

Abstract
αIIbβ3 activation in platelets is followed by activation of the tyrosine kinase c-Src associated with the carboxyl terminus of the β3 cytosolic tail. Exogenous peptides designed to interact with the αIIb transmembrane (TM) domain activate single αIIbβ3 molecules in platelets by binding to the αIIb TM domain and causing separation of the αIIbβ3 TM domain heterodimer. Here we asked whether directly activating single αIIbβ3 molecules in platelets using the designed peptide anti-αIIb TM also initiates αIIbβ3-mediated outside-in signaling by causing activation of β3-associated c-Src. Anti-αIIb TM caused activation of β3-associated c-Src and the kinase Syk, but not the kinase FAK, under conditions that precluded extracellular ligand binding to αIIbβ3. c-Src and Syk are activated by trans-autophosphorylation, suggesting that activation of individual αIIbβ3 molecules can initiate αIIbβ3 clustering in the absence of ligand binding. Consistent with this possibility, incubating platelets with anti-αIIb TM resulted in the redistribution of αIIbβ3 from a homogenous ring located at the periphery of discoid platelets into nodular densities consistent with clustered αIIbβ3. Thus, these studies indicate that not only is resting αIIbβ3 poised to undergo a conformational change that exposes its ligand-binding site, but it is poised to rapidly assemble into intracellular signal-generating complexes as well.
Keywords: integrin, platelet, signal transduction, Src, transmembrane domain
Introduction
αIIbβ3 activation in platelets is followed by αIIbβ3 clustering (1) and αIIbβ3-mediated “outside-in” signal transduction (2) that is initiated by activation of the tyrosine kinase c-Src associated with the carboxyl terminus of the β3 cytosolic tail (CT)3 (3–5). Activated c-Src then initiates an intracellular signaling cascade culminating in the reorganization of the platelet cytoskeleton, platelet spreading, and fibrin clot retraction, events important for efficient hemostasis in the hemodynamic environment of flowing blood (6).
Recently, we reported studies of the interaction of c-Src with the β3 CT (5). We detected little to no interaction in unstimulated platelets, but following platelet stimulation with thrombin, c-Src associated with β3 in a time-dependent manner and underwent transient activation. We also found that the β3 CT binds to the c-Src SH3 domain at a site that is occupied by the linker connecting the c-Src SH2 and kinase domains in inactive c-Src. Following platelet activation, c-Src is “unlatched,” allowing β3 CT binding to the SH3 domain when the linker binding site is vacated. However, the full catalytic activity of c-Src requires phosphorylation of Tyr419 (numbered according to the UniProtKB/Swiss-Prot entry P12931 for human c-Src), located in a loop between the two lobes of the c-Src kinase domain (7). In platelets, this occurs by trans-autophosphorylation when αIIbβ3 clustering brings β3 CT-bound c-Src into proximity (3). Thus, αIIbβ3 clustering is an essential step in the outside-in signaling that follows αIIbβ3 activation.
Transmembrane (TM) helix-helix interactions play an important role in maintaining αIIbβ3 in its inactive state, and disrupting these interactions is sufficient to cause αIIbβ3 activation (8, 9). Membrane-soluble peptides corresponding to the αIIb TM domain (10) or designed to interact strongly with the αIIb TM domain (11) cause αIIbβ3 activation, as well as platelet aggregation, by directly disrupting the TM domain interactions, mimicking the effect of the physiologic αIIbβ3 activator talin (Fig. 1) (12). Moreover, these peptides activate αIIbβ3 in platelets in the absence of inside-out signaling. Thus, they provide a way to test whether directly activating individual αIIbβ3 molecules can also cause αIIbβ3 clustering. Here we used the computationally designed peptide anti-αIIb TM (11) to directly activate single αIIbβ3 molecules in platelets and used the trans-autoactivation of β3-associated c-Src as a probe for αIIbβ3 clustering. We found that anti-αIIb TM caused αIIbβ3-dependent c-Src activation under conditions that preclude ligand binding to αIIbβ3 and platelet actin polymerization. These results suggest that not only is resting αIIbβ3 poised to undergo a conformational change that exposes its ligand binding site, but it is poised to rapidly oligomerize into intracellular signal-generating complexes as well.
FIGURE 1.
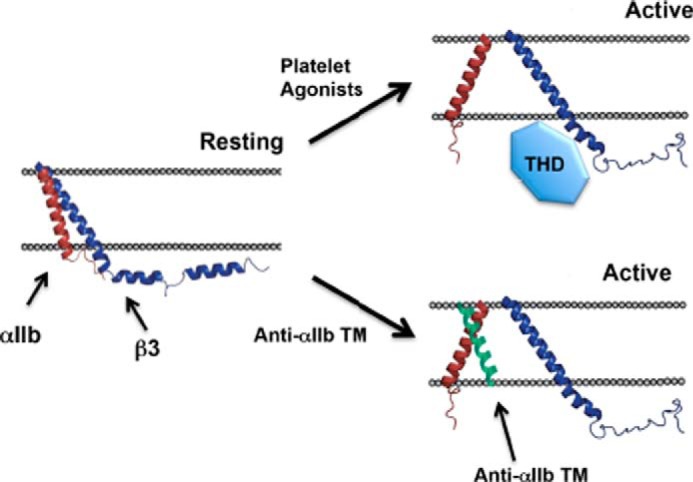
The talin head domain and the membrane-soluble peptide anti-αIIb TM each induce αIIbβ3 activation by causing separation of the αIIbβ3 TM domain heterodimer. The talin head domain (THD) sterically disrupts the TM domain heterodimer (52). Anti-αIIb TM disrupts the TM domain heterodimer by binding to the face of the αIIb TM helix that would otherwise interact with the β3 TM helix (11, 53).
Experimental Procedures
Peptide Synthesis
The peptides anti-αIIb TM (11) (KKAYVMLLPFFIGLLLGLIFGGAFWGPARHLKK) and MS1 (13) (BQLLIAVLLLIAVNLILLIAVARLRYLVG, where B represents β-alanine) were synthesized with a Discover microwave peptide synthesizer (CEM, Matthews, NC) using the standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) protection strategy and on a Rink Amide AM resin (200–400 mesh) (Nova Biochem) with a substitution level of 0.71 mmol/g. Five equivalents of HBTU coupling reagent (Pepnet) and five equivalents of amino acid (ChemPep) were used per coupling. Each coupling was performed in duplicate with a coupling time of 2 min. After cleavage and workup, peptides with an amidized C terminus and a free amino N terminus were characterized using a Perseptive Biosystems MALDI-TOF mass spectrometer. Purification of peptides was carried out on a Waters 600E HPLC equipped with a SepaxGP-C8 reverse-phase, 21.2 × 250 mm column over a 50:50 to 0:100 (water, 0.1% TFA:acetonitrile, 0.1% TFA) gradient for 30 min. Fractions were characterized with MALDI mass spectrometry and lyophilized to dryness. The purity of each product was verified by HPLC.
Measurements of Platelet Function
Human platelet studies were approved by the Institutional Review Board of the University of Pennsylvania Office of Regulatory Affairs. Platelet aggregation and ATP secretion were measured using a Chrono-Log model 700 lumi-aggregometer (10). Human platelets, obtained from blood anticoagulated with sodium citrate (65 mm), citric acid (77 mm), and glucose (95 mm) (pH 4.4) were washed with 10 mm HEPES buffer (pH 6.5) containing 150 mm NaCl, 3 mm EDTA, 1 μm PGE1, and 0.3 units/ml apyrase and resuspended in modified Tyrode's buffer (20 mm HEPES (pH 7.35) containing 135 mm NaCl, 2.7 mm KCl, 3 mm NaH2PO4, 5 mm glucose, and 0.1% BSA). 450-μl aliquots of the platelet suspension supplemented with 200 μg/ml human fibrinogen and 1 mm CaCl2 were stirred at 1200 rpm in siliconized aggregometer cuvettes. Secreted ATP secretion was measured using 50 μl of CHRONO-LUMETM reagent (firefly luciferin-luciferase) and analyzed using AGGRO/LINK8® software. Membrane-soluble peptides were dissolved in DMSO solution, and final DMSO concentrations were no greater than 0.5%.
FITC-conjugated fibrinogen binding to washed platelets was measured as described previously (14). Briefly, 500-μl aliquots of the washed platelets were stimulated in the presence of 200 μg/ml FITC-conjugated fibrinogen. The platelets were then fixed with 0.37% formalin in PBS buffer for 10 min, washed, and examined by FACS.
Immunoprecipitation and Immunoblotting
500-μl aliquots of washed or gel-filtered human platelets (15) were incubated with 10 μg/ml collagen, 1 unit/ml thrombin, or 2 μm anti-αIIb TM for 1 min at 37 °C, after which the platelets were lysed with 125 μl of 50 mm Tris buffer containing 1% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, and protease and phosphatase inhibitors (protease inhibitor mixture, Sigma-Aldrich; phosphatase inhibitor mixture set III, Millipore). In some experiments, proteins were immunoprecipitated from the lysates, separated in 4–12% NuPAGE Bis-Tris gels (Life Technologies), and transferred to nitrocellulose membranes (IB301001, Life Technologies) for immunoblotting. Immunoblotted proteins were detected using horseradish peroxidase-conjugated anti-IgG and ECL Western blotting detection reagent (GE Healthcare Life Sciences). β3 was immunoprecipitated and immunoblotted with either the β3-specific mAb SSA6 (16) or the mAb D-11 (Santa Cruz Biotechnology). c-Src was immunoblotted with the antibody 36D10 (Cell Signaling Technology), and phosphorylated c-Src residue 419 (Tyr(P)419) was detected with antibody 2101 (Cell Signaling Technology). Syk was immunoprecipitated using the mAb G-2 (Santa Cruz Biotechnology) and FAK using the mAb 4.47 (Merck Millipore). Tyrosine-phosphorylated Syk and FAK were detected by immunoblotting with 4G10 (Merck Millipore). Talin was immunoblotted using mAb TA205 (Merck Millipore). Kindlin-3 antibodies were prepared against a kindlin-3 C-terminal peptide, C-GEELDEDLFLQLTG (residues 645–658), containing an N-terminal cysteine residue for coupling to maleimide-activated blue carrier protein (Pierce/Thermo Fisher). Monospecific polyclonal antibodies were then produced in rabbits by Cocalico Biologicals (Reamstown, PA). After initial immunoblotting, membranes were stripped of protein-bound antibody using Restore Western blot stripping buffer (Thermo Fisher Scientific), blocked with 5% nonfat dry milk (Bio-Rad), and immunoblotted again for either β3 using D-11, c-Src using 36D10, or FAK using 4.47.
Immunostaining of Platelet αIIbβ3
To visualize αIIbβ3 on the surface of resting platelets and platelets incubated with anti-αIIb TM, washed human platelets were resuspended at a concentration of 2.5 × 108 platelets/ml in Tyrode's buffer containing 5 mm EDTA but without BSA. 500-μl aliquots of the platelet suspension were then transferred to BSA-blocked centrifuge tubes and incubated for 1 min at 37 °C in the presence or absence of 2 μm anti-αIIb TM (8). Following the incubation, the platelets were fixed by adding 500 μl of 4% paraformaldehyde for 15 min at room temperature and transferred to poly-lysine-coated chamber glass slides. The platelet-containing chambers were then incubated sequentially with 10% goat serum for 1 h at room temperature and with the anti-β3 mAb SSA6 labeled with Alexa Fluor® 488 (Thermo Fisher Scientific) for 1 h at 37 °C. After washing with PBS, the stained platelets were mounted with Fluoromount G (Southern Biotech) and visualized using a Zeiss Axioplan upright microscope with a Zeiss FluoArc mercury lamp. Images were captured using a Zeiss AxioCam HRm high-resolution monochrome charge-coupled device camera and AxioVision 4.5 software (Zeiss).
Results
Role of c-Src in Anti-αIIb TM-induced Platelet Aggregation and Secretion
To address whether c-Src is involved in platelet function stimulated by anti-αIIb TM, we compared the effects of the peptide to the effects of low and high concentrations of thrombin on the function of normal platelets in the absence and presence of the c-Src inhibitor PP2. As shown in Fig. 2A, 2 μm anti-αIIb TM caused both platelet aggregation and ATP secretion. However, 10 μm PP2 attenuated anti-αIIb TM-induced platelet aggregation and abrogated peptide-induced platelet ATP secretion. Likewise, both platelet aggregation and ATP secretion stimulated by 0.1 units/ml thrombin were inhibited by 10 μm PP2. By contrast, when platelets were stimulated by thrombin at 5 units/ml, 10 μm PP2 had no effect on ATP secretion, although platelet aggregation was modestly attenuated. Thus, as noted previously by Liu et al. (17), when platelets were stimulated by γ-thrombin, c-Src activation appears to potentiate the effects of weaker platelet stimuli such as anti-αIIb TM and 0.1 units/ml thrombin, but increasing the strength of the platelet stimulus, in this case increasing the thrombin concentration to 5 units/ml, largely bypasses the c-Src requirement.
FIGURE 2.

Anti-αIIb TM causes platelet aggregation and c-Src-dependent ATP secretion by causing αIIbβ3 activation. A, simultaneous platelet aggregation and ATP secretion stimulated by 2 μm anti-αIIb TM, 0.1 unit/ml thrombin, or 5 units/ml thrombin was measured in the presence or absence of the Src kinase inhibitor 10 μm PP2. These experiments were repeated four times using platelets from four different blood donors with identical results. B, comparison of anti-αIIb TM- and thrombin-induced ATP secretion from the platelets of a well characterized patient with GT. These experiments were repeated using platelets from another patient with GT with identical results. C, thrombin (Tb) causes c-Src phosphorylation in GT and WT platelets. GT and WT platelets were stimulated with 1 unit/ml thrombin in the presence or absence of PP2. c-Src was then immunoprecipitated from platelet lysates and immunoblotted for c-Src pTyr419. MW, molecular weight. D, platelet aggregation and ATP secretion following platelet incubation with the membrane-soluble peptide MS1. These experiments were performed five times with platelets from different blood donors with similar results. E, anti-αIIb TM causes fibrinogen binding to αIIbβ3 on human platelets. Human platelets were incubated with 2 μm anti-αIIb TM for 1 min in the presence of 200 μg/ml FITC-labeled human fibrinogen, after which fibrinogen binding to αIIbβ3 was measured by fluorescence-activated cell sorting. The specificity of fibrinogen binding to αIIbβ3 was verified using either 5 mm EDTA or 10 μm eptifibatide. The contribution of secreted ADP or activated c-Src to fibrinogen binding was tested by adding either 10 units/ml apyrase or 10 μm PP2 to the incubation. These measurements were repeated using platelets from a different platelet donor with identical results.
Next we asked whether the effects of anti-αIIb TM on platelets require the presence of αIIbβ3 by measuring ATP secretion from the platelets of a well characterized patient with Glanzmann thrombasthenia (GT) whose platelets lack αIIbβ3 (18). Although 0.1 units/ml thrombin caused brisk ATP secretion from the GT platelets, an anti-αIIb TM concentration that caused robust ATP secretion from normal platelets, had no effect on GT platelets (Fig. 2B). On the other hand, c-Src in both wild-type control and GT platelets undergoes phosphorylation when the platelets are stimulated by thrombin, confirming that c-Src in GT platelets can be activated by a stimulus that is independent of αIIbβ3 (Fig. 2C). Studies using platelets from another well characterized patient with GT (19) produced identical results (data not shown). Nonetheless, it is still possible that the functional effects of anti-αIIb TM are due to its membrane solubility. To test this possibility, we studied the effects of an unrelated membrane-soluble peptide on platelet aggregation and secretion. The membrane-soluble peptide MS1 was designed using the two-stranded coiled-coil peptide GCN4-P1 as a template and is insoluble in water but readily solubilized in a variety of detergents (13) and inserts into lipid bilayers (20). As seen in Fig. 2D, incubating platelets with 2 μm MS1 caused neither platelet aggregation nor ATP release. Thus, these experiments indicate that the ability of anti-αIIb TM to initiate platelet function requires the presence of αIIbβ3.
To test this conclusion and to address whether anti-αIIb TM-induced αIIbβ3 activation requires c-Src kinase activity, we measured anti-αIIb TM-induced fibrinogen binding to platelet αIIbβ3 in the absence or presence of PP2. As shown by the flow cytometry histograms in Fig. 2E, 2 μm anti-αIIb TM caused FITC-fibrinogen binding to washed human platelets. The platelet-associated fibrinogen was bound to αIIbβ3 because binding was inhibited by the αIIbβ3 antagonist eptifibatide (21) and prevented by the divalent cation chelator EDTA (10, 11). On the other hand, fibrinogen binding was only slightly decreased by the ADP-metabolizing enzyme apyrase, indicating that anti-αIIb TM-stimulated αIIbβ3 activation is not mediated by secreted platelet ADP (10, 11). The small attenuation is likely due to the lack of signal amplification caused by secondary stimulation from secreted ADP. Furthermore, fibrinogen binding was unaffected by 10 μm PP2, indicating that it did not require c-Src kinase activity. Thus, these results indicate that anti-αIIb TM initiates platelet activation by binding and altering the conformation of αIIbβ3 and that this effect on αIIbβ3 is independent of platelet secretion and c-Src activity. Nonetheless, the attenuated platelet aggregation and lack of ATP secretion we observed in the presence of PP2 demonstrates that c-Src-stimulated platelet ADP secretion potentiates the platelet aggregation induced by the peptide.
Anti-αIIb TM Causes the Activation of β3-bound c-Src in Platelets
Platelet agonists like thrombin cause the rapid and transient activation of β3-bound c-Src (5). To determine whether anti-αIIb TM has the same effect, we measured the anti-αIIb TM-stimulated interaction of c-Src with the β3 CT. Although we found no detectable c-Src associated with β3 in unstimulated platelets, c-Src binding to β3 was detected within 10 s after platelet exposure to anti-αIIb TM (Fig. 3). Similar to platelets stimulated by thrombin (5), the amount of β3-bound c-Src increased progressively as the duration of exposure to anti-αIIb TM increased, reaching a maximum at 40 s, after which it declined. Phosphorylation of the β3-bound c-Src was first detected at 10 s. Like c-Src binding, it increased progressively, reaching a maximum at 40–60 s, after which it also declined (Fig. 3). Thus, like conventional platelet agonists, anti-αIIb TM causes rapid c-Src binding to β3 and its subsequent phosphorylation.
FIGURE 3.

Anti-αIIb TM causes c-Src binding to the β3 cytosolic tail and the subsequent phosphorylation of c-Src residue Tyr419. A, time course of c-Src binding to β3 and c-Src phosphorylation when platelets were incubated with 2 μm anti-αIIb TM. At the indicated times, platelets were lysed with 1% Nonidet P-40, β3 was immunoprecipitated (IP), and the immunoprecipitated β3 was immunoblotted (IB) for c-Src, phosphorylated c-Src residue 419 (pTyr419), and β3. The time course experiments were performed five times. B and C, the bar graphs were generated from densitometry of the immunoblots from the five experiments and represent the mean ± S.E. of β3-bound c-Src (B) and β3-bound c-Src Tyr(P)419 (C).
c-Src represents 0.2–0.4% of total platelet protein (22), but only 3% is associated with the β3 CT (3). Thus, it is conceivable that the c-Src phosphorylation caused by anti-αIIb TM could be an off-target effect, unrelated to the ability of anti-αIIb TM to bind to αIIb, thereby releasing β3 to interact with cytoplasmic proteins (Fig. 1). To test this possibility, we compared anti-αIIb TM-induced c-Src phosphorylation in GT and WT control platelets. Although c-Src was readily detected in both the GT and WT platelets, we did not detect phosphorylated Tyr419 when the GT platelets were stimulated with either 10 μg/ml collagen or 2 μm anti-αIIb TM (Fig. 4A). Thus, these results indicate that anti-αIIb TM-induced phosphorylation of c-Src on Tyr419 occurs in an αIIbβ3-specific manner. However, it remains possible that c-Src activation is a nonspecific consequence of simply incubating platelets with a hydrophobic membrane-soluble peptide. To address this possibility, we incubated platelets with the membrane-soluble peptide MS1 as a negative control. However, as shown in Fig. 4B, incubating platelets with MS1 did not cause c-Src phosphorylation. Taken together, these results confirm that anti-αIIb TM-induced c-Src phosphorylation occurs when anti-αIIb TM interacts with αIIbβ3.
FIGURE 4.

c-Src phosphorylation induced by anti-αIIb TM requires the presence of αIIbβ3. A, WT and GT platelets were incubated for 1 min with 10 μg/ml collagen, 2 μm anti-αIIb TM, or 2 μm anti-αIIb TM plus 10 μm PP2. Platelet lysates were then immunoblotted (IB) for β3, c-Src Tyr(P)419, and c-Src. MW, molecular weight. B, washed platelets were incubated with buffer, 2 μm anti-αIIb TM, 1 units/ml thrombin, or 2 μm MS1 for 1 min. Platelet lysates were then immunoblotted for c-Src and c-Src Tyr(P)419. C, washed platelets were incubated with buffer or 2 μm anti-αIIb TM for 1 min in the absence or the presence of 5 mm EDTA, 10 μm eptifibatide, 4 μm R406, 10 μm cytochalasin D (Cyto D), 2 μm latrunculin (Lat A), 10 μm PP2, or 10 μm PP3. c-Src was immunoprecipitated from platelet lysates and immunoblotted for c-Src and c-Src Tyr(P)419. Top panel, c-Src Tyr(P)419. Bottom panel, c-Src loading control. The experiments were performed three times. D, the bar graph was generated from densitometry of the immunoblots from the three experiments and represents the mean ± S.E. of c-Src Tyr(P)419 normalized using the densitometry of the corresponding c-Src band.
Next, to address whether anti-αIIb TM-induced c-Src activation requires ligand binding to αIIbβ3, we incubated washed platelets suspended in buffer lacking fibrinogen and containing αIIbβ3 antagonists with 2 μm anti-αIIb TM for 1 min. c-Src was then immunoprecipitated from platelet lysates and immunoblotted for the phosphorylated c-Src residue Tyr419. As shown in Fig. 4, C and D, anti-αIIb TM induced c-Src phosphorylation despite the presence of EDTA or eptifibatide, implying that it occurred in the absence of αIIbβ3-bound ligand. It also occurred when platelets were preincubated with the actin polymerization inhibitors cytochalasin D or latrunculin, indicating that c-Src phosphorylation was independent of platelet cytoskeletal rearrangement. On the other hand, anti-αIIb TM-induced c-Src phosphorylation was prevented by the c-Src kinase inhibitor PP2 but not by its inactive congener PP3 or by the Syk kinase inhibitor R406. Thus, these data indicate that anti-αIIb TM induces c-Src phosphorylation by a process that is independent of both extracellular ligand binding to αIIbβ3 and platelet cytoskeletal rearrangement but requires c-Src enzymatic activity and likely results from c-Src trans-autophosphorylation.
Anti-αIIb TM Causes Syk, but not FAK, Phosphorylation Independent of Ligand Binding to αIIbβ3
The tyrosine kinase Syk is rapidly and transiently phosphorylated following platelet stimulation in a c-Src-dependent reaction (23–25). Although Syk was initially reported to interact directly with the β3 CT of ligand-occupied αIIbβ3 (26), subsequent studies suggest that Syk binds instead to the c-Src phosphorylated tyrosines of immunoreceptor tyrosine-based activation motif (ITAM)-containing proteins such as platelet FcγRIIa (27). Regardless, we found that incubating human platelets with anti-αIIb TM caused the phosphorylation of Syk on tyrosine residues (Fig. 5, A and B). Anti-αIIb TM-induced Syk phosphorylation was unaffected by EDTA, eptifibatide, cytochalasin D, and latrunculin A, indicating that it did not require ligand binding to αIIbβ3 or platelet cytoskeletal rearrangement, but it was prevented PP2, indicating that it was initiated by c-Src kinase activation (25, 28). It was also diminished by the Syk inhibitor R406, indicating that it results in part from trans-autophosphorylation of Syk as well (25).
FIGURE 5.

A. Anti-αIIb TM causes phosphorylation of Syk in platelets. Gel-filtered human platelets were incubated for 1 min with buffer, 10 μg/ml collagen, or 2 μm anti-αIIb TM in the absence or presence of 5 mm EDTA, 10 μm eptifibatide, 4 μm R406, 10 μm PP2, 10 μm PP3, 10 μm cytochalasin D (CytoD), or 2 μm latrunculin (LatA). Syk was immunoprecipitated from platelet lysates and immunoblotted with 4G10, a monoclonal antibody specific for phosphotyrosine. Top panel, phosphorylated Syk (p-Syk). Bottom panel, Syk loading control. The experiment was performed three times. B, the bar graphs were generated from densitometry of the immunoblots from the three experiments and represent the mean ± S.E. of phosphorylated Syk normalized using the densitometry of the corresponding Syk band. C, gel-filtered human platelets were incubated for 1 min with buffer, 10 μg/ml collagen, 1 unit/ml thrombin, or 2 μm anti-αIIb TM, the latter in the absence or presence of 5 mm EDTA, 10 μm PP2, 10 μm PP3, 10 μm eptifibatide, or 50 μg/ml abciximab. FAK was immunoprecipitated from platelet lysates, and phosphorylated FAK was detected by immunoblotting using 4G10.
FAK also undergoes Src-mediated phosphorylation following platelet stimulation by agonists such as thrombin and collagen (29). However, in contrast to the rapidity of agonist-stimulated c-Src and Syk phosphorylation, FAK phosphorylation occurs minutes after platelet stimulation, coincident with the onset of platelet aggregation (24). Furthermore, although there is no conclusive evidence that FAK binds to β3 (30), agonist-stimulated FAK phosphorylation is prevented by αIIbβ3 antagonists, implying that it depends on the binding of macromolecular multivalent ligands to αIIbβ3 (29). We found that incubating platelets with anti-αIIb TM caused c-Src-dependent FAK phosphorylation (Fig. 5C). However, unlike anti-αIIb TM-induced c-Src and Syk phosphorylation, anti-αIIb TM-induced FAK phosphorylation was inhibited by EDTA and by the αIIbβ3 antagonists eptifibatide and abciximab, indicating anti-αIIb TM-induced FAK phosphorylation requires ligand binding to αIIbβ3. Furthermore, although small-molecule αIIbβ3 antagonists such as eptifibatide have been shown to cause αIIbβ3 activation (31), incubating platelets with eptifibatide was not sufficient to cause FAK phosphorylation.
Effect of Mn2+ on c-Src Phosphorylation in Platelets
Mn2+ activates integrins by perturbing the conformation of their extracellular domains (32). However, this appears to be a relatively local perturbation induced by the metal ion because it does not propagate to induce separation of the TM domains of the leukocyte integrin αLβ2 (33). We used this property of Mn2+ to address whether anti-αIIb TM-induced c-Src phosphorylation requires separation of the αIIb and β3 stalks. We found that, although thrombin and anti-αIIb TM caused platelet aggregation and ATP secretion, incubating platelets with 2 mm MnCl2 caused neither (Fig. 6A). Moreover, both thrombin and anti-αIIb TM caused the phosphorylation of β3-bound c-Src but MnCl2 did not (Fig. 6B). Thus, inducing an active conformation in the αIIbβ3 extracellular domain alone is insufficient to cause c-Src phosphorylation, and TM domain separation at a minimum is required for phosphorylation to occur.
FIGURE 6.

Thrombin and anti-αIIb TM, but not Mn2+, cause platelet aggregation, platelet ATP secretion, and c-Src phosphorylation. A, platelets were incubated for 1 min with either 1 unit/ml thrombin, 2 μm anti-αIIb TM, or 2 mm MnCl2 before platelet aggregation and ATP secretion were measured. B, c-Src binding to β3 induced by 2 μm anti-αIIb TM or 2 mm MnCl2 and its subsequent phosphorylation were measured as described in Fig. 3. DMSO, the solvent for anti-αIIb TM; Buffer, platelet suspension buffer without Mn2+; +EDTA, 2 μm anti-αIIb TM plus 5 mm EDTA; +PP2, 2 μm anti-αIIb TM plus 10 μm PP2. IP, immunoprecipitation; IB, immunoblot.
Anti-αIIb TM Causes Clustering of αllbβ3 in Platelets
Because both c-Src and Syk are activated by trans-autophosphorylation, it is likely that anti-αIIb TM causes c-Src phosphorylation by inducing αIIbβ3 clustering. To test this possibility, we incubated washed human platelets for 60 s at 37 °C in the presence or absence of 2 μm anti-αΙIb TM, after which the platelets were fixed with 4% paraformaldehyde and stained with the Alexa Fluor® 488-labeled β3-specific monoclonal antibody SSA6. In the absence of anti-αIIb TM, αllbβ3 was present in a homogenous ring at the periphery of the discoid platelets (Fig. 7, A and B). By contrast, when the platelets were incubated with anti-αIIb TM, αIIbβ3 was redistributed into discrete nodular densities (Fig. 7, C and D). Classifying 270 resting platelets in 12 microscopic fields revealed that αIIbβ3 was homogeneously distributed in 80% of the platelets and as nodular densities in the remaining 20%. Conversely, when 262 anti-αIIb TM-incubated platelets were classified in six microscopic fields, αIIbβ3 was present exclusively as discrete nodular densities in 85% of the platelets and was homogeneously distributed in the remaining 15%. The differences in the distribution of αIIbβ3 in resting and anti-αIIb TM-incubated platelets were highly significant by chi-square testing (p = 1.82E-50). Thus, incubating platelets with anti-αIIb TM not only induces αIIbβ3 activation but causes the formation of αIIbβ3 clusters. In turn, αIIbβ3 clustering increases the local concentration of β3-bound c-Src (5), thereby facilitating c-Src trans-autophosphorylation.
FIGURE 7.
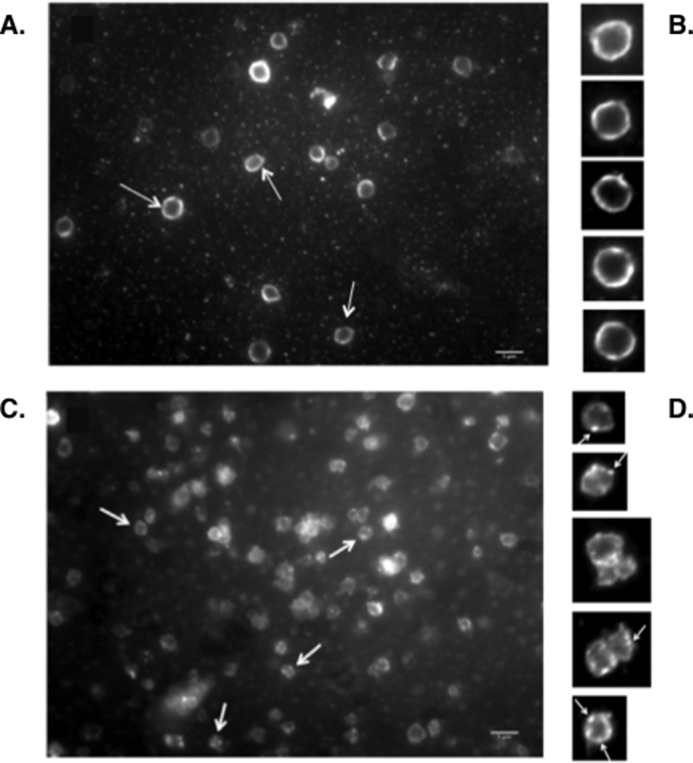
Anti-αIIb TM induces ligand binding-independent clustering of αIIbβ3 in platelets. Washed human platelets were incubated with buffer or 2 μm anti-αIIb TM in the presence of EDTA for 1 min. The platelets were then fixed with 4% paraformaldehyde, incubated with the Alexa Fluor 488-labeled anti-β3 mAb SSA6 for 1 h at 37 °C and examined by fluorescence microscopy. The experiments were performed three times. A is representative of 12 fields containing 270 resting platelets. C is representative of six fields containing 262 anti-αIIb TM-stimulated platelets. The arrows in A indicate resting platelets, and the arrows in C indicate examples of anti-αIIb TM-stimulated platelets. B and D are enlargements of resting and anti-αIIb TM-stimulated platelets, respectively. The arrows in D point to nodular clusters of αIIbβ3. Scale bars = 5 μm.
Anti-αIIb TM-induced αIIbβ3 Clustering Does Not Require Stable Binding of Talin-1 and Kindlin-3 to the β3 CT
How anti-αIIb TM drives αIIbβ3 clustering is unclear, but it is possibly indirect, mediated by proteins bound to the β3 CT. Physiologic αIIbβ3 activation occurs when the 4.1/ezrin/radixin/moesin domains of the focal adhesion protein kindlin-3 and the cytoskeletal protein talin-1 bind to the β3 CT (12, 34, 35). Talin-1 is predominantly present in platelets as an antiparallel homodimer (36, 37). Thus, β3-bound talin-1 could cross-link adjacent activated αIIbβ3 molecules (35). It has also been suggested that β3-bound kindlin-3 supports talin-induced αIIbβ3 activation by promoting αIIbβ3 clustering (38). To test whether either protein mediates anti-αIIb TM-induced αΙIbβ3 clustering, we incubated washed human platelets with 2 μm anti-αIIb TM or 1 unit/ml thrombin for specified intervals up to 180 s, after which the platelets were lysed using 1% Nonidet P-40. β3 was then immunoprecipitated from the lysates, and the immunoprecipitates were immunoblotted for talin-1 and kindlin-3. We detected little talin-1 associated with β3 in unstimulated platelets (Fig. 8, A and B). However, we found that the amount of β3-associated talin-1 increased 2.50 ± 0.63-fold (n = 4) after platelets were stimulated for 90 s with 1 unit/ml thrombin (Fig. 8C). By contrast, the amount of β3-associated talin-1 increased only 0.63 ± 0.26-fold when platelets were exposed to 2 μm anti-αIIb TM for 90 s. Kindlin-3 appeared to be constitutively associated with β3 in unstimulated platelets (Fig. 8A), and there was a small, non-significant, 0.55 ± 0.10-fold increase in the amount of β3-associated kindlin-3 when platelets were stimulated for 90 s with 1 unit/ml thrombin (p = 0.12, t test for pair samples, n = 4). On the other hand, the amount of β3-associated kindlin-3 actually decreased 0.42 ± 0.11-fold below resting levels when platelets were incubated for 90 s with 2 μm anti-αIIb TM (Fig. 8C). In summary, although we cannot rule out a very weak or transient interaction with kindlin-3 and talin-1, these interactions appear much weaker when platelets are treated with anti-αIIb TM than when platelets are activated by thrombin. Therefore, we consider it unlikely that either talin-1 and kindlin-3 binding to β3 accounts for the αIIbβ3 clustering that initiates c-Src trans-autophosphorylation when platelets are incubated with anti-αIIb TM.
FIGURE 8.

Anti-αIIb TM- and thrombin-stimulated talin-1 and kindlin-3 binding to β3. Washed platelets were incubated with 2 μm anti-αIIb TM or 1 unit/ml thrombin for 30, 90, and 180 s, after which the platelets were lysed with 1% Nonidet P-40, β3 was immunoprecipitated from the platelet lysates, and the β3 immunoprecipitates were immunoblotted for talin-1 and kindlin-3. A, time course of talin-1 and kindlin-3 binding to β3 following platelet simulation by 1 unit/ml thrombin and 2 μm anti-αIIb TM. These experiments were repeated four times. The lower molecular weight (MW) band seen in several of the kindlin-3 lanes is nonspecific and is present sporadically when kindlin-3 is immunoblotted with polyclonal anti-kindlin-3 antibodies. B, densitometry using National Institutes of Health ImageJ software of the immunoblots shown in A. To facilitate comparisons, the densitometry for β3-bound talin-1 and kindlin-3 at each time point was normalized using the densitometry for the 0 time point. C, comparison of the amount of β3-associated talin-1 and kindlin-3 at 90 s after platelet stimulation by 1 units/ml thrombin or 2 μm anti-αIIb TM. The data shown are the mean ± S.E. of the four separate experiments and were generated from the 90-s time point of the four experiments.
Discussion
Activation of the c-Src associated with the β3 CT in platelets triggers a protein phosphorylation cascade that results in outside-in platelet signaling (4). Here we report that this cascade can be initiated by activating individual αIIbβ3 molecules using a membrane-soluble peptide that binds to the αIIb TM domain. The membrane-soluble peptide, anti-αIIb TM, was designed computationally to bind with high affinity and specificity to the αIIb TM domain (8, 39). When added to suspensions of washed platelets, anti-αIIb TM spontaneously inserts into the platelet plasma membrane, where it binds to the αIIb TM domain and causes αIIbβ3 activation by disrupting the TM domain heterodimer of resting αIIbβ3 (8, 40). In this respect, anti-αIIb TM mimics the physiologic αIIbβ3 activator talin, which causes αIIbβ3 TM domain separation when it binds to the membrane-proximal region of β3 CT (Fig. 1) (12).
As we have reported recently (5), c-Src does not interact specifically with the β3 CT of resting platelets but binds to the CT and is phosphorylated within seconds following platelet stimulation by agonists such as thrombin. The time courses of anti-αIIb TM-induced c-Src binding to the β3 CT and its subsequent phosphorylation mimic the response to thrombin, implying that the mechanisms may be the same. Nevertheless, it is possible that incubating platelets with a hydrophobic peptide like anti-αIIb TM could have off-target effects that cause c-Src activation by a mechanism unrelated to αIIbβ3. To address this possibility, we added apyrase to platelet suspensions to metabolize any platelet ADP that might be nonspecifically released by anti-αIIb TM, tested platelets from two patients with GT to confirm that αIIbβ3 must be present to observe an anti-αIIb TM effect, and used an unrelated membrane-soluble peptide, MS1, to further control for nonspecific peptide effects. Because anti-αIIb TM caused c-Src phosphorylation, despite the presence of apyrase, did not cause c-Src phosphorylation in GT platelets, and MS1 neither caused platelet aggregation, platelet secretion, or c-Src phosphorylation, anti-αIIb TM appears to initiate platelet outside-in signaling by directly interacting with αIIbβ3.
Besides activating c-Src, the platelet agonists thrombin, collagen, and ADP cause phosphorylation and subsequent activation of the non-receptor tyrosine kinase Syk (24). Initially, Syk was thought to directly interact with the distal β3 CT at a site overlapping the c-Src binding site (26). However, more recent observations suggest that Syk is activated after binding via its SH2 domains to ITAM-containing proteins such as FcγRIIa previously phosphorylated by activated c-Src (27, 41). We found that anti-αIIb TM, like conventional platelet agonists, causes Syk phosphorylation on tyrosine residues. Moreover, anti-αIIb TM-stimulated Syk phosphorylation was prevented by the Src-kinase inhibitor PP2, consistent with a proximal requirement for c-Src activation, and was attenuated by an inhibitor of Syk kinase activity, suggesting that, like c-Src, Syk undergoes trans-autophosphorylation (25).
The tyrosine kinase FAK is also phosphorylated by activated c-Src in agonist-stimulated platelets (29). Although it is clear that FAK phosphorylation in platelets requires αIIbβ3 (29) and can be induced by forced αIIbβ3 clustering (42), there is no conclusive evidence that FAK actually associates with the integrin (30). We found that anti-αIIb TM initiates the c-Src-dependent phosphorylation of FAK, but like FAK phosphorylation induced by conventional agonists, it was prevented by αIIbβ3 antagonists and actin polymerization inhibitors (data not shown). Thus, although direct activation of single αIIbβ3 molecules alone appears sufficient to activate c-Src and Syk, events subsequent to αIIbβ3 activation appear to be required to cause FAK activation.
The observation that anti-αIIb TM-induced αIIbβ3 activation causes c-Src trans-autophosphorylation implies that anti-αIIb TM-activated αIIbβ3 undergoes oligomerization. Moreover, because anti-αIIb TM-induced c-Src phosphorylation was not impaired by EDTA or by αIIbβ3 antagonists, it appears that the necessary αIIbβ3 oligomerization was not driven by extracellular ligand binding. The redistribution of αIIbβ3 into discrete nodular densities we observed by immunofluorescence when suspended platelets were incubated with anti-αIIb TM in the presence of EDTA supports this possibility.
How activated αIIbβ3 might undergo spontaneous oligomerization is unclear. It is likely that oligomerization is indirect (43–46), mediated by proteins bound to the β3 CT of activated αIIbβ3. Physiologic αIIbβ3 activation occurs when the head domain of the cytoskeletal protein talin binds to the membrane-proximal β3 CT (12, 34). Talin, a 270-kDa protein, consists of a 50-kDa N-terminal 4.1/ezrin/radixin/moesin domain connected to a 200-kDa rod domain by a largely unstructured flexible linker (36). The C terminus of the talin rod also contains a dimerization domain, and, in platelets, talin is predominantly present as an antiparallel homodimer (36, 37). Thus, β3-bound talin would seem a likely candidate to cross-link adjacent activated αIIbβ3 molecules (35). However, we found that, although thrombin stimulation caused an increase the amount of β3-associated talin-1, there was essentially no change in the amount of β3-associated when platelets were incubated with anti-αIIb TM. This makes it improbable that talin-1 is responsible for the apparent αIIbβ3 oligomerization that follows αIIbβ3 activation by anti-αIIb TM.
In addition to talin, kindlin-3, a member of the kindlin family of focal adhesion proteins, is required for agonist-stimulated αIIbβ3 activation in platelets (35). Kindlin-3 binds to the β3 CT via its 4.1/ezrin/radixin/moesin domain at a site distinct from talin (47). Although kindlin-3 binding to β3 alone is not sufficient to cause αIIbβ3 activation, agonist-stimulated αIIbβ3 activation in platelets does not occur when kindlin-3 is absent, despite the presence of talin (48). The mechanism by which kindlin-3 “primes” talin-induced αIIbβ3 activation is unknown, but it has been suggested that kindlin-3 increases the avidity of αIIbβ3 for multivalent, but not univalent, soluble ligands by promoting αIIbβ3 clustering (38). However, kindlins are monomeric (35), so their ability to cause αIIbβ3 clustering would have to be indirect, and additional kindlin binding partners such as migfilin (49) and integrin-linked kinase (ILK) (50, 51) would be required. We detected the constitutive association of kindlin-3 with β3 in resting platelets. Although there was a small increase in the amount of β3-associated kindlin-3 when platelets were stimulated by thrombin, the amount of β3-associated kindlin-3 progressively declined when platelets were incubated with anti-αIIb TM. Thus, like talin-1, these results indicate that kindlin-3 binding to the β3 CT does not account for anti-αIIb TM-induced αIIbβ3 clustering.
In summary, we found that anti-αIIb TM, a computationally designed peptide that binds with high affinity to the αIIb TM domain, causes αIIbβ3 activation, platelet aggregation, and ATP secretion as well as the phosphorylation of β3-associated c-Src. Because β3-associated c-Src is activated by trans-autophosphorylation, these observations imply that activation of individual αIIbβ3 molecules can initiate αIIbβ3 oligomerization even when ligand binding is absent. Thus, our studies indicate that not only is resting αIIbβ3 poised to undergo a conformational change that exposes its ligand binding site, but it is poised to rapidly assemble into intracellular signal-generating complexes as well.
Author Contributions
H. Z. and K. P. F. performed the majority of the experiments with the assistance of L. M. S., K. Y., and D. T. M. R. T. synthesized anti-αIIb TM and MS1, supervised by H. Y. W. F. D. analyzed the data and assisted with the preparation of the manuscript: J. S. B. designed the experiments, analyzed the data, and prepared the manuscript.
Acknowledgments
We thank Dr. Mortimer Poncz for help with obtaining platelets from patients with Glanzmann thrombasthenia.
This work was supported by National Institutes of Health Grants HL40387, HL120846, GM54616, and GM103843; National Science Foundation Grant CHEM0954819; and bridge funding from the American Society of Hematology. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CT
- cytoplasmic tail
- TM
- transmembrane
- SH
- Src homology
- GT
- Glanzmann thrombasthenia
- FAK
- focal adhesion kinase.
References
- 1. Fox J. E., Shattil S. J., Kinlough-Rathbone R. L., Richardson M., Packham M. A., and Sanan D. A. (1996) The platelet cytoskeleton stabilizes the interaction between aIIbb3 and its ligand and induces selective movements of ligand-occupied integrin. J. Biol. Chem. 271, 7004–7011 [DOI] [PubMed] [Google Scholar]
- 2. Shattil S. J., and Newman P. J. (2004) Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood 104, 1606–1615 [DOI] [PubMed] [Google Scholar]
- 3. Arias-Salgado E. G., Lizano S., Sarkar S., Brugge J. S., Ginsberg M. H., and Shattil S. J. (2003) Src kinase activation by direct interaction with the integrin β cytoplasmic domain. Proc. Natl. Acad. Sci. U.S.A. 100, 13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Senis Y. A., Mazharian A., and Mori J. (2014) Src family kinases: at the forefront of platelet activation. Blood 124, 2013–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu Y., Span L. M., Nygren P., Zhu H., Moore D. T., Cheng H., Roder H., DeGrado W. F., and Bennett J. S. (2015) The tyrosine kinase c-Src specifically binds to the active integrin αIIbβ3 to initiate outside-in signaling in platelets. J. Biol. Chem. 290, 15825–15834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lam W. A., Chaudhuri O., Crow A., Webster K. D., Li T. D., Kita A., Huang J., and Fletcher D. A. (2011) Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat. Mater. 10, 61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu W., Doshi A., Lei M., Eck M. J., and Harrison S. C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638 [DOI] [PubMed] [Google Scholar]
- 8. Li R., Mitra N., Gratkowski H., Vilaire G., Litvinov R., Nagasami C., Weisel J. W., Lear J. D., DeGrado W. F., and Bennett J. S. (2003) Activation of integrin aIIbb3 by modulation of transmembrane helix associations. Science 300, 795–798 [DOI] [PubMed] [Google Scholar]
- 9. Li W., Metcalf D. G., Gorelik R., Li R., Mitra N., Nanda V., Law P. B., Lear J. D., Degrado W. F., and Bennett J. S. (2005) A push-pull mechanism for regulating integrin function. Proc. Natl. Acad. Sci. U.S.A. 102, 1424–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin H., Litvinov R. I., Vilaire G., Zhu H., Li W., Caputo G. A., Moore D. T., Lear J. D., Weisel J. W., Degrado W. F., and Bennett J. S. (2006) Activation of platelet alphaIIbbeta3 by an exogenous peptide corresponding to the transmembrane domain of αIIb. J. Biol. Chem. 281, 36732–36741 [DOI] [PubMed] [Google Scholar]
- 11. Yin H., Slusky J. S., Berger B. W., Walters R. S., Vilaire G., Litvinov R. I., Lear J. D., Caputo G. A., Bennett J. S., and DeGrado W. F. (2007) Computational design of peptides that target transmembrane helices. Science 315, 1817–1822 [DOI] [PubMed] [Google Scholar]
- 12. Wegener K. L., Partridge A. W., Han J., Pickford A. R., Liddington R. C., Ginsberg M. H., and Campbell I. D. (2007) Structural basis of integrin activation by talin. Cell 128, 171–182 [DOI] [PubMed] [Google Scholar]
- 13. Choma C., Gratkowski H., Lear J. D., and DeGrado W. F. (2000) Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7, 161–166 [DOI] [PubMed] [Google Scholar]
- 14. Basani R. B., D'Andrea G., Mitra N., Vilaire G., Richberg M., Kowalska M. A., Bennett J. S., and Poncz M. (2001) RGD-containing peptides inhibit fibrinogen binding to platelet α(IIb)β3 by inducing an allosteric change in the amino-terminal portion of α(IIb). J. Biol. Chem. 276, 13975–13981 [DOI] [PubMed] [Google Scholar]
- 15. Bennett J. S., and Vilaire G. (1979) Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Invest. 64, 1393–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weisel J. W., Nagaswami C., Vilaire G., and Bennett J. S. (1992) Examination of the platelet membrane glycoprotein IIb/IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J. Biol. Chem. 267, 16637–16643 [PubMed] [Google Scholar]
- 17. Liu J., Jackson C. W., and Gartner T. K. (2008) The Src requirement for washed platelet aggregation and dense granule secretion in response to stimulation by a low level of γ-thrombin. J. Thromb. Haemost. 6, 1035–1037 [DOI] [PubMed] [Google Scholar]
- 18. Poncz M., Rifat S., Coller B. S., Newman P. J., Shattil S. J., Parrella T., Fortina P., and Bennett J. S. (1994) Glanzmann thrombasthenia secondary to a Gly273ÆAsp mutation adjacent to the first calcium-binding domain of platelet glycoprotein IIb. J. Clin. Invest. 93, 172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basani R. B., Vilaire G., Shattil S. J., Kolodziej M. A., Bennett J. S., and Poncz M. (1996) Glanzmann thrombasthenia due to a two amino acid deletion in the fourth calcium-binding domain of aIIb: demonstration of the importance of calcium-binding domains in the conformation of aIIbb3. Blood 88, 167–173 [PubMed] [Google Scholar]
- 20. Cristian L., Nanda V., Lear J. D., and DeGrado W. F. (2005) Synergistic interactions between aqueous and membrane domains of a designed protein determine its fold and stability. J. Mol. Biol. 348, 1225–1233 [DOI] [PubMed] [Google Scholar]
- 21. Bennett J. S. (2001) Novel platelet inhibitors. Annu Rev Med 52, 161–184 [DOI] [PubMed] [Google Scholar]
- 22. Golden A., and Brugge J. S. (1989) Thrombin treatment induces rapid changes in tyrosine phosphorylation in platelets. Proc. Natl. Acad. Sci. U.S.A. 86, 901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taniguchi T., Kitagawa H., Yasue S., Yanagi S., Sakai K., Asahi M., Ohta S., Takeuchi F., Nakamura S., and Yamamura H. (1993) Protein-tyrosine kinase p72syk is activated by thrombin and is negatively regulated through Ca2+ mobilization in platelets. J. Biol. Chem. 268, 2277–2279 [PubMed] [Google Scholar]
- 24. Clark E. A., Shattil S. J., Ginsberg M. H., Bolen J., and Brugge J. S. (1994) Regulation of the protein tyrosine kinase pp72syk by platelet agonists and the integrin aIIbb3. J. Biol. Chem. 269, 28859–28864 [PubMed] [Google Scholar]
- 25. Gao J., Zoller K. E., Ginsberg M. H., Brugge J. S., and Shattil S. J. (1997) Regulation of the pp72syk protein tyrosine kinase by platelet integrin aIIbb3. EMBO J. 16, 6414–6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woodside D. G., Obergfell A., Leng L., Wilsbacher J. L., Miranti C. K., Brugge J. S., Shattil S. J., and Ginsberg M. H. (2001) Activation of Syk protein tyrosine kinase through interaction with integrin β cytoplasmic domains. Curr. Biol. 11, 1799–1804 [DOI] [PubMed] [Google Scholar]
- 27. Boylan B., Gao C., Rathore V., Gill J. C., Newman D. K., and Newman P. J. (2008) Identification of FcγRIIa as the ITAM-bearing receptor mediating αIIbβ3 outside-in integrin signaling in human platelets. Blood 112, 2780–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Obergfell A., Eto K., Mocsai A., Buensuceso C., Moores S. L., Brugge J. S., Lowell C. A., and Shattil S. J. (2002) Coordinate interactions of Csk, Src, and Syk kinases with αIIbβ3 initiate integrin signaling to the cytoskeleton. J. Cell Biol. 157, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lipfert L., Haimovich B., Schaller M. D., Cobb B. S., Parsons J. T., and Brugge J. S. (1992) Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J. Cell Biol. 119, 905–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parsons J. T. (2003) Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 31. Zhu J., Choi W. S., McCoy J. G., Negri A., Zhu J., Naini S., Li J., Shen M., Huang W., Bougie D., Rasmussen M., Aster R., Thomas C. J., Filizola M., Springer T. A., and Coller B. S. (2012) Structure-guided design of a high-affinity platelet integrin αIIbβ3 receptor antagonist that disrupts Mg2+ binding to the MIDAS. Sci. Transl. Med. 4, 125ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J., Salas A., and Springer T. A. (2003) Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat. Struct. Biol. 10, 995–1001 [DOI] [PubMed] [Google Scholar]
- 33. Kim M., Carman C. V., and Springer T. A. (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301, 1720–1725 [DOI] [PubMed] [Google Scholar]
- 34. Calderwood D. A., Zent R., Grant R., Rees D. J., Hynes R. O., and Ginsberg M. H. (1999) The Talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071–28074 [DOI] [PubMed] [Google Scholar]
- 35. Calderwood D. A., Campbell I. D., and Critchley D. R. (2013) Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Critchley D. R. (2009) Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 38, 235–254 [DOI] [PubMed] [Google Scholar]
- 37. Goult B. T., Xu X. P., Gingras A. R., Swift M., Patel B., Bate N., Kopp P. M., Barsukov I. L., Critchley D. R., Volkmann N., and Hanein D. (2013) Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: implications for talin activation. J. Struct. Biol. 184, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye F., Petrich B. G., Anekal P., Lefort C. T., Kasirer-Friede A., Shattil S. J., Ruppert R., Moser M., Fässler R., and Ginsberg M. H. (2013) The mechanism of kindlin-mediated activation of integrin IIbβ3. Curr. Biol. 23, 2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walters R. F., and DeGrado W. F. (2006) Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 13658–13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tang J., Yin H., Qiu J., Tucker M. J., DeGrado W. F., and Gai F. (2009) Using two fluorescent probes to dissect the binding, insertion, and dimerization kinetics of a model membrane peptide. J. Am. Chem. Soc. 131, 3816–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grädler U., Schwarz D., Dresing V., Musil D., Bomke J., Frech M., Greiner H., Jäkel S., Rysiok T., Müller-Pompalla D., and Wegener A. (2013) Structural and biophysical characterization of the Syk activation switch. J. Mol. Biol. 425, 309–333 [DOI] [PubMed] [Google Scholar]
- 42. Hato T., Pampori N., and Shattil S. J. (1998) Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin aIIbb3. J. Cell Biol. 141, 1685–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim M., Carman C. V., Yang W., Salas A., and Springer T. A. (2004) The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J. Cell Biol. 167, 1241–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luo B. H., Carman C. V., Takagi J., and Springer T. A. (2005) Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc. Natl. Acad. Sci. U.S.A. 102, 3679–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim C., Lau T. L., Ulmer T. S., and Ginsberg M. H. (2009) Interactions of platelet integrin αIIb and β3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood 113, 4747–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang W., Zhu J., Springer T. A., and Luo B. H. (2011) Tests of integrin transmembrane domain homo-oligomerization during integrin ligand binding and signaling. J. Biol. Chem. 286, 1860–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bledzka K., Liu J., Xu Z., Perera H. D., Yadav S. P., Bialkowska K., Qin J., Ma Y. Q., and Plow E. F. (2012) Spatial coordination of kindlin-2 with talin head domain in interaction with integrin β cytoplasmic tails. J. Biol. Chem. 287, 24585–24594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moser M., Nieswandt B., Ussar S., Pozgajova M., and Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 49. Brahme N. N., Harburger D. S., Kemp-O'Brien K., Stewart R., Raghavan S., Parsons M., and Calderwood D. A. (2013) Kindlin binds migfilin tandem LIM domains and regulates migfilin focal adhesion localization and recruitment dynamics. J. Biol. Chem. 288, 35604–35616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Böttcher R. T., Lange A., and Fässler R. (2009) How ILK and kindlins cooperate to orchestrate integrin signaling. Curr. Opin. Cell Biol. 21, 670–675 [DOI] [PubMed] [Google Scholar]
- 51. Jones C. I., Tucker K. L., Sasikumar P., Sage T., Kaiser W. J., Moore C., Emerson M., and Gibbins J. M. (2014) Integrin-linked kinase regulates the rate of platelet activation and is essential for the formation of stable thrombi. J Thromb. Haemost. 12, 1342–1352 [DOI] [PubMed] [Google Scholar]
- 52. Anthis N. J., Wegener K. L., Ye F., Kim C., Goult B. T., Lowe E. D., Vakonakis I., Bate N., Critchley D. R., Ginsberg M. H., and Campbell I. D. (2009) The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28, 3623–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caputo G. A., Litvinov R. I., Li W., Bennett J. S., Degrado W. F., and Yin H. (2008) Computationally designed peptide inhibitors of protein-protein interactions in membranes. Biochemistry 47, 8600–8606 [DOI] [PMC free article] [PubMed] [Google Scholar]