

Abstract
Examples of dynamic polymerase exchange have been previously characterized in model systems provided by coliphages T4 and T7. Using a dominant negative D403E polymerase (Pol) III α that can form initiation complexes and sequester primer termini but not elongate, we investigated the possibility of exchange at the Escherichia coli replication fork on a rolling circle template. Unlike other systems, addition of polymerase alone did not lead to exchange. Only when D403E Pol III was bound to a τ-containing DnaX complex did exchange occur. In contrast, addition of Pol IV led to rapid exchange in the absence of bound DnaX complex. Examination of Pol III* with varying composition of τ or the alternative shorter dnaX translation product γ showed that τ-, τ2-, or τ3-DnaX complexes supported equivalent levels of synthesis, identical Okazaki fragment size, and gaps between fragments, possessed the ability to challenge pre-established replication forks, and displayed equivalent susceptibility to challenge by exogenous D403E Pol III*. These findings reveal that redundant interactions at the replication fork must stabilize complexes containing only one τ. Previously, it was thought that at least two τs in the trimeric DnaX complex were required to couple the leading and lagging strand polymerases at the replication fork. Possible mechanisms of exchange are discussed.
Keywords: clamp loader, DNA helicase, DNA polymerase, genomic instability, nucleic acid enzymology, protein-DNA interaction, DnaX complex, dynamic processivity, polymerase exchange, replication fork
Introduction
As with all cellular replicases, the DNA polymerase (Pol)2 III holoenzyme (HE) of Escherichia coli is tripartite with a sliding clamp processivity factor (β2), a clamp loader (DnaX complex, DnaX3δδ'χψ), and an associated replicative polymerase (Pol III, αϵθ) (for a review, see Ref. 2). In E. coli and many other bacteria, DnaX encodes two products: a shorter γ protein that has ATPase activity and the ability to support clamp loading on single-stranded DNA and a longer τ protein that has additional domains that interact with the DnaB6 replicative helicase and Pol III. Cells that cannot express γ exhibit defects in UV viability and Pol IV-mediated mutagenesis (3). A proposal has been made that γ may be required to avoid the presence of a third Pol III at the replication fork that might outcompete other polymerases, such as Pol IV, that must enter the replication fork to resolve unrepairable lesions and to perform stress-induced mutagenesis functions (2, 3).
An E. coli rolling circle replication system has been developed that exploits a 409-nt flapped circular template that has a 50:1 asymmetric GC content, allowing convenient distinction, quantification, and labeling of leading and lagging strands. The asymmetric GC distribution allows specific slowing of lagging strand synthesis by substitution of dGDPNP for dGTP. The Vmax for insertion of this analog is lower than the natural nucleotide, and the Km is higher, allowing “dialing in” a desired elongation rate without reducing the nucleotide to a level where its concentration is depleted during the progress of the reaction. Because the lagging strand half of the Pol III HE cycles when a new primer is made, even when the preceding Okazaki fragment is incomplete, gaps are left between fragments when the lagging strand rate is decreased to a level where cycling is induced before fragment synthesis is complete (4).
The concept of dynamic processivity and dynamic polymerase exchange was first established in the bacteriophage T4 system (5, 6). T4 replication is highly processive, but exogenous polymerase can exchange into the fork and inhibit coupled rolling circle replication more rapidly than the dissociation rate of the T4 DNA polymerase, gp43. These results suggested that gp43-D408N actively displaces gp43 rather than passively binding template after dissociation of wild-type gp43. It was proposed that the C terminus of the incoming polymerases binds the interdomain loop of one subunit of the sliding clamp and displaces the polymerase at the replication fork (6).
Similarly, with bacteriophage T7, a complex of wild-type DNA polymerase (gp5) and its processivity factor, E. coli thioredoxin, readily exchanges with excess mutant gp5-thioredoxin complexes (gp5-Y526F/thioredoxin) without affecting processivity. Replacing tyrosine 526 with phenylalanine in the nucleotide binding site makes the mutant gp5 resistant to inhibition of ddNTPs but does not affect its ability to bind to other protein components and elongate. Both strand displacement synthesis and leading strand synthesis in a coupled reaction initiated by gp5-Y526F/trx are inhibited upon addition of gp5/trx and ddNTPs, indicating polymerase exchange (7). Exchange was not observed in a single-stranded DNA replication reaction where helicase was not included. Thus, it was proposed that an exogenous polymerase binds the hexameric T7 helicase and exchanges with polymerase that transiently dissociates (7).
YFP-tagged E. coli Pol III α proteins have been detected by single-molecule microscopy in live cells bound near the replication fork (8). Repetitive bursts of fluorescent Pol III α were observed in living cells that were interpreted as new polymerases continuously exchanging with the DnaX complex organizer at replication forks with the synthesis of each Okazaki fragment. This appeared to be inconsistent with the 4.9-h mean lifetime (koff = 5.7 × 10−5) observed for α-τ DnaX dissociation (10) and is the primary subject of this investigation. In addition to a putative exchange of Pol III into the replication fork, exchange of other polymerases, particularly Pol IV, has been well documented (11–16). Two different mechanisms have been proposed for Pol IV exchange. One, called the toolbelt model, predicts that Pol IV is recruited through contact with one of the two canonical clamp binding sites, whereas the other predicts recruitment mediated by a direct contact of Pol IV with both Pol III* and an accessory site on the rim of the β2 sliding clamp, subsequently displacing Pol III to allow binding by Pol IV (12–15, 17–19). If operative, the toolbelt model will require modification in light of a recent elegant cryo-EM structure that shows that both of the canonical binding sites within β2 are occupied by interacting Pol III α and ϵ subunits of the Pol III HE (20). We pursued this issue in this work and compared the requirements for exchange with those of Pol III to determine whether similar mechanisms were used.
Recently, we have established that the major cellular form of Pol III HE is Pol III2τ2γδδ'χψ, disproving proposals that the cellular replicase contains three τs and three polymerases (3). One of the observations that was initially used to support the three-polymerase model was that three Pol III assemblies synthesized DNA in a coupled fork system with smaller gaps between Okazaki fragments (21). We reinvestigated this observation using a minicircle replication system we developed (4). Our findings are presented here.
Experimental Procedures
Nucleotide Analogs and Radioactive Nucleotides
dGDPNP was custom-synthesized for this project by TriLink Biotechnologies. The radioactive nucleotides α-[32P]dATP (3000 Ci/mmol), α-[32P]dCTP (3000 Ci/mmol), and α-[32P]dGTP (3000 Ci/mmol) were from PerkinElmer Life Sciences.
Proteins
The following proteins used were purified as described: D403E Pol III α (22), ϵ (23), θ (24); Pol III (25); wild-type and mutant DnaX complexes, including τ3, τ2γ1, τγ2, and τ2γ1M (26); Pol IV (DinB) (27); β2 (28); SSB4 (29); DnaB6 (30); DnaC (4); DnaG (29); DnaT3 (4); PriA (4); and PriB2 (4). The purifications of the various forms of the DnaX complexes were performed by high-resolution FPLC chromotography on Mono S columns, providing baseline resolution between the forms with no cross-contamination, as revealed by rechromotography (26). D403E Pol III was assembled by incubating D403E Pol III α, ϵ, and θ at a molar ratio of 1:1:1 on ice for 15 min. Three forms of Pol III* containing D403E Pol III α were assembled: D403E Pol III3τ3δδ'χψ, D403E Pol III2τ2γδδ'χψ, and D403E Pol III1τγ2δδ'χψ. In each case, they were assembled by incubating D403E Pol III and the corresponding DnaX complex (τ3, τ2γ, or τγ2) on ice for 15 min at a ratio of 6:1 for D403E Pol III3τ3δδ'χψ, at a ratio of 4:1 for D403E Pol III2τ2γδδ'χψ, and at a ratio of 2:1 for D403E Pol III1τγ2δδ'χψ. Likewise, wild-type Pol III* (Pol III2τ2γδδ'χψ) and mutant Pol III* bearing the ATPase K51E mutation in the γ subunit (Pol III2τ2γMδδ'χψ) were assembled by incubating Pol III and the corresponding DnaX complex (τ2γδδ'χψ, or τ2γMδδ'χψ) on ice for 15 min at a ratio of 4:1.
Rolling Circle Reactions
Rolling circle reactions were performed as described previously (4). 20 nm minicircle DNA template, 2 μm SSB4, 100 nm β2, 72 nm DnaB6, 100 nm DnaG, 2.5 nm Pol III*, 160 nm PriA, 50 nm PriB2, 333 nm DnaT3, and 108 nm DnaC were incubated with 5 μm ATPγS, 200 μm CTP, 200 μm UTP, and 200 μm GTP for 5 min at 30 °C to form an initiation complex. The reaction buffer was 10 mm magnesium acetate, 70 mm KCl, 50 mm Hepes (pH 7.5), 100 mm potassium glutamate, 20% glycerol, 200 μg/ml bovine serum albumin, 0.02% Nonidet P-40, and 10 mm dithiothreitol. The reaction was started by addition of 1 mm ATP and 100 μm dNTPs. After 4 min, α-[32P]dCTP or α-[32P]dGTP were added to quantify leading and lagging strand synthesis, respectively. After an additional 4 min, the reaction was quenched with EDTA (140 mm final concentration), and samples were analyzed by liquid scintillation counting after trichloroacetic acid precipitation as described previously (31).
Competition Experiments with D403E Pol III
For control experiments that analyzed competition of D403E Pol III, wild-type Pol III and varying D403E Pol III were combined first and then mixed with the other components of the rolling circle reaction to form an initiation complex as described in the preceding paragraph. Wild-type Pol III was added at a 2-fold molar excess of the DnaX complex (2.5 nm final concentration) and depended on the stoichiometry of the τ subunit. For τ3 complex, Pol III was added at 15 nm; for τ2γ complex, Pol III was added at 10 nm; and for τγ2 complex, Pol III was added at 5 nm. After 5 min, 1 mm ATP, 100 μm dNTPs, and α-[32P]dCTP or α-[32P]dGTP were added to start the reaction. After 8 min, the reaction was quenched with 140 mm EDTA final concentration. For experiments that analyzed competition of D403E Pol III added to an ongoing reaction, reactions were started with non-radioactive nucleotides. After 4 min of elongation to establish coupled replication, competitor D403E Pol III was added along with α-[32P]dCTP or α-[32P]dGTP, and the reaction was allowed to continue for an additional 4 min. The data shown in most figures represent single points. However, all experiments were conducted in a similar format multiple times with the same conclusions.
Competition Experiments with Pol IV
Reactions were set up the same as experiments performed with D403E Pol III, except that Pol IV was used as the challenge. Concentrations for Pol IV competitor were 0, 15.6, 31.3, 62.5, 125, 250, 500, and 1000 nm.
Rolling Circle Reactions with Pol III* Reconstituted with DnaX Complexes Containing τ3, τ2γ, and τγ2
Rolling circle reactions containing either α-[32P]dCTP or α-[32P]dGTP were used to label the leading and lagging strands in reactions containing Pol III* reconstituted with four different DnaX complexes: τ3δδ'χψ, τ2γδδ'χψ, τγ2δδ'χψ, and γ3δδ'χψ. The reaction products were analyzed for strand-specific incorporation by liquid scintillation counting of trichloroacetic acid-precipitable products. Rolling circle reactions were also conducted in the presence of either 100 μm dGTP or dGDPNP (30, 60, 120, and 240 μm) using Pol III* reconstituted with four different DnaX complexes (τ3δδ'χψ, τ2γδδ'χψ, τγ2δδ'χψ, and γ3δδ'χψ) using α-[32P]dATP to label both strands. The reaction products were analyzed by alkaline-agarose gel electrophoresis.
Rolling Circle Replication with Pol III* Containing Wild-type and Mutant γ DnaX Subunits
Rolling circle reactions were set up using two different forms of Pol III*: one with a wild-type form of the γ subunit (τ2γ) and one with K51Eγ (τ2γM, ATPase-defective). Both forms of the polymerase were functional in holoenzyme reconstitution experiments (26). For incorporation experiments, the amount of dGTP was varied (0, 1.6, 3.1, 6.3, 12.5, 25, 50, and 100 μm). For analysis of rolling circle reaction products on alkaline-agarose gels, reactions were set up in the presence of α-[32P]dCTP, α-[32P]dGTP, or α-[32P]dATP. The nucleotide α-[32P]dCTP specifically labels the leading strand, α-[32P]dGTP specifically labels the lagging strand, and α-[32P]dATP labels both the leading and lagging strands. A total of 50,000 cpm was loaded to each lane.
Alkaline-Agarose Gel Electrophoresis
For the analysis of leading and lagging strand products, samples were mixed with 30 mm NaOH, 2 mm EDTA, 2% glycerol, and 0.02% bromphenol blue and fractionated on 0.6% alkaline-agarose gels (10 × 14 cm) for 16 h at 60 mA (∼20 V) in a running buffer of 30 mm NaOH and 2 mm EDTA. Gels were fixed in 8% (w/v) trichloroacetic acid, dried onto DEAE paper, autoradiographed on storage phosphor screens, and scanned with a PhosphorImager. Okazaki fragment length was determined as described previously (4).
Pol III* Exchange Assay on a Single-stranded Template
This assay measures the ability of a Pol III* competitor to exchange with a Pol III* undergoing DNA synthesis on a single-stranded template. Holoenzyme reactions (22) were assembled on ice using WT Pol III* (40 nm, experimentally determined to be at saturation), β2 (6 nm), DnaG primase (60 nm), SSB4 (0.6 μm), M13Gori DNA (2.3 nm as circle), four rNTPs (0.2 mm each), and 1 mm ATP. The reaction buffer was 10 mm magnesium acetate, 70 mm KCl, 50 mm Hepes (pH 7.5), 100 mm potassium glutamate, 20% glycerol, 200 μg/ml bovine serum albumin, 0.02% Nonidet P-40, and 10 mm dithiothreitol. Initiation complexes were formed at 30 °C for 3 min. Control reactions demonstrated that initiation complexes were completely formed by 1 min. The reaction was initiated by adding dNTPs (48 μm each dATP, dCTP, dGTP; 18 μm dTTP; 100 cpm [3H]/pmol dNTPs) plus competitor Pol III* (D403E Pol III*, WT Pol III*, or buffer control) at various concentrations (0, 24, 48, 94, 188, 375, and 750 nm). Reactions were performed in triplicate. DNA synthesis was quenched after 30 s by the addition of EDTA (140 mm), and samples were analyzed by liquid scintillation counting after trichloroacetic acid precipitation. 1 unit of Pol III* activity is 1 pmol of total nucleotide incorporated into acid-insoluble DNA per minute.
Results
Exogenous D403E Pol III Does Not Exchange with Pol III at the Replication Fork
A proposal has been made that free Pol III can exchange with the replication fork by dissociation and reassociation with τ (8). This observation seemed to be in conflict with the observation that the Pol III α-τ interaction has a lifetime of 4.9 h (10). We employed a dominant negative mutant E. coli Pol III α (α-D403E) that is able to form an initiation complex at the replication fork but cannot elongate (22, 32) to test this proposal. When D403E Pol III was premixed with wild-type E. coli Pol III to form an initiation complex, rolling circle DNA replication was inhibited in proportion to the concentration of D403E Pol III as expected (Fig. 1A). When D403E Pol III was added to an ongoing rolling circle DNA replication reaction, inhibition was not observed (Fig. 1B), indicating that exchange of Pol III does not occur over the 8-min time course of the reaction.
FIGURE 1.
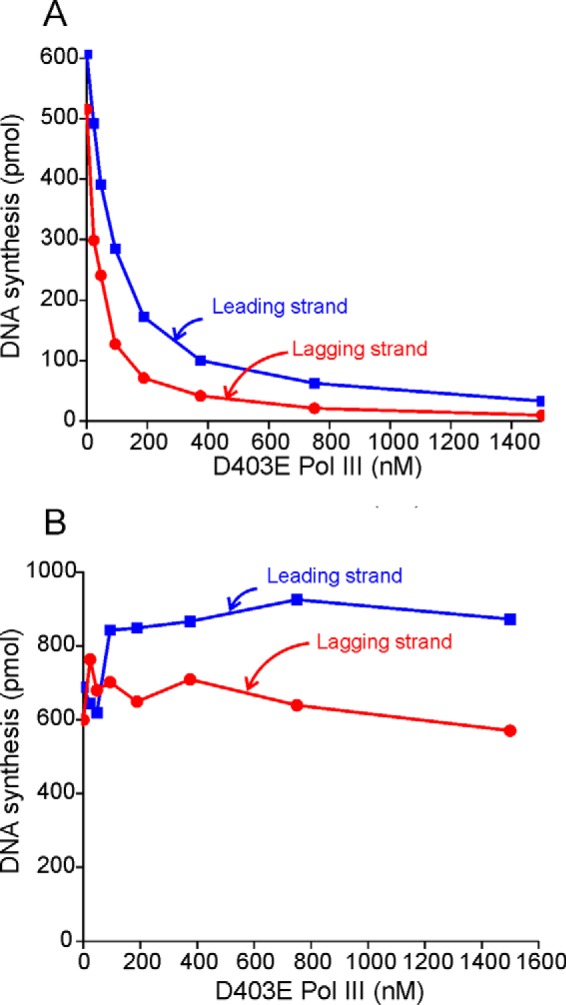
Pol III cannot exchange into the replication fork. A, protein components required for a rolling circle reaction were added to a premixed solution of wild-type Pol III and D403E Pol III. After initiation complex formation, the rolling circle replication reaction was started by addition of 1 mm ATP, 100 μm dNTPs, and α-[32P]dCTP or α-[32P]dGTP. B, D403E Pol III was added at the designated final concentration to an ongoing rolling circle reaction.
Exogenous D403E Pol III* Exchanges with Pol III* at the Replication Fork
We next tested whether exogenous D403E Pol III* was able to compete with wild-type Pol III* at the replication fork. In a control reaction, D403E Pol III* inhibited a rolling circle reaction when premixed with wild-type Pol III* to form the initiation complex (Fig. 2A). Surprisingly, D403E Pol III*, when added after initiation, inhibited an ongoing rolling circle reaction. Lagging strand synthesis was marginally more susceptible to challenge, being reduced 50% with half the level of D403E Pol III* challenge compared with leading strand synthesis. (Fig. 2B).
FIGURE 2.
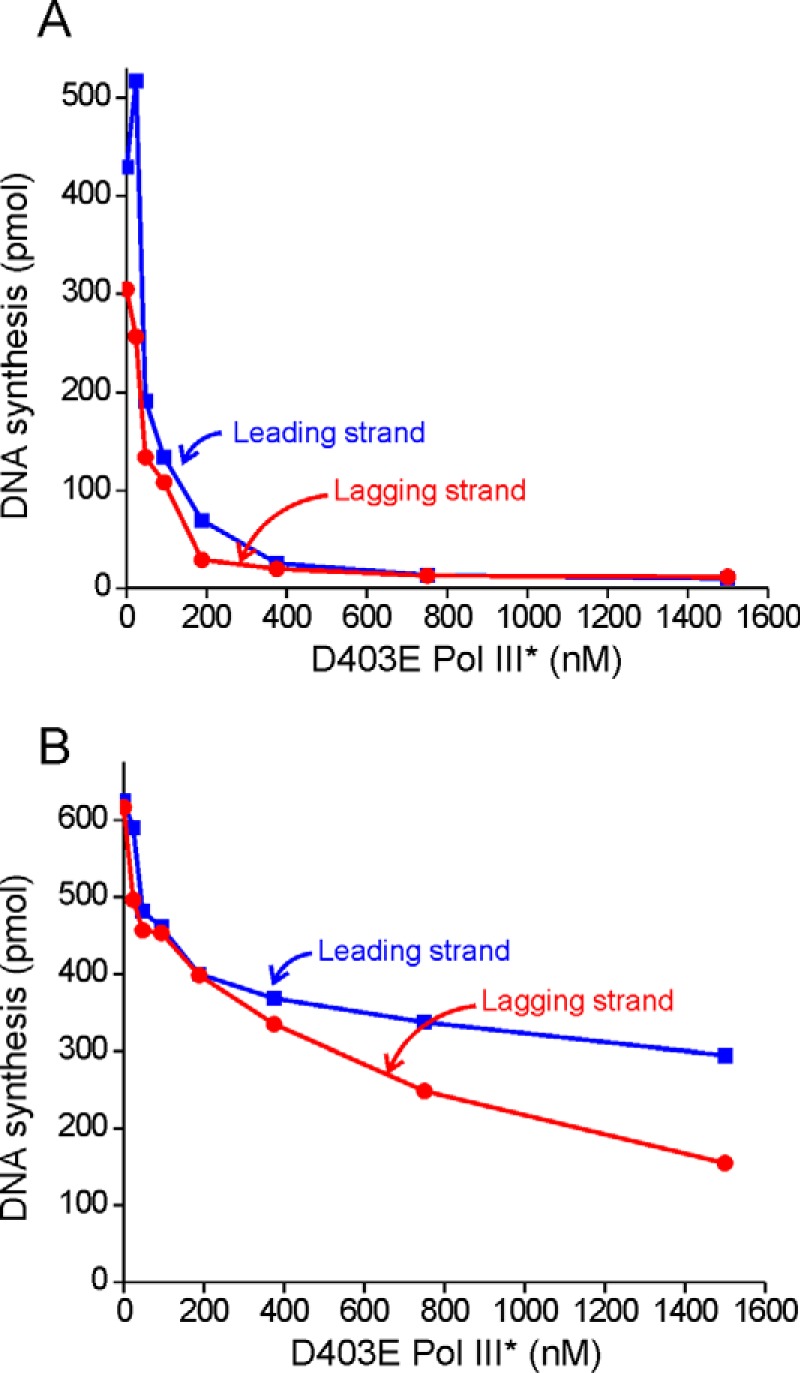
Pol III* can exchange into the replication fork. A, protein components required for a rolling circle reaction were added to a premixed solution of wild-type Pol III* and D403E Pol III*. After initiation complex formation, the rolling circle replication reaction was started by addition of 1 mm ATP, 100 μm dNTPs, and α-[32P]dCTP or α-[32P]dGTP. B, D403E Pol III* was added at the designated final concentration to an ongoing rolling circle reaction.
DNA Pol IV Exchanges into the Replication Fork with Different Protein Requirements than Pol III
We repeated the experiments we performed to assess exchange of free Pol III, substituting Pol IV. Pol IV incorporates nucleotides very slowly, so inhibition of elongation can be used as an assay for exchange (13, 14, 19). We observed that Pol IV exchanges just as effectively whether added before or after initiation complex formation by Pol III HE (Fig. 3). In both cases, lagging strand synthesis is more susceptible to a challenge by Pol IV than leading strand synthesis. Because Pol IV alone can exchange into ongoing replication forks and Pol III requires bound DnaX complex, the protein requirements and perhaps mechanism are different from that of Pol III.
FIGURE 3.
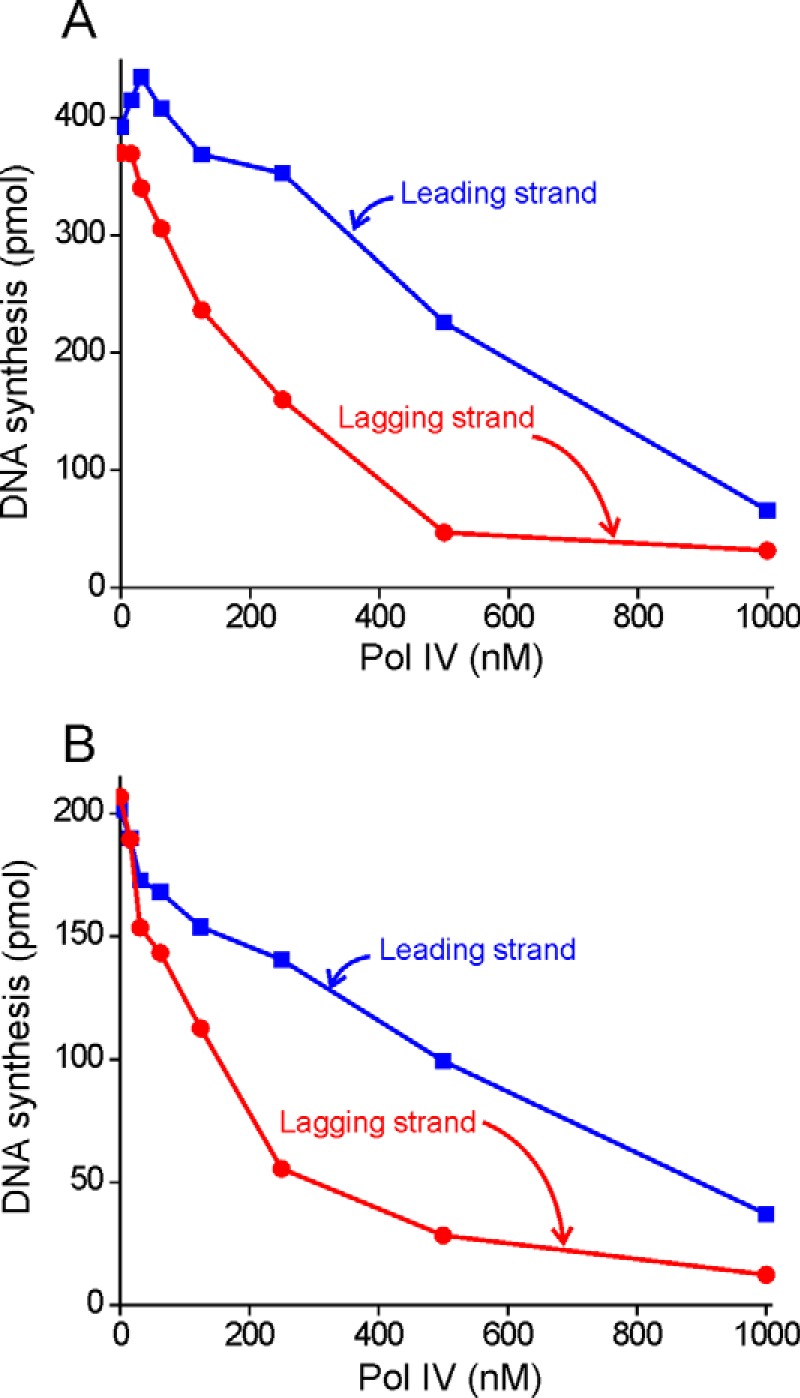
Pol IV can exchange into the replication fork under conditions where Pol III cannot. A, protein components required for a rolling circle reaction were added to a premixed solution of wild-type Pol III* and Pol IV. After initiation complex formation, the rolling circle replication reaction was started by addition of 1 mm ATP, 100 μm dNTPs, and α-[32P]dCTP or α-[32P]dGTP. B, Pol IV was added to an ongoing rolling circle reaction.
Okazaki Fragment Size and Gaps between Fragments Are the Same, Regardless of the τ Content of Pol III*
Because DnaX complexes contain a total of three γ and/or τ subunits, four different stoichiometries are possible: τ3δδ'χψ, τ2γδδ'χψ, τγ2δδ'χψ, and γ3δδ'χψ. We tested the function of all four DnaX complexes in a minicircle replication system. We found that all τ-containing DnaX complexes functioned equivalently in terms of the level of leading and lagging strand synthesis (Fig. 4A). All τ-containing DnaX complexes synthesized Okazaki fragments of the same length (Fig. 4B). The extent to which Okazaki fragments were decreased when made shorter by decreasing dGDPNP concentration was identical for all τ-containing complexes (Fig. 4B). DnaX complex lacking τ (γ complex) was unable to function under the experimental conditions used (substoichiometric Pol III*, Fig. 4).
FIGURE 4.

Pol III* reconstituted with DnaX complexes containing τ3, τ2γ, and τγ2 produce the same-length lagging strand products. A, an optimized rolling circle reaction was conducted with Pol III* reconstituted with the designated DnaX complexes. α-[32P]dCTP or α-[32P]dGTP labeled the leading and lagging strands, respectively. B, optimized rolling circle reactions were conducted in the presence of either 100 μm dGTP or the indicated amount of dGDPNP. α-[32P]dATP (2000 cpm/pmol) was used to label both the leading and lagging strands, which could be distinguished by their length after alkaline-agarose gel electrophoresis.
The observation that Pol III HE containing only one τ can function in coupled synthesis was surprising. We previously thought that two τs were required to couple leading and lagging strand synthesis. In light of this finding, we further explored the stability of replication forks reconstituted with single τ Pol III HE by testing whether they are more susceptible to challenge by D403E Pol III or Pol III* (supplemental Figs. 1 and 2). We observed that, like other forms of Pol III HE, the single τ form was resistant to a challenge by Pol III alone and similarly susceptible to a challenge by D403E Pol III*. However, a single τ Pol III HE is not sufficiently stable to function in a flow system, presumably because it dissociates sufficiently frequently to be carried away in the flow solution, removing it from an immobilized template (33). In a DnaB-dependent leading strand-tethered bead single-molecule experiment, it was observed that DnaX complex containing only one τ was functional (34).
We also observe that forks reconstituted with single-τ Pol III* are just as resistant to a challenge by D403E Pol III* as τ3 forms (supplemental Fig. 1). Similarly, D403E Pol III*s containing one or three τs are similarly effective as a challenge (supplemental Fig. 2), but “Pol III*” reconstituted with γ complex (no τ) is unable to exchange into replication forks (supplemental Fig. 3).
DnaX Complex with One Inactive ATPase Is Not Competent for Rolling Circle DNA Replication
To determine whether one inactive DnaX ATPase among the three present in DnaX complex affects coupled leading and lagging strand synthesis, a rolling circle reaction was conducted with reconstituted τ2γ Pol III* containing either one wild-type γ DnaX subunit or one mutant K51E γ DnaX subunit. The mutation occurs within the Walker A motif and abolishes ATP binding (26). In the presence of Pol III* containing mutant γ, the total level of rolling circle DNA synthesis is decreased 7-fold (Fig. 5A). Specific examination of leading and lagging synthesis using alkaline gel electrophoresis shows that some leading strand synthesis occurs but no detectable lagging strand synthesis (Fig. 5B).
FIGURE 5.
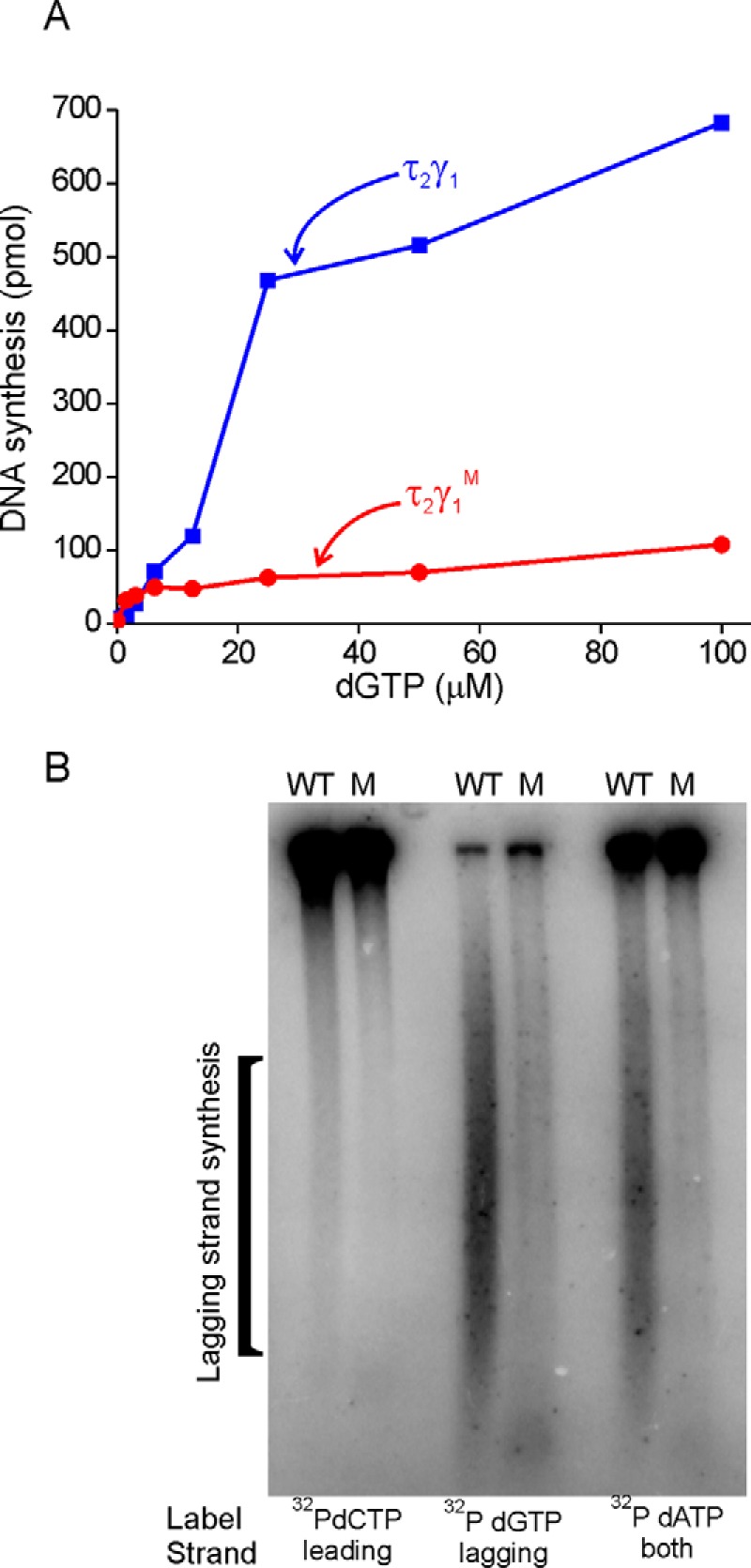
Pol III* with an inactive ATPase cannot support lagging strand DNA synthesis. A, total amount of DNA synthesis was compared between rolling circle reactions containing wild-type Pol III* (DnaX complex (τ2γ)) and reactions containing mutant Pol III* (DnaX complex (τ2γM)). Rolling circle reactions were conducted in the presence of α-[32P]dATP and the indicated amount of dGTP. B, α-[32P]dCTP-, α-[32P]dGTP-, and α-[32P]dATP-labeled reaction products from rolling circle reactions were loaded onto an alkaline-agarose gel to assess lagging strand synthesis from wild-type Pol III* (DnaX complex (τ2γδδ'χψ)) and mutant Pol III* (DnaX complex (τ2γM δδ'χψ)). α-[32P]dCTP selectively labels the leading strand, α-[32P]dGTP selectively labels the lagging strand, and α-[32P]dATP labels both the leading and lagging strands. M, mutant.
D403E Pol III* Can Also Exchange into Elongating Pol III HE on Long Single-stranded Templates
To determine whether DnaB6 or other components unique to replication forks were required for exchange, we challenged Pol III HE elongating a long single-stranded template. We preformed initiation complexes using wild-type Pol III* and β2 on DnaG primase-synthesized primers on SSB-encoated M13 circles containing a G4 origin and then added challenge D403E Pol III* together with dNTPs to initiate the reaction. We observe the same level of inhibition on these simpler single-stranded templates as we do in the coupled rolling circle assays (Fig. 6). Thus, the DnaB6 helicase is not a required component to enable exchange.
FIGURE 6.
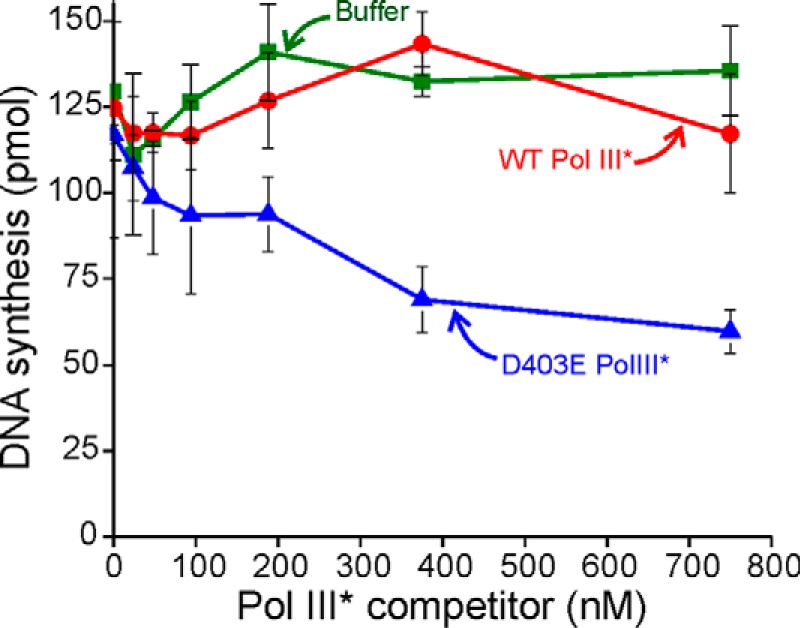
D403E Pol III* can exchange into Pol III HE during elongation on a long single-stranded template in the absence of helicase. Protein components required for a holoenzyme reconstitution experiment were added to an M13Gori template. Initiation complexes were allowed to form for 3 min at 30 °C in the presence of 1 mm ATP. The reaction was started by addition of 18 μm dNTPs, [3H]dTTP, and a competitor Pol III* (0, 24, 48, 94, 188, 375, and 750 nm). DNA synthesis was quenched after 30 s by the addition of EDTA to 140 mm. Reactions were run in triplicate. Shown are competitor mutant D403E Pol III* (blue), WT Pol III* (red), and buffer control (green). The buffer control samples were prepared identically to the Pol III* samples except that no Pol III* was added.
Discussion
Unlike the model T4 and T7 systems, core Pol III from E. coli cannot exchange into replication forks. It requires association with a τ-containing DnaX complex. Thus, the E. coli system is fundamentally different from these two systems. Further contrasting the model systems, T7 dynamic exchange is thought to be mediated by a polymerase-helicase interaction (7). In our E. coli system, exchange occurs on single-stranded templates in the absence of helicase (Fig. 6). Exchange of Pol III with Pol IV or Pol II also occurs on single-stranded templates independently of helicase (13–15, 35, 36).
We examined the ability of Pol III HE to function in coupled rolling circle replication with variable τ content. The τ3, τ2, and τ1 forms functioned equivalently in terms of levels of synthesis but also Okazaki fragment size distribution, even in the presence of decreasing dGDPNP concentrations (Fig. 4). At low dGDPNP concentration, lagging strand synthesis is slowed specifically, whereas leading strand synthesis progresses at a normal rate along with the synthesis of new primers by DnaG primase. This induces the lagging strand polymerase to cycle before synthesis is complete, leaving large gaps (4). That the size of fragments is identical regardless of τ content suggests that all three forms of Pol III HE experience the same dynamics at the replication fork, initiating and cycling between Okazaki fragment synthesis at nearly equivalent rates, resulting in the same length of gaps between fragments.
It has been proposed that τ3 Pol III HE leaves shorter gaps between Okazaki fragments than Pol III HE containing two τs. These experiments were performed by a low-resolution single-molecule technique where a single pixel covered 600 nucleotides, approaching the size of the 1- to 2-kb Okazaki fragments found under physiological conditions. To surmount this obstacle, conditions were altered to increase Okazaki fragment size to 12 kb (21). Our results do not contradict these results obtained under non-physiological conditions but do show that gap size is the same under physiological conditions. Furthermore, a trimeric polymerase is not the primary form of Pol III HE in cells (3), making the single-molecule results less relevant.
Pol III HE-like activity, reconstituted in the presence of γ complex (no τ), was inactive in rolling circle synthesis under the experimental conditions employed: substoichiometric Pol III*. This is consistent with the requirement for a τ-DnaB helicase association to stabilize replication forks (37). We were initially surprised by the ability of Pol III HE that contained only one copy of τ to function efficiently (Fig. 4). When free in solution, Pol III HE must contain at least two τs to interact with DnaB6 under physiologically relevant concentrations (38). However, we have observed3 that the presence of ATPγS and an associated replication fork significantly strengthens the interaction of Pol III HE with τ. Perhaps the synergy of a multimeric complex stabilizes the interaction sufficiently to permit single τ HE interaction. We do not believe that this form of HE exists in cells to a significant extent, but its function may be a reflection of a redundancy of stabilizing interactions at the replication fork.
Our finding that Pol III* can exchange into replication forks may reconcile the conflict between our finding that free polymerase cannot exchange with Pol III at the replication fork and the single molecule experiments that were interpreted to indicate that free Pol III could dynamically exchange at replication fork (8). The bursts of fluorescence they observed could have resulted from Pol III* exchange at the replication fork because it is not possible to distinguish between Pol III and Pol III* from the fluorescence assay they conducted. Alternatively, they could have observed exogenous Pol III required for other processes associated with the nascent replication product, such as mismatch repair. Involvement of the DnaX complex in Pol III* exchange indicates that interactions mediated by the DnaX complex are important for polymerase exchange.
DnaX complexes containing a mixture of inactive and wild-type DnaX subunits can function on long single-stranded templates in 5-min reactions (26). Examination of the kinetics of initiation complex formation reveals a 3500-fold lower rate for DnaX complexes containing a single inactive ATPase subunit (39). We exploited the rolling circle system we recently developed that permits use of substoichiometric Pol III* (4) to provide a sensitive test for function under physiological conditions. We observe a modest level of leading strand synthesis, consistent with the ability of mixed mutant complexes to function on long single-stranded templates, but could not detect any lagging strand synthesis, suggesting that Pol III HE must have three active ATPases to support the rapid initiation complex formation and cycling required for lagging strand synthesis (Fig. 5). Because of the frequent initiation complex formation on the lagging strand in a rolling circle reaction, slowing the rate of initiation complex formation by the inactive ATPase exerts a much more severe effect than in an single-stranded DNA replication reaction.
For E. coli Pol III to exchange into a replication fork requires sequestered DnaX complex. γ Complex, which can load β2 clamps onto DNA but cannot bind Pol III α, does not substitute. Thus, some component of τ-containing DnaX complexes must be responsible for polymerase exchange. Because τ complexes are functional and γ complexes are not, the simplest explanation would be that domain IV and/or V, present only in τ, interacts with elongating Pol III HE and triggers exchange. However, we have discovered situations where τ is required only as a bridge to hold χψ together in the same complex with Pol III; for example, enabling replication through SSB on a single-stranded template in the absence of β2 and for stabilizing leading strand polymerase on a forked template so that it can strand-displace, in the absence of helicase, by interaction with SSB bound to the displaced strand (31, 40). These findings have been confirmed and extended (41, 42). It has also been observed that only τ-containing DnaX complexes will support initiation complex formation using slowly hydrolyzable ATPγS (32, 43), likely by stabilizing intermediates during slow initiation complex formation. The δ subunit is involved in either opening β2 complexes or trapping open complexes from solution as part of the initiation complex formation reaction. The δ subunit could be involved in modulating β2 structures to enable access by other proteins. Thus, components of the DnaX complex other than τ could facilitate exchange.
We have recently demonstrated that the signaling model is exclusively operational as a mechanism to drive Pol III* release and recycling during Okazaki fragment synthesis (4). The availability of a new primer is required as the signal to initiate cycling. DnaG primase is not required when primers are provided exogenously. In a simpler model system, we observed that τ-containing DnaX complex, exogenous primer template, and ATP, but not ATPγS, could modestly accelerate Pol III* release from chip-bound oligonucleotide templates (44). τ-Containing DnaX complexes can rapidly chaperone Pol III onto nascently loaded β2 and accelerate the overall initiation complex formation reaction 100-fold, a requisite for the physiological rate of Okazaki fragment formation (39). Combining this information, we have proposed a working model that τ-containing DnaX complexes may also chaperone Pol III off of nascent Okazaki fragments when a primer becomes available for the next fragment (4). It is possible that the DnaX complex in exogenous Pol III* complexes competes in the process. This could explain the increased sensitivity of lagging strands to exchange by a challenging polymerase (Fig. 2).
Other mechanisms might be responsible for permitting access to primer termini on the leading strand. Because the leading strand polymerase is not free to rotate around the template strand because it is linked to the lagging strand polymerase, DNA behind the polymerase will be underwound. The polymerase may need to transiently release the primer terminus to relieve this negative superhelical tension, allowing the DNA to rotate within the channel of the bound, processively elongating Pol III HE (9, 45–47). This might grant access to the primer terminus, enabling an exchange reaction.
Alternatively, direct protein contacts between the invading Pol III* with the displaced Pol III HE might be required. Clearly, interactions of Pol IV with β2 are important to enable exchange (13–15, 19). Mutant Pol IV that lacks its β2 interacting motif can still exchange, albeit at higher concentrations, suggesting an interaction between Pol IV and a component of Pol III* (19). It is not clear whether these interactions act simultaneously or sequentially to mediate exchange. Interaction of Pol IV with Pol III* may trigger a conformational change so that at least one of the two mutual binding sites within β2, sequestered by Pol III α and ϵ during elongation (20), become accessible. Consistent with this possibility, residues within or near the start of α helix 5 in Pol IV, which are well removed from the two distinct β2 clamp binding motifs, are required to displace Pol III* from β2 assembled on DNA (17). Pol III* exchange could proceed by a similar mechanism, except the primary interacting motif would have to reside within τ-containing DnaX complexes. Alternatively, the mechanism of Pol III* exchange could be different from that used by Pol IV, consistent with different protein requirements for exchange.
These issues surrounding polymerase exchange at replication forks are general and important. Unpublished results4 show that Bacillus subtilis Pol C actively evicts an otherwise highly processive DnaE starting ∼35 nucleotides after initiating synthesis from a nascent primer during Okazaki fragment synthesis. Similarly, Pol δ must displace Pol α during eukaryotic Okazaki fragment synthesis. Very importantly, a host of error-prone or bypass polymerases must exchange with their normal replicative counterparts to maintain genome stability in all cell types. Establishing general mechanisms for exchange should be a challenging and fruitful area of investigation for future investigators.
Author Contributions
C. M. designed the study. Q. Y. and P. R. D. performed the experiments. C. M. and M. D. S. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Diane Hager for preparation of the figures and references.
This work was supported by National Science Foundation Award MCB-1329285 (to C. S. M.) and a Public Health Service Award R01 GM066094 from the NIGMS/National Institutes of Health (to M. D. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This is the last original research paper based on work initiated in the McHenry laboratory during our 40 years of investigating mechanisms of DNA replication. It is particularly fitting that the first paper from our laboratory (1) was also published in this Journal.

This article contains supplemental Figs. 1–3.
L. Wilson, B. Glover, and C. McHenry, unpublished observations.
J. Zinder and C. McHenry, unpublished results.
- Pol
- polymerase
- HE
- holoenzyme
- dGDPNP
- 5′-guanylyl beta-gamma-imidodiphosphate
- SSB
- single-stranded DNA-binding protein
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
References
- 1. McHenry C. S., and Crow W. (1979) DNA polymerase III of Escherichia coli: purification and identification of subunits. J. Biol. Chem. 254, 1748–1753 [PubMed] [Google Scholar]
- 2. McHenry C. S. (2011) DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80, 403–436 [DOI] [PubMed] [Google Scholar]
- 3. Dohrmann P. R., Correa R., Frisch R. L., Rosenberg S. M., and McHenry C. S. (2016) The DNA polymerase III holoenzyme contains γ and is not a trimeric polymerase. Nucleic Acids Res. 44, 1285–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan Q., and McHenry C. S. (2014) Cycling of the E. coli lagging strand polymerase is triggered exclusively by the availability of a new primer at the replication fork. Nucleic Acids Res. 42, 1747–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaboord B. F., and Benkovic S. J. (1995) Accessory proteins function as matchmakers in the assembly of the T4 DNA polymerase holoenzyme. Curr. Biol. 5, 149–157 [DOI] [PubMed] [Google Scholar]
- 6. Yang J., Zhuang Z., Roccasecca R. M., Trakselis M. A., and Benkovic S. J. (2004) The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl. Acad. Sci. U.S.A. 101, 8289–8294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson D. E., Takahashi M., Hamdan S. M., Lee S. J., and Richardson C. C. (2007) Exchange of DNA polymerases at the replication fork of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 104, 5312–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lia G., Michel B., and Allemand J. F. (2012) Polymerase exchange during Okazaki fragment synthesis observed in living cells. Science 335, 328–331; Retraction (2014) Science346, 1466 [DOI] [PubMed] [Google Scholar]
- 9. McHenry C. S. (2015) Cycling of the lagging strand replicase during Okazaki fragment synthesis. In Molecular Life Sciences: An Encyclopedic Reference (Bell E., Bond J., Klinman J., Masters B. S., and Wells R., eds.) Springer, New York [Google Scholar]
- 10. Kim D. R., and McHenry C. S. (1996) Biotin tagging deletion analysis of domain limits involved in protein-macromolecular interactions: mapping the τ binding domain of the DNA polymerase III α subunit. J. Biol. Chem. 271, 20690–20698 [DOI] [PubMed] [Google Scholar]
- 11. Burnouf D. Y., Olieric V., Wagner J., Fujii S., Reinbolt J., Fuchs R. P., and Dumas P. (2004) Structural and biochemical analysis of sliding clamp/ligand interactions suggest a competition between replicative and translesion DNA polymerases. J. Mol. Biol. 335, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 12. Tang M., Pham P., Shen X., Taylor J. S., O'Donnell M., Woodgate R., and Goodman M. F. (2000) Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 404, 1014–1018 [DOI] [PubMed] [Google Scholar]
- 13. Kath J. E., Jergic S., Heltzel J. M., Jacob D. T., Dixon N. E., Sutton M. D., Walker G. C., and Loparo J. J. (2014) Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc. Natl. Acad. Sci. U.S.A. 111, 7647–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Indiani C., McInerney P., Georgescu R., Goodman M. F., and O'Donnell M. (2005) A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 19, 805–815 [DOI] [PubMed] [Google Scholar]
- 15. Heltzel J. M., Maul R. W., Scouten Ponticelli S. K., and Sutton M. D. (2009) A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc. Natl. Acad. Sci. U.S.A. 106, 12664–12669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ikeda M., Furukohri A., Philippin G., Loechler E., Akiyama M. T., Katayama T., Fuchs R. P., and Maki H. (2014) DNA polymerase IV mediate efficient and quick recovery of replication forks stalled at N(2)-dG adducts. Nucleic Acids Res. 42, 8461–8472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scotland M. K., Heltzel J. M., Kath J. E., Choi J. S., Berdis A. J., Loparo J. J., and Sutton M. D. (2015) A genetic selection for dinB mutants reveals an interaction between DNA polymerase IV and the replicative polymerase that is required for translesion synthesis. PLoS Genet. 11, e1005507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gabbai C. B., Yeeles J. T., and Marians K. J. (2014) Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms. J. Biol. Chem. 289, 32811–32823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furukohri A., Goodman M. F., and Maki H. (2008) A dynamic polymerase exchange with Escherichia coli polymerase IV replacing polymerase III on the sliding clamp. J. Biol. Chem. 283, 11260–11269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernandez-Leiro R., Conrad J., Scheres S. H., and Lamers M. H. (2015) cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife 4, e11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Georgescu R. E., Kurth I., and O'Donnell M. E. (2012) Single-molecule studies reveal the function of a third polymerase in the replisome. Nat. Struct. Mol. Biol. 19, 113–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pritchard A. E., and McHenry C. S. (1999) Identification of the acidic residues in the active site of DNA polymerase III. J. Mol. Biol. 285, 1067–1080 [DOI] [PubMed] [Google Scholar]
- 23. Wieczorek A., and McHenry C. S. (2006) The NH(2)-terminal php domain of the α subunit of the E. coli replicase binds the ϵ proofreading subunit. J. Biol. Chem. 281, 12561–12567 [DOI] [PubMed] [Google Scholar]
- 24. Carter J. R., Franden M. A., Aebersold R., Kim D. R., and McHenry C. S. (1993) Isolation, sequencing and overexpression of the gene encoding the θ subunit of DNA polymerase III holoenzyme. Nucleic Acids Res. 21, 3281–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D. R., and McHenry C. S. (1996) In vivo assembly of overproduced DNA polymerase III: overproduction, purification, and characterization of the α, α-ϵ, and α-ϵ-θ subunits. J. Biol. Chem. 271, 20681–20689 [DOI] [PubMed] [Google Scholar]
- 26. Wieczorek A., Downey C. D., Dallmann H. G., and McHenry C. S. (2010) Only one ATP-binding DnaX subunit is required for initiation complex formation by the E. coli DNA polymerase III holoenzyme. J. Biol. Chem. 285, 29049–29053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maul R. W., Ponticelli S. K., Duzen J. M., and Sutton M. D. (2007) Differential binding of Escherichia coli DNA polymerases to the β-sliding clamp. Mol. Microbiol. 65, 811–827 [DOI] [PubMed] [Google Scholar]
- 28. Johanson K. O., Haynes T. E., and McHenry C. S. (1986) Chemical characterization and purification of the β subunit of the DNA polymerase III holoenzyme from an overproducing strain. J. Biol. Chem. 261, 11460–11465 [PubMed] [Google Scholar]
- 29. Griep M. A., and McHenry C. S. (1989) Glutamate overcomes the salt inhibition of DNA polymerase III holoenzyme. J. Biol. Chem. 264, 11294–11301 [PubMed] [Google Scholar]
- 30. Marians K. J. (1995) φ X174-type primosomal proteins: purification and assay. Methods Enzymol. 262, 507–521 [DOI] [PubMed] [Google Scholar]
- 31. Yuan Q., and McHenry C. S. (2009) Strand displacement by DNA polymerase III occurs through a τ-ψ-χ link to SSB coating the lagging strand template. J. Biol. Chem. 284, 31672–31679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Downey C. D., and McHenry C. S. (2010) Chaperoning of a replicative polymerase onto a newly-assembled DNA-bound sliding clamp by the clamp loader. Mol. Cell 37, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanner N. A., Tolun G., Loparo J. J., Jergic S., Griffith J. D., Dixon N. E., and van Oijen A. M. (2011) E. coli DNA replication in the absence of free β clamps. EMBO J. 30, 1830–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanner N. A., Hamdan S. M., Jergic S., Loscha K. V., Schaeffer P. M., Dixon N. E., and van Oijen A. M. (2008) Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol. 15, 170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heltzel J. M., Maul R. W., Wolff D. W., and Sutton M. D. (2012) Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III* replicase. J. Bacteriol. 194, 3589–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kath J. E., Chang S., Scotland M. K., Wilbertz J. H., Jergic S., Dixon N. E., Sutton M. D., and Loparo J. J. (2016) Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res. 44, 1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim S., Dallmann H. G., McHenry C. S., and Marians K. J. (1996) Coupling of a replicative polymerase and helicase: a τ-DnaB interaction mediates rapid replication fork movement. Cell 84, 643–650 [DOI] [PubMed] [Google Scholar]
- 38. Gao D., and McHenry C. S. (2001) τ Binds and organizes Escherichia coli replication proteins through distinct domains: domain IV, located within the unique C terminus of τ, binds the replication fork helicase, DnaB. J. Biol. Chem. 276, 4441–4446 [DOI] [PubMed] [Google Scholar]
- 39. Downey C. D., Crooke E., and McHenry C. S. (2011) Polymerase chaperoning and multiple ATPase sites enable the E. coli DNA polymerase III holoenzyme to rapidly form initiation complexes. J. Mol. Biol. 412, 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glover B. P., and McHenry C. S. (1998) The χψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of a SSB-coated template. J. Biol. Chem. 273, 23476–23484 [DOI] [PubMed] [Google Scholar]
- 41. Jergic S., Horan N. P., Elshenawy M. M., Mason C. E., Urathamakul T., Ozawa K., Robinson A., Goudsmits J. M., Wang Y., Pan X., Beck J. L., van Oijen A. M., Huber T., Hamdan S. M., and Dixon N. E. (2013) A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 32, 1322–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marceau A. H., Bahng S., Massoni S. C., George N. P., Sandler S. J., Marians K. J., and Keck J. L. (2011) Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 30, 4236–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glover B. P., and McHenry C. S. (2001) The DNA polymerase III holoenzyme: an asymmetric dimeric replicative complex with leading and lagging strand polymerases. Cell 105, 925–934 [DOI] [PubMed] [Google Scholar]
- 44. Dohrmann P. R., Manhart C. M., Downey C. D., and McHenry C. S. (2011) The rate of polymerase release upon filing the gap between Okazaki fragments is inadequate to support cycling during lagging strand synthesis. J. Mol. Biol. 414, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ullsperger C. J., Vologodskii A. V., and Cozzarelli N. R. (1995) In Nucleic Acids and Molecular Biology (Eckstein F., and Lilley D. M. J., eds.), pp. 115–142, Springer, Berlin [Google Scholar]
- 46. McHenry C. S., Tomasiewicz H., Griep M. A., Fürste J. P., and Flower A. M. (1988) In DNA Replication and Mutagenesis (Moses R., and Summers W., eds.), pp. 14–26, American Society for Microbiology, Washington, D.C. [Google Scholar]
- 47. Postow L., Peter B. J., and Cozzarelli N. R. (1999) Knot what we thought before: the twisted story of replication. BioEssays 21, 805–808 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.