [Google Scholar]
- 42. Rizzo W. B., Craft D. A., Somer T., Carney G., Trafrova J., and Simon M. (2008) Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J. Lipid Res. 49, 410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gorgas K., Teigler A., Komljenovic D., and Just W. W. (2006) The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim. Biophys. Acta 1763, 1511–1526 [DOI] [PubMed] [Google Scholar]
- 44. Ashibe B., Hirai T., Higashi K., Sekimizu K., and Motojima K. (2007) Dual subcellular localization in the endoplasmic reticulum and peroxisomes and a vital role in protecting against oxidative stress of fatty aldehyde dehydrogenase are achieved by alternative splicing. J. Biol. Chem. 282, 20763–20773 [DOI] [PubMed] [Google Scholar]
- 45. Ikeda M., Kihara A., and Igarashi Y. (2004) Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 325, 338–343 [DOI] [PubMed] [Google Scholar]
- 46. Ohkuni A., Ohno Y., and Kihara A. (2013) Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem. Biophys. Res. Commun. 442, 195–201 [DOI] [PubMed] [Google Scholar]
- 47. Catalá A. (2009) Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 157, 1–11 [DOI] [PubMed] [Google Scholar]
- 48. Beauchamp R. O. Jr., Andjelkovich D. A., Kligerman A. D., Morgan K. T., and Heck H. D. (1985) A critical review of the literature on acrolein toxicity. Crit. Rev. Toxicol. 14, 309–380 [DOI] [PubMed] [Google Scholar]
- 49. Jagell S., and Lidén S. (1982) Ichthyosis in the Sjögren-Larsson syndrome. Clin. Genet. 21, 243–252 [DOI] [PubMed] [Google Scholar]
- 50. Dreger H., Westphal K., Wilck N., Baumann G., Stangl V., Stangl K., and Meiners S. (2010) Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc. Res. 85, 395–403 [DOI] [PubMed] [Google Scholar]
- 51. Mitsuishi Y., Motohashi H., and Yamamoto M. (2012) The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front. Oncol. 2, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobayashi A., Kang M. I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., and Yamamoto M. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abiko Y., Mizokawa M., and Kumagai Y. (2014) Activation of the Kelch-like ECH-associated protein 1 (Keap1)/NF-E2-related factor 2 (Nrf2) pathway through covalent modification of the 2-alkenal group of aliphatic electrophiles in Coriandrum sativum L. J. Agric. Food Chem. 62, 10936–10944 [DOI] [PubMed] [Google Scholar]
- 54. Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S., Imakado S., Kotsuji T., Otsuka F., Roop D. R., Harada T., Engel J. D., and Yamamoto M. (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245 [DOI] [PubMed] [Google Scholar]
- 55. Schäfer M., Farwanah H., Willrodt A. H., Huebner A. J., Sandhoff K., Roop D., Hohl D., Bloch W., and Werner S. (2012) Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol. Med. 4, 364–379 [DOI] [PMC free article] [PubMed] [Google Scholar]