

Abstract
Folliculin (FLCN) is the tumor suppressor associated with Birt-Hogg-Dubé (BHD) syndrome that predisposes patients to incident of hamartomas and cysts in multiple organs. Its inactivation causes deregulation in the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway. However, the underlying mechanism is poorly defined. In this study, we show that FLCN is a ciliary protein that functions through primary cilia to regulate mTORC1. In response to flow stress, FLCN associates with LKB1 and recruits the kinase to primary cilia for activation of AMPK resided at basal bodies, which causes mTORC1 down-regulation. In cells depleted of FLCN, LKB1 fails to accumulate in primary cilia and AMPK at the basal bodies remains inactive, thus nullifying the inhibitory effect of flow stress on mTORC1 activity. Our results demonstrate that FLCN is part of a flow sensory mechanism that regulates mTORC1 through primary cilia.
Keywords: AMP-activated kinase (AMPK), mammalian target of rapamycin (mTOR), mTOR complex (mTORC), primary cilium, tumor suppressor gene, FLCN, LKB1, folliculin
Introduction
Birt-Hogg-Dubé (BHD)4 syndrome is a rare autosomal dominant genetic disorder characterized by development of hamartomas and cysts in multiple organs, including skin, lung, colon, and kidney (1, 2). The syndrome is caused by germ-line mutations in the BHD gene, which encodes the folliculin protein (FLCN), a 64-kDa polypeptide that shares little sequence similarity with any other known proteins (3). FLCN is found to complex with AMPK and FNIP1, although the significance of the complex for FLCN function remains unclear (4). In mouse models FLCN deficiency leads to development of polycystic kidneys and renal cell carcinoma that are characteristically similar to those found in BHD patients (5, 6). Inactivation of FNIP1, together with its homolog FNIP2, in mice also produces similar phenotypes as does by FLCN deficiency (7), suggesting that FNIP1 may be required for FLCN function. Analyses of tumors derived from BHD patients and FLCN deficient animals have revealed deregulation in mammalian target of rapamycin complex 1 (mTORC1) signaling, a key event in tumorigenesis (5, 8–12). This abnormality in mTORC1 signaling is believed to be a major contributor to the pathological conditions in BHD, as inhibition of mTORC1 with rapamycin has been found to reduce the BHD tumor growth in animal models (5, 6). However, the mechanism by which FLCN regulates mTORC1 remains poorly understood.
At non-cycling resting state, most eukaryotic cells possess a microtubule-based membranous protrusion from cell surface termed as primary cilium (13). This unique structure plays a critical role in maintaining tissue homeostasis by functioning as a sensor for extracellular fluidic shear stress and chemicals (14, 15). Many signaling pathways involved in cell growth and proliferation are regulated by this environmental sensor, among which is the mTORC1 pathway (16–18). Several upstream regulators of mTORC1 have been found to localize to primary cilia, including the tuberous sclerosis complex proteins, LKB1 and AMPK (19–21). A recent study has shown that primary cilia are able to act through a LKB1- and AMPK-dependent mechanism to down-regulate mTORC1 signaling activity and cell size in response to flow stress, demonstrating mTORC1 as a key signaling event mediated by primary cilia (19).
Primary cilium is formed by extension of axonemal microtubules from the centriole derived basal body. Its assembly and maintenance require the intraflagellar transport that is driven by kinesin-2 motor (22). The motor transports proteins toward the “plus” end of microtubule and is comprised of two motor subunits, KIF3A and KIF3B, and an associated protein, KAP3 (23, 24). In a yeast two hybrid screen aiming to isolate FLCN-interacting proteins, we identified one of the motor subunits, KIF3A, as a FLCN-interacting protein, which raised a possibility that FLCN was a ciliary protein. In the present study we show that tumor suppressor FLCN is part of a flow sensory mechanism that regulates mTORC1 through primary cilia.
Experimental Procedures
Cell Lines and Culture
HKC-8 cells were cultured in DMEM/F12 medium supplemented with 10% FBS and 100 units ml−1 penicillin/streptomycin. FLCN null line UOK257 and its corresponding FLCN restored line UOK257–2 were kind gifts from Laura Schmidt and Marston Linehan at NCI and have been described before (25). To induce cilium formation, cells were cultured for 3 additional days after cell density reaching confluence. Antibodies for FLCN (Cat. # 3697), AKT (Cat. # 9272), phospho-AKT(Thr-308) (Cat. # 9275), phospho-AKT(S473), LKB1(Cat. # 3050), AMPKα (Cat. # 2793), phospho-AMPKα(T172) (Cat. # 2535), S6 (Cat. # 2317), phospho-S6 (S235/236) (Cat. # 2211), 4EBP1, phospho-4EBP1(T37/46) (Cat. # 2855), Tsc2 (Cat. # 4308), and phospho-Tsc2(S1387) (Cat. # 5584) were purchased from Cell Signaling Technology. Anti-KIF3A (Cat. # 376680) and KIF3B (Cat. # sc-50456) antibodies were from Santa Cruz Biotechnology. Anti-γ-tubulin (Cat. # T5326) and acetylated-tubulin (Cat. # T7451) antibodies were from Sigma and anti-IFT88 antibody (Cat. # 13967-1-AP) from Proteintech Group Inc.
shRNAs and Inducible Knockdown Cell Lines
The targeting sequences of the shRNAs are: FLCN (5′-GATGGAGAAGCTCGCTGATTT-3′), IFT88 (5′-GCTTGGAGCCTATTACATTGA-3′), and LKB1 (5′-GCCAACGTGAAGAAGGAAATT-3′). The shRNAs were designed and cloned into Tet-pLKO-Puro vector (Addgene). The resulting plasmids were transfected into HKC-8 cells and selected against puromycin. Clones stably expressing the doxycycline-inducible shRNAs were isolated. The inducible cells were cultured in the presence of puromycin and the expression of shRNA was induced by including 100 ng ml−1 of doxycycline in the culture medium. Lentiviral vectors expressing FNIP1 specific shRNAs, pLKO-puro-FNIP1 shRNAs, were purchased from Sigma. Two of the shRNAs (TRCN0000239412 and TRCN0000239413) with similar knockdown efficiency were used. A vector expressing a scrambled shRNA from Addgene was used as the control. The vectors were packaged in lentiviral particles and used for infection of HKC-8 cells following the instruction from pLKO-puro vector supplier (Addgene). The two FNIP1-specific shRNAs produced comparable results. Only the data from TRCN0000239412 were shown (Fig. 3E).
FIGURE 3.

FLCN regulates mTORC1 and AMPK activity in response to flow stress. FLCN-deficient and their wild type control cells were subjected to no-flow or flow stress. Total and phosphorylation levels of indicated proteins in the cells were examined by Western blotting. A, ciliated HKC-8 cells stably expressing doxycycline inducible FLCN shRNA were grown in the absence (Dox −) or presence (Dox +) of doxycycline under flow or no-flow condition. B, quantitative presentation of the relative phosphorylation levels of S6 shown in A (*, p < 0.01). The relative phosphorylation level was expressed as the ratio between the phosphorylation and protein levels that was normalized against that of the control cells grown in the absence of doxycycline and flow stress. Data were from four independent experiments. C, ciliated UOK257–2 (FLCN +) and UOK257 (FLCN −) cells were grown under flow or no-flow condition. D, ciliated HKC-8 cells stably expressing doxycycline inducible IFT88 shRNA were grown in the presence (Dox +) or absence (Dox −) under flow or no-flow condition. E, ciliated HKC-8 cells stably expressing scrambled control (FNIP1 shRNA−) or FNIP1 specific (FNIP1 shRNA+) were subjected to flow or no-flow condition. F, quantitative presentation of the relative phosphorylation levels of AMPK shown in A (*, p < 0.01). The relative phosphorylation level was expressed as the ratio between the phosphorylation and protein levels that was normalized against that of the control cells grown in the absence of doxycycline and flow stress. Data were from four independent experiments. G, HKC-8 cells were grown under flow (Flow +) or no-flow (Flow −) condition for 5 days and treated with (Comp C +) or without (Comp C −) AMPK inhibitor Compound C at a concentration of 10 μm (EMD Millipore) for 24 h. H, HKC-8 cells stably expressing doxycycline inducible FLCN shRNA were grown in the presence (Dox +) or absence (Dox −) for 3 days followed by treatment with 20 mm of 2-deoxyglucose (2DG +) or vehicle control (2DG−) for 2 h.
Flow Assay
2.5 × 105 cells in 320 μl of culture medium were seeded into a μ-slide I0.8 luer chamber (50 × 5× 0.8 mm) pre-coated with poly-l-lysine (ibidi). Cells were allowed to adhere for 8 h before being subjected to flow at a shear stress at 1.0 dyn per cm2 for 6 days. The flow system was set up as described before (19), which contained an air pressure pump and a two-way switch valve that pumped 40 ml of culture medium unidirectionally between two reservoirs through the flow chamber. The whole system except the air pump was hosted in a CO2 incubator. Under this condition, it took 3 days for HKC-8 cells, UOK257, and UOK257-2 cells to reach maximal ciliation. The no-flow control was set up in the same type luer chamber that was placed between two reservoirs containing 40 ml of medium. The two reservoirs were connected through a peristaltic pump controlled by an electrical controller, which turned the pump on for 5 s every hour, allowing ∼0.2 ml of medium to pass through the chamber. The whole no-flow system was hosted in a CO2 incubator.
Co-immunoprecipitation and Western Blotting
Cells were lysed with lysis buffer containing 50 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1 mm PMSF, and 1× protease inhibitor mixture (Roche). Cell debris and unbroken cells were removed by centrifugation at 10,000 × g for 10 min. Clarified lysates were incubated with antibodies overnight followed by addition of Protein A conjugated agarose beads (Life Technologies). After incubation for additional 1.5 h with agitation, beads were washed four times with lysis buffer, once with 20 mm Tris-HCl (pH 7.4), and boiled for 5 min in 60 μl of 2× SDS sample buffer. Samples were subjected to SDS-PAGE. Western blotting was performed by standard protocols and developed using ECL 2 reagents (Pierce). Antibody concentrations were optimized with various dilutions to ensure that the blotting signals are linear to the levels of loaded antigens. Quantitative analysis of the blots was performed with densitometry scanning. Data from at least three independent experiments were analyzed.
Immunofluorescence Microscopy
Cells cultured on glass coverslips or in flow chambers were fixed with 4% paraformaldehyde at room temperature and permeabilized with 0.02% saponin in PBS buffer containing 2% BSA and 1% fish skin gelatin. Cells were incubated overnight at 4 °C with primary antibody followed by 1 h incubation at room temperature with Alexa 488 or Cy3-conjugated secondary antibodies (Life Technologies). DNA was stained with sytox orange (Life Technologies) or DAPI (Fisher Scientific). Fluorescent samples were visualized using Olympus BX61WI FluoviewTM FV1000 confocal microscope with an Olympus PlanAbo 60×/1.45 oil objective. Images were analyzed using Image J (NIH) software to determine the percentage of ciliated cells, cilium length, and fluorescent signal intensity.
Statistical Analysis
Data are presented as means ± S.D., and comparison analyses were done using Student's t test. A p value < 0.05 is considered statistically significant.
Results
FLCN Interacts with Kinesin-2 in a Cilium-dependent Manner
Because kinesin-2 functions mainly in primary cilia, we examined whether the interaction of FLCN with KIF3A was cilium-dependent. Using co-immunoprecipitation, we were able to detect the interaction between the endogenously expressed FLCN and KIF3A in cultured cells (Fig. 1). Importantly, the interaction was detectable only in ciliated resting cells but not in cycling cells, which do not contain primary cilia (Fig. 1, A and B). Removal of primary cilia by shRNA-mediated depletion of IFT88, a gene required for formation of primary cilia (Fig. 1C) (26), abrogated the interaction of FLCN with KIF3A (Fig. 1D), indicating that the interaction takes place in primary cilia. FLCN was also found to associate with the other motor subunit of kinesin-2, KIF3B. Again, the association was cilium-dependent (Fig. 1E). Using an in vitro binding assay, we demonstrated that purified bacterially expressed recombinant FLCN bound directly to the C-terminal globular domain of KIF3A (Fig. 1F), the region involved in interaction with the cargo proteins (23), indicating that FLCN may be a cargo protein for kinesin-2 motor.
FIGURE 1.
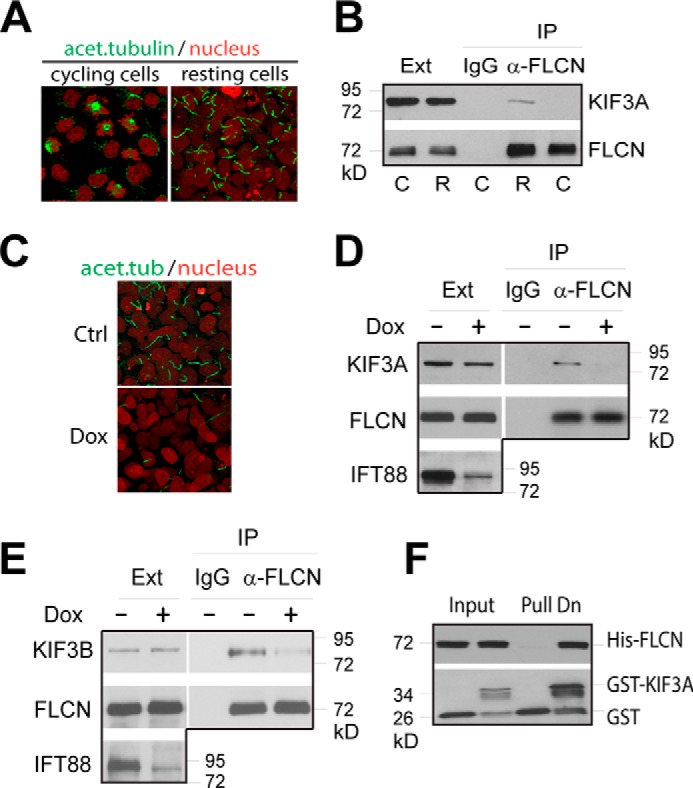
FLCN interacts with kinesin-2. A, HKC-8 cells were harvested before (cycling) or 3 days after cell density reaching confluence (resting). Cells were stained with anti-acetylated tubulin antibody (green) and nuclei with sytox (red) and examined with confocal microscopy. B, extracts from cycling (C) and resting (R) cells were immunoprecipitated with anti-FLCN antibody (α-FLCN) or control IgG. The levels of KIF3A and FLCN in the cell extracts (Ext) and precipitates (IP) were analyzed by Western blotting. C, HKC-8 cells stably expressing a doxycycline inducible IFT88 shRNA were grown in the presence (Dox) or absence (Ctrl) of doxycycline until 3 days after cell density reaching confluence. The ciliation of the cells was shown by fluorescent imaging as in A. D, extracts from cells treated as in C were immunoprecipitated with anti-FLCN antibody (α-FLCN). The levels of KIF3A, FLCN and IFT88 in the extracts (Ext) and precipitates (IP) were examined by Western blotting. E, extracts from cells treated as in C were precipitated with anti-FLCN antibody (α-FLCN) or control IgG. The levels of KIF3B and FLCN in the extracts (Ext) and precipitates (IP) were analyzed by Western blotting. F, purified GST or GST fused C-terminal globular domain (amino acids 597–701) of KIF3A (GST-KIF3A) was incubated with purified His-tagged full length FLCN (His-FLCN). The amounts of His-FLCN co-purified with GST or GST-KIF3A (Pull Dn) were analyzed by Western blotting.
FLCN Localizes to Primary Cilia
The finding that FLCN was able to associate with kinesin-2 indicated that the tumor suppressor might reside within primary cilia. Indeed, FLCN was detected in the primary cilia of UOK257–2 cells that expressed wild type FLCN but not those of FCLN-null UOK257 cells derived from renal tumors of a BHD patient (Fig. 2A) (25). FLCN was also visible in the primary cilia of normal HKC-8 cells but not in those with FLCN expression down-regulated by doxycycline inducible shRNA (Fig. 2B). The ciliary localization of FLCN is consistent with a recent study from van Steensel's group examining the intracellular distribution of the protein (27). In addition, we found that flow stress did not affect the ciliary presentation of the protein in HKC-8 cells (Fig. 2B, lower panels). Collectively, our findings demonstrate that FLCN localizes to primary cilia.
FIGURE 2.

FLCN localizes to primary cilia. A, ciliary localization of FLCN was examined by confocal microscopy in ciliated FLCN wild type (UOK257–2) and null (UOK257) cells grown under no-flow condition. Cilia were stained with anti-acetylated tubulin antibody (green), FLCN with anti-FLCN antibody (red), and nuclei with DAPI (blue). Scale bar, 5 μm. B, HKC-8 cells stably expressing doxycycline inducible FLCN shRNA were grown in the presence (Dox) and absence (Ctrl) of doxycycline under flow (lower panels) or no-flow (upper panels) condition. The ciliary localization of FLCN in the cells was analyzed by confocal microscopy. Scale bar, 5 μm. C and D, length of the primary cilia in the ciliated UOK257–2 (FLCN+) and UOK257 (FLCN−) cells shown in A (C) and the HKC-8 cells shown in B (D) was measured with Image J (NIH) software. A total of 150 ciliated cells from three independent experiments was analyzed for each indicated condition. *, p > 0.05; **, p < 0.05. E and F, frequency of the ciliated cells shown in A (E) and B (F) was analyzed by confocal microscopy. The frequency was expressed as the ratio between the numbers of cilium and those of nucleus. For each of the indicated conditions, the data were collected from a total of 400 cells in three independent experiments and each time cells were sampled at six different fields.
A comparison of the primary cilia in FLCN-null UOK257 cells with those in the paired wild-type control cells revealed that the absence of FLCN did not affect the length of the cilia under no-flow condition (Fig. 2C). When UOK257 cells were grown under flow condition, there was a small but statistically significant decrease in the length of the primary cilia (Fig. 2C). A similar result was seen in HKC-8 cells with FLCN down-regulated by stably expressed doxycycline inducible shRNA. However, the reduction in the cilium length under flow condition was not statistically significant (Fig. 2D). In addition, FLCN deficiency exhibited little effect on the frequency of the ciliated cells under flow and no-flow conditions (Fig. 2, E and F). These observations suggest that FLCN is not required for formation of primary cilia under no-flow and flow condition. Nevertheless, it may have a minor effect on the stability of the cilia under flow condition.
FLCN Regulates mTORC1 Activity in Response to Flow Stress
Primary cilia have been recently shown to play a critical role in repressing mTORC1 activity in noncycling cells in response to flow stress (19). The observation that FLCN is a ciliary protein hence raised a possibility that FLCN might act through primary cilia to control mTORC1 activity. To test this notion, we examined the effect of FLCN depletion on mTORC1 activity in cells grown under flow stress, which bends primary cilia. We found that flow stress effectively reduced mTORC1 activity in HKC-8 cells as measured by the mTORC1-directed phosphorylation of S6 and 4E-BP1 (Fig. 3A). However, the inhibitory effect of flow stress diminished when FLCN was depleted by doxycycline induced shRNA (Fig. 3, A and B). Similarly, flow stress inhibited phosphorylation of S6 and 4E-BP1 in FLCN-expressing UOK257–2 cells but was ineffective in FLCN-null UOK257 cells (Fig. 3C). When formation of primary cilia was blocked by shRNA-mediated knockdown of IFT88, the inhibitory effect of the flow stress on mTORC1 was largely attenuated (Fig. 3D), demonstrating that the effect of flow stress requires primary cilia. Depletion of FNIP1, which forms a complex with FLCN (4), also produced similar effect on mTORC1 activity as did by depletion of FLCN (Fig. 3E), indicating that FNIP1 is involved in the ciliary function of FLCN. Interestingly, when FNIP1 level was reduced by shRNA, the level of FLCN also decreased, indicating that FNIP1 may stabilize FLCN. Under no-flow condition, however, neither knockdown nor absence of FLCN had obvious effect on mTORC1 activity (Fig. 3, A and C). Collectively, the above findings demonstrate that FLCN is essential for the flow stress-mediated down-regulation of mTORC1 in ciliated cells, and suggest that FLCN is part of a flow-sensing mechanism in primary cilia.
FLCN Is Required for Flow Stress-induced AMPK Activation
In response to flow stress, a reduction in AKT activity was observed in HKC-8 cells as measured by its phosphorylation levels at Thr-308 and Ser-473. However, FLCN did not appear to play a role in the process, since its depletion did not impart a difference in the phosphorylation (Fig. 3A). In the BHD tumor-derived UOK257–2 cells, AKT activity appeared to be insensitive to flow stress, yet mTORC1-dependent phosphorylation of S6 and 4E-BP1 remained responsive to the stress (Fig. 3C). These findings suggest that the flow-induced mTORC1 down-regulation is independent of AKT activity. On the other hand, in both HKC-8 and UOK257–2 cells grown under flow conditions, there was an increase in AMPK activity as indicated by the changes in the levels of activation-dependent phosphorylation of the kinase at Thr-172 (Fig. 3, A and C). This increase correlated with an increase in the AMPK-dependent phosphorylation of Tsc2 at serine 1387 (Fig. 3, A and C), as well as a decrease in mTORC1 activity (Fig. 3, B and F). When the flow-induced activation of AMPK was inhibited by an AMPK inhibitor, the Tsc2 phosphorylation and mTORC1 activity became insensitive to flow stress (Fig. 3G), demonstrating that flow stress represses mTORC1 activity through activation of AMPK and phosphorylation of Tsc2. However, when FLCN was depleted by shRNA in HKC-8 cells or was absent as in UOK257 cells, flow stress was unable to activate AMPK, and correlatively, the AMPK-dependent phosphorylation of Tsc2 was not increased and mTORC1 remained largely active (Fig. 3, A and C). This observation suggests that FLCN is required for flow-induced AMPK activation. In contrast, FLCN was not needed for ATP depletion-induced activation of AMPK, since the activation was not affected in FLCN-deficient cells treated with glycolysis inhibitor 2-deoxy-d-glucose (Fig. 3H), which blocks ATP production.
FLCN Recruits LKB1 for AMPK Activation under Flow Stress
FLCN has been previously shown to associate with AMPK under normal growth conditions (4). We thus examined whether flow controls AMPK activation by modulating the association of FLCN with the kinase. We found that the amounts of AMPK co-immunoprecipitated with FLCN were similar under flow and no-flow conditions (Fig. 4A), suggesting that flow does not affect the association. However, we found that the amount of phosphorylated AMPK associated with FLCN was greater under flow condition than that under no-flow condition (Fig. 4A). This observation indicates that FLCN is involved in recruiting an activator of AMPK for its activation in response to flow stress. We thus examined whether the AMPK activation kinase, LKB1, was required for FLCN-mediated AMPK activation. We found that reducing LKB1 levels by shRNA-mediated knockdown attenuated flow-induced AMPK activation (Fig. 4B). To determine whether FLCN is required for recruiting LKB1 to AMPK under flow condition, we examined the association of LKB1 with FLCN. We found that LKB1 was not in complex with FLCN in the absence of flow stress but became associated with it under flow condition (Fig. 4C). Flow also induced association of LKB1 with AMPK (Fig. 4C), which was reduced when FLCN expression was knocked down by shRNA (Fig. 4D), suggesting that the association requires FLCN. When formation of primary cilia was blocked by shRNA-mediated down-regulation of IFT88, the association of LKB1 with FLCN and AMPK also decreased (Fig. 4E), indicating that the association is mediated through primary cilia. Collectively, these results show that FLCN, in response to flow stress, recruits LKB1 to AMPK for its activation.
FIGURE 4.

FLCN recruits LKB1 for AMPK activation in response to flow stress. A, extracts from HKC-8 cells grown under flow (Flow +) or no-flow (Flow −) condition were immunoprecipitated with anti-FLCN antibody (α-FLCN). The levels of the indicated proteins in the extracts (Ext) and precipitates (IP) were determined by Western blotting. B, HKC-8 cells stably expressing doxycycline inducible LKB1 shRNA were grown under flow or no-flow condition in the presence (Dox +) or absence (Dox −). The expression levels of LKB1, AMPK, actin, and phosphorylation of AMPK were analyzed by Western blotting. C, extracts from HKC-8 cells grown under flow (Flow +) or no-flow (Flow −) condition were immunoprecipitated with anti-LKB1 antibody (α-LKB1) or control IgG. D, HKC-8 cells stably expressing doxycycline inducible FLCN shRNA were grown under flow condition in the presence (Dox +) or absence (Dox −) of doxycycline. Extracts from the treated cells were immunoprecipitated with anti-LKB1 antibody (α-LKB1) or control IgG. E, HKC-8 cells stably expressing doxycycline inducible IFT88 shRNA were grown under flow condition in the presence (Dox +) or absence (Dox −) of doxycycline. Extracts from the treated cells were immunoprecipitated with anti-LKB1 antibody (α-LKB1) or control IgG.
FLCN Is Required for Accumulation of LKB1 in Primary Cilia under Flow Stress
To determine whether FLCN recruits LKB1 through primary cilia, we examined the role of FLCN in ciliary localization of LKB1 and AMPK in response to flow. We found that in HKC-8 cells active AMPK accumulated at the basal bodies after the cells were subjected to flow stress. However, in cells depleted of FLCN, the flow-induced accumulation of the active AMPK at the site was abolished (Fig. 5, A–C). In contrast to the active AMPK, LKB1 was not found at the basal bodies. In normal HKC-8 cells without subjecting to flow stress, LKB1 was undetectable in the primary cilia of a majority of the cells (Fig. 5D), with only ∼5 ± 2% of ciliated cells showed a weak LKB1 staining in their cilia (Fig. 5E). Following flow treatment, LKB1 became visible in the cilia of a significant portion (∼45 ± 9%) of the cells (Fig. 5, D and E). In cells depleted of FLCN by inducible shRNA knockdown, however, the flow-induced accumulation of LKB1 in primary cilia was blocked (Fig. 5, D and E). Similar results were obtained with FLCN-null UOK257 cells (data not shown). These observations suggest that FLCN is required for accumulation of LKB1 in primary cilia in response to flow stress. Because primary cilia are required for the association of LKB1 with AMPK under flow condition (Fig. 4E), it is conceivable that FLCN recruits LKB1 through primary cilia before targeting it to basal bodies for AMPK activation.
FIGURE 5.

FLCN is required for ciliary localization of LKB1 and AMPK activation at basal bodies. HKC-8 cells stably expressing doxycycline inducible FLCN shRNA were grown in the absence (Ctrl) or presence (Dox) of doxycycline under flow or no-flow condition. A, cells were stained with anti-phospho-AMPK antibody (red), anti-acetylated tubulin (green) antibody that marked primary cilia, and DAPI (blue). B, cells were stained with anti-phospho-AMPK antibody (red), anti-γ tubulin antibody (green) that marked basal bodies, and DAPI (blue). C, quantitative presentation of phosphorylated AMPK shown in B. The density of the antibody staining at the basal bodies was analyzed with Image J (NIH) software. The relative levels of phosphorylated AMPK at the basal bodies were expressed as the ratio between the fluorescent intensities of phospho-AMPK and tubulin staining. A total of 100 pairs of basal bodies from three independent experiments performed under identical experimental settings was analyzed for each indicated condition. *, p < 0.01. D, cells were stained with anti-acetylated tubulin antibody (green), anti-LKB1 antibody (red), and DAPI (blue). The stained cells were imaged by confocal microscopy. Scale bar, 5 μm. E, percentage of the ciliated cells displayed detectable LKB1 under the indicated conditions was determined. A total of 200 ciliated cells from three independent experiments performed under identical experimental settings was examined for each condition. #, p < 0.001.
Discussion
Since its identification more than a decade ago, FLCN has been implicated in regulation of mTORC1. However, the underlying molecular mechanism remains unsolved (2). Although FLCN has been found in complex with AMPK in cycling cells, the significance of the association is unclear, as FLCN does not appear to affect AMPK activity or the response of AMPK to changes in intracellular energy levels (4). A recent study demonstrates that FLCN affects cellular energy homeostasis through an AMPK-dependent manner. However, it is unclear how FLCN regulates AMPK (28). In animal studies conflicting evidence exist showing opposite effects of FLCN on mTORC1 activity (5, 8–12), which has led to the suggestion that the function of FLCN may be cell-content dependent (12). In the present study we show that FLCN is a ciliary protein acting in a cilium-dependent manner, which provides an explanation for its elusive action in mTORC1 regulation. Our findings identify FLCN as an upstream regulator of LKB1 that is responsible for recruiting the kinase to primary cilia under flow stress. How FLCN recruits LKB1 to primary cilia remains unclear. It is possible that the recruitment involves other proteins. Alternatively, FLCN may act to retain LKB1 within primary cilia. While additional studies are needed to define the underlying mechanism, our findings demonstrate that the FLCN mediated recruitment is responsible for bringing LKB1 to its downstream target, AMPK, localized at basal bodies. The association of LKB1 with AMPK results in its activation and consequently phosphorylation of Tsc2, a negative regulator of mTORC1 (Fig. 6). A previous study has shown that Tsc1 is localized to basal bodies in ciliated cells. It is likely that Tsc2, the binding and functional partner of Tsc1, is also associated with basal bodies, allowing phosphorylation by AMPK. In addition to Tsc2, AMPK has also been shown to phosphorylate Raptor, the regulatory subunit of mTORC1, when cells are subjected to energy stress (29, 30). However, we did not observe any obvious effect of FLCN deficiency on the AMPK-dependent phosphorylation of Raptor under flow and no-flow conditions (data not shown). This result is consistent with the observation that FLCN did not affect AMPK activation under metabolic stress in cycling cells (Fig. 3H), suggesting that FLCN controls mTORC1 through AMPK-dependent phosphorylation of Tsc2 but not of Raptor. Taken together, our findings unveil the action mechanism of the tumor suppressor in the cilium-dependent regulation of mTORC1.
FIGURE 6.
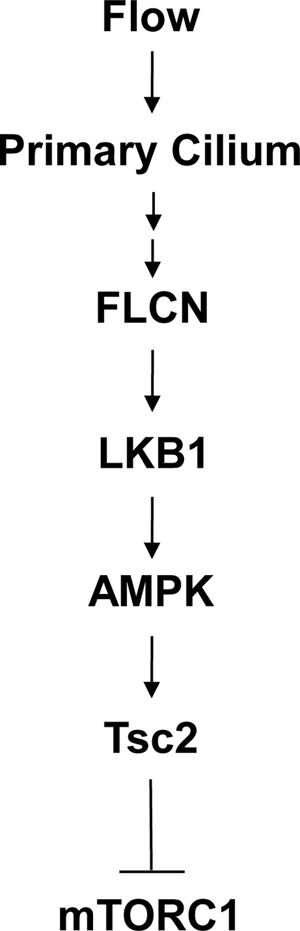
A schematic illustration of the ciliary action of FLCN in mTORC1 regulation (See text for detail).
The ability of FLCN to interact directly with kinesin-2 motor suggests that this protein is involved in intraflagellar trafficking either as a transporter or as a cargo protein. However, the nonessential role of FLCN in formation of primary cilia distinguishes it from the intraflagellar transport proteins (IFT), such as IFT88, that supply materials for axonemal growth (31). The fact that the activity of FLCN is manifested only under flow condition further indicates FLCN as a part of the mechanosensory machinery than a structural component of primary cilia. It is likely that FLCN is involved in trafficking signaling molecules or maintaining their steady state levels within the organelle in response to flow and other environmental signals. In this regard, the FLCN-mediated accumulation of LKB1 in primary cilia represents a unique signaling mechanism for the function of primary cilia, as it suggests that flow stress signal controls cilium signaling through regulating the ciliary traffic of signaling molecules. A key question remained to be answered is how FLCN, a soluble intracellular protein, is able to sense flow stress. On this point, a potential link between FLCN and ciliary mechanosensory proteins, such as polycystin-1 and 2, warrants further studies.
The primary cilium is a signaling hub for noncycling resting cells and plays an important role in maintaining tissue homeostasis (32). Many tumor suppressors, including polycystin-1, LKB1, Ptch1, and pVHL, are found to function through this organelle (33). Defects in cilium formation and signaling have been associated with tumorigenesis in several organs, including kidney, skin, and lung (34, 35), which are manifested in BHD syndrome. Our findings that FLCN controls LKB1 accumulation and mTORC1 activation through primary cilia suggest that FLCN, like many other tumor suppressors, may elicit its anti-tumor activity through this organelle in quiescent cells.
Author Contributions
M. Z. conducted the imaging studies shown in Figs. 2 and 5 and part of biochemical analyses shown in Figs. 1 and 3. X. Z. did the biochemical analyses shown in Figs. 1, 3, and 4. J. L., G. Y., M. T., S. G., Y. Z., and Y. L. performed experiments. Yong J., Y. L., and Yu J. conceived and planned the experiments and interpreted data. Yu J. wrote the manuscript.
Acknowledgments
We thank Laura Schmidt and Marston Linehan at NIH for providing FLCN plasmid, UOK257, and UOK257–2 cell lines and appreciate other laboratory members for comments and discussion during the course of the study.
This work was supported by National Institutes of Health Grant CA169186 (to Yu J.) and National Natural Science Foundation of China Grants 81272269 (to Yu J.) and 81372030 (to Yong J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BHD
- Birt-Hogg-Dubé
- FLCN
- folliculin
- AMPK
- AMP-activated kinase
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex.
References
- 1. Birt A. R., Hogg G. R., and Dubé W. J. (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol. 113, 1674–1677 [PubMed] [Google Scholar]
- 2. Schmidt L. S. (2013) Birt-Hogg-Dube syndrome: from gene discovery to molecularly targeted therapies. Familial Cancer 12, 357–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nickerson M. L., Warren M. B., Toro J. R., Matrosova V., Glenn G., Turner M. L., Duray P., Merino M., Choyke P., Pavlovich C. P., Sharma N., Walther M., Munroe D., Hill R., Maher E., Greenberg C., Lerman M. I., Linehan W. M., Zbar B., and Schmidt L. S. (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2, 157–164 [DOI] [PubMed] [Google Scholar]
- 4. Baba M., Hong S. B., Sharma N., Warren M. B., Nickerson M. L., Iwamatsu A., Esposito D., Gillette W. K., Hopkins R. F. 3rd, Hartley J. L., Furihata M., Oishi S., Zhen W., Burke T. R. Jr., Linehan W. M., Schmidt L. S., and Zbar B. (2006) Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 15552–15557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baba M., Furihata M., Hong S. B., Tessarollo L., Haines D. C., Southon E., Patel V., Igarashi P., Alvord W. G., Leighty R., Yao M., Bernardo M., Ileva L., Choyke P., Warren M. B., Zbar B., Linehan W. M., and Schmidt L. S. (2008) Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 100, 140–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen J., Futami K., Petillo D., Peng J., Wang P., Knol J., Li Y., Khoo S. K., Huang D., Qian C. N., Zhao P., Dykyma K., Zhang R., Cao B., Yang X. J., Furge K., Williams B. O., and Teh B. T. (2008) Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PloS one 3, e3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hasumi H., Baba M., Hasumi Y., Lang M., Huang Y., Oh H. F., Matsuo M., Merino M. J., Yao M., Ito Y., Furuya M., Iribe Y., Kodama T., Southon E., Tessarollo L., Nagashima K., Haines D. C., Linehan W. M., and Schmidt L. S. (2015) Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc. Natl. Acad. Sci. U.S.A. 112, E1624–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baba M., Keller J. R., Sun H. W., Resch W., Kuchen S., Suh H. C., Hasumi H., Hasumi Y., Kieffer-Kwon K. R., Gonzalez C. G., Hughes R. M., Klein M. E., Oh H. F., Bible P., Southon E., Tessarollo L., Schmidt L. S., Linehan W. M., and Casellas R. (2012) The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dube syndrome is required for murine B-cell development. Blood 120, 1254–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takagi Y., Kobayashi T., Shiono M., Wang L., Piao X., Sun G., Zhang D., Abe M., Hagiwara Y., Takahashi K., and Hino O. (2008) Interaction of folliculin (Birt-Hogg-Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene 27, 5339–5347 [DOI] [PubMed] [Google Scholar]
- 10. Hasumi Y., Baba M., Ajima R., Hasumi H., Valera V. A., Klein M. E., Haines D. C., Merino M. J., Hong S. B., Yamaguchi T. P., Schmidt L. S., and Linehan W. M. (2009) Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. U.S.A. 106, 18722–18727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hartman T. R., Nicolas E., Klein-Szanto A., Al-Saleem T., Cash T. P., Simon M. C., and Henske E. P. (2009) The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene 28, 1594–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hudon V., Sabourin S., Dydensborg A. B., Kottis V., Ghazi A., Paquet M., Crosby K., Pomerleau V., Uetani N., and Pause A. (2010) Renal tumour suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J. Med. Genet. 47, 182–189 [DOI] [PubMed] [Google Scholar]
- 13. Satir P., Pedersen L. B., and Christensen S. T. (2010) The primary cilium at a glance. J. Cell Sci. 123, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singla V., and Reiter J. F. (2006) The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629–633 [DOI] [PubMed] [Google Scholar]
- 15. Pazour G. J., and Witman G. B. (2003) The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol. 15, 105–110 [DOI] [PubMed] [Google Scholar]
- 16. Berbari N. F., O'Connor A. K., Haycraft C. J., and Yoder B. K. (2009) The primary cilium as a complex signaling center. Curr. Biol. 19, R526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goetz S. C., and Anderson K. V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huber T. B., Walz G., and Kuehn E. W. (2011) mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int. 79, 502–511 [DOI] [PubMed] [Google Scholar]
- 19. Boehlke C., Kotsis F., Patel V., Braeg S., Voelker H., Bredt S., Beyer T., Janusch H., Hamann C., Gödel M., Müller K., Herbst M., Hornung M., Doerken M., Köttgen M., Nitschke R., Igarashi P., Walz G., and Kuehn E. W. (2010) Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 12, 1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dere R., Wilson P. D., Sandford R. N., and Walker C. L. (2010) Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PloS one 5, e9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartman T. R., Liu D., Zilfou J. T., Robb V., Morrison T., Watnick T., and Henske E. P. (2009) The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 18, 151–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S., and Dynlacht B. D. (2013) Assembling a primary cilium. Curr. Opin. Cell Biol. 25, 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scholey J. M. (2013) Kinesin-2: a family of heterotrimeric and homodimeric motors with diverse intracellular transport functions. Annu. Rev. Cell Dev. Biol. 29, 443–469 [DOI] [PubMed] [Google Scholar]
- 24. Verhey K. J., Dishinger J., and Kee H. L. (2011) Kinesin motors and primary cilia. Biochem. Soc. Trans. 39, 1120–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Y., Padilla-Nash H. M., Vira M. A., Abu-Asab M. S., Val D., Worrell R., Tsokos M., Merino M. J., Pavlovich C. P., Ried T., Linehan W. M., and Vocke C. D. (2008) The UOK 257 cell line: a novel model for studies of the human Birt-Hogg-Dube gene pathway. Cancer Genet. Cytogenet. 180, 100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pazour G. J., Dickert B. L., Vucica Y., Seeley E. S., Rosenbaum J. L., Witman G. B., and Cole D. G. (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 151, 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luijten M. N., Basten S. G., Claessens T., Vernooij M., Scott C. L., Janssen R., Easton J. A., Kamps M. A., Vreeburg M., Broers J. L., van Geel M., Menko F. H., Harbottle R. P., Nookala R. K., Tee A. R., Land S. C., Giles R. H., Coull B. J., and van Steensel M. A. (2013) Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum. Mol. Genet. 22, 4383–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan M., Gingras M. C., Dunlop E. A., Nouët Y., Dupuy F., Jalali Z., Possik E., Coull B. J., Kharitidi D., Dydensborg A. B., Faubert B., Kamps M., Sabourin S., Preston R. S., Davies D. M., Roughead T., Chotard L., van Steensel M. A., Jones R., Tee A. R., and Pause A. (2014) The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J. Clin. Invest. 124, 2640–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoki K., Zhu T., and Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 30. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scholey J. M. (2008) Intraflagellar transport motors in cilia: moving along the cell's antenna. J. Cell Biol. 180, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lancaster M. A., and Gleeson J. G. (2009) The primary cilium as a cellular signaling center: lessons from disease. Curr. Opin. Genet. Dev. 19, 220–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan J., Seeger-Nukpezah T., and Golemis E. A. (2013) The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell. Mol. Life Sci. 70, 1849–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hildebrandt F., Benzing T., and Katsanis N. (2011) Ciliopathies. New Engl. J. Med. 364, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quinlan R. J., Tobin J. L., and Beales P. L. (2008) Modeling ciliopathies: Primary cilia in development and disease. Curr. Top. Dev. Biol. 84, 249–310 [DOI] [PubMed] [Google Scholar]