

Abstract
Bok is a member of the Bcl-2 protein family that governs the intrinsic apoptosis pathway, although the role that Bok plays in this pathway is unclear. We have shown previously in cultured cell lines that Bok interacts strongly with inositol 1,4,5-trisphosphate receptors (IP3Rs), suggesting that it may contribute to the structural integrity or stability of IP3R tetramers. Here we report that Bok is similarly IP3R-assocated in mouse tissues, that essentially all cellular Bok is IP3R bound, that it is the helical nature of the Bok BH4 domain, rather than specific amino acids, that mediates binding to IP3Rs, that Bok is dramatically stabilized by binding to IP3Rs, that unbound Bok is ubiquitinated and degraded by the proteasome, and that binding to IP3Rs limits the pro-apoptotic effect of overexpressed Bok. Agents that stimulate IP3R activity, apoptosis, phosphorylation, and endoplasmic reticulum stress did not trigger the dissociation of mature Bok from IP3Rs or Bok degradation, indicating that the role of proteasome-mediated Bok degradation is to destroy newly synthesized Bok that is not IP3R associated. The existence of this unexpected proteolytic mechanism that is geared toward restricting Bok to that which is bound to IP3Rs, implies that unbound Bok is deleterious to cell viability and helps explain the current uncertainty regarding the cellular role of Bok.
Keywords: apoptosis, B-cell lymphoma 2 (Bcl-2) family, inositol trisphosphate receptor (InsP3R), proteasome, ubiquitin
Introduction
Bok is a member of the Bcl-2 protein family that controls the intrinsic apoptosis pathway (1–3). Bok contains four Bcl-2 homology domains (BH1–4)2 and shares greatest sequence homology with the pro-apoptotic proteins Bak and Bax (1–4). However, unlike Bak and Bax, which have clearly defined roles in mediating mitochondrial outer membrane permeabilization (5, 6), the cellular role of Bok is unclear. Key observations that pertain to our current understanding of the function of Bok are (i) that the atypical C-terminal transmembrane (TM) domain of Bok localizes it to membranes of the endoplasmic reticulum (ER) and Golgi (7), (ii) that Bok over-expression leads to apoptosis (7–10) if Bak or Bax are present (7), indicating that Bok lies upstream of Bak and Bax, (iii) that Bok−/− mice are phenotypically normal (4, 11, 12), while Bax−/−Bak−/− mice exhibit multiple severe defects (1), indicating that Bok cannot substitute for Bak and Bax, and (iv) that ER stress-induced apoptosis (13) can be suppressed in Bok−/− mouse cells in vitro and in vivo (12), although this result has not been seen by all groups (7). Overall, these data suggest that Bok plays a very different role from Bax and Bak, and that it may participate in the pathway between ER stress and apoptosis.
Another intriguing facet of Bok cell biology is that it binds very strongly to inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) (14), proteins that form tetrameric, IP3-, and Ca2+-gated Ca2+ channels in ER membranes and play a key role in vertebrate cell signaling (15, 16). Of the three mammalian IP3R types, Bok binding is strongest to IP3R1 and IP3R2 (14) and the binding site for Bok can be localized to a regulatory hotspot within the IP3R ARM3 domain, part of the region that links the N-terminal IP3-binding domain to the C-terminal channel domain (14, 16). Bok binds via its BH4 domain, since deletion of that domain abolishes binding to IP3R1 (14). Interestingly, Bcl-2 also binds to IP3R1 via its BH4 domain, albeit much less strongly than Bok and to a different IP3R region (17–19). Nevertheless, Bcl-2 binding modulates IP3R-mediated Ca2+ mobilization (17–19) and a peptide derived from the binding site in IP3R1 reverses the suppressive effect of Bcl-2 on Ca2+ signaling and apoptosis (17, 20). In contrast, Bok binding does not appear to dramatically alter the Ca2+-mobilizing function of IP3Rs, but does protect IP3Rs against proteolysis within the ARM3 domain in vitro and in vivo, apparently by steric hindrance of protease access (14).
Here we report that Bok stability is highly dependent upon binding to IP3Rs and that “free” (non-IP3R bound) Bok is ubiquitinated and is degraded by the proteasome. This novel mechanism restricts cellular Bok to that which is bound to IP3Rs, implies that unbound Bok is deleterious to cell viability, and helps explain the current lack of clarity regarding the cellular role of Bok.
Experimental Procedures
Materials
αT3 cells and HEK 293T cells were cultured as described (14). Antibodies raised in rabbits were: anti-IP3R1 (21), anti-erlin2 (22), anti-IP3R1–3 (23), anti-HA epitope (24), anti-Mcl-1 D35A5, anti-Bcl-xL 54H6, anti-Bcl-2 50E3, anti-caspase-3 9662, and anti-poly (ADP-ribose) polymerase (PARP) 9542 (Cell Signaling Technology), anti-Bak 06–536 (Millipore), anti-Bax N-20 (Santa Cruz Biotechnology Inc.), and anti-Bok, raised against amino acids 19–32 of mouse Bok (4, 7). Mouse monoclonal antibodies were: anti-ubiquitin clone FK2 (BioMol International), anti-HA epitope clone HA11 (Covance), anti-FLAG epitope clone M2 (Sigma), and anti-p97 (Research Diagnostics Inc.). Horseradish peroxidase-conjugated secondary antibodies, gonadotropin-releasing hormone (GnRH), N-ethylmaleimide, protease inhibitors, Triton X-100, CHAPS, cycloheximide (CHX), thapsigargin, and forskolin were purchased from Sigma. DTT, Precision PlusTM Protein Standards, and SDS-PAGE reagents were from Bio-Rad. Protein A-Sepharose CL-4B was from GE Healthcare. MG132 was from Biomol. Staurosporine was from Enzo Life Sciences. Linear, MW∼25,000 polyethylenimine (PEI) was from Polysciences Inc.
Cell Lysis, Immunoprecipitation (IP), and SDS-PAGE
To prepare lysates, cells or brain tissues were harvested with ice-cold lysis buffer containing either 1% CHAPS or 1% Triton X-100 (14). CHAPS was used for all experiments involving IP, except when exogenous Bok ubiquitination was assessed, when cells were harvested with DTT-free Triton X-100 lysis buffer supplemented with 5 mm N-ethylmaleimide, followed by addition of 5 mm DTT 30 min later (25). Lysates were incubated on ice for 30 min and clarified by centrifugation at 16,000 × g for 10 min at 4 °C. For IP, clarified lysates were incubated with antisera and Protein A-Sepharose CL-4B for ∼16 h at 4 °C, and IPs were washed thoroughly with lysis buffer, were resuspended in gel-loading buffer, were incubated at 37 °C for 30 min or 100 °C for 3 min, were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose for probing as described (14).
Analysis of Exogenous Bok in HEK and HeLa Cells
Vectors encoding mouse Bok tagged at the N terminus with a triple FLAG epitope (3F-Bok) and mouse IP3R1 tagged at the C terminus with an HA epitope have been described (14). Additional constructs were created encoding 3F-Bok-HA (3F-Bok with an HA epitope, GYPYDVPDYAG, added to the C terminus), 3F-Bak (14), and untagged mouse Bok inserted between the KpnI/BglII sites of pCAG (26). HEK 293T cells seeded at 4.5 × 105/9.6 cm2 well, were transfected ∼24 h later with 0.125–2.125 μg cDNAs and 6 μl of 1 mg/ml PEI (pre-mixed in 50 μl of serum-free culture medium), and ∼24 h later were harvested with ∼ 0.4 ml/well lysis buffer. HeLa cells were seeded at 7 × 105/9.6 cm2 well, were transfected ∼24 h later with 0.5 μg of cDNAs and 3.5 μl of Lipofectamine (Invitrogen), and ∼24 h later were harvested with ∼ 0.2 ml/well lysis buffer.
Generation and Analysis of IP3R1 Knock-out (KO) Lines
The CRISPR/hCas9 system (27) was used to target exons within the mouse IP3R1 gene and create αT3IP3R1KO cell lines as described (25). Oligonucleotides targeting exons 5 or 13 (GGTGCGGAGTATCGATTCAT and GCACCTCCACGCAGAGTCGT, respectively) were introduced into αT3 cells and clones were screened by immunoblotting with anti-IP3R1 (25). Of the cell lines screened, ∼25% lacked IP3R1, and 2 lines for each targeted exon were characterized further with essentially identical results. Cells (106/9.6 cm2 well) were transiently transfected with IP3R1HA constructs (0.25–1.2 μg of cDNAs and 6 μl of 1 mg/ml PEI) and were harvested 48 h later.
Measurement of mRNA Levels
RNA was extracted using the RNeasy Mini Kit (Qiagen) and was reverse transcribed into cDNA using the Reverse Transcription System (Promega). qRT-PCR was performed using the forward primer GATGGACGGATGTCCTCAAG and the reverse primer TCTCTGGCAACAACAGGAAG, a Taq master mix developed in-house containing SYTO-82 (Molecular Probes), 3 mm MgCl2 and TaqDNA Polymerase (Thermo Fisher), a Stratagene MX3000P real time qPCR thermal cycler, and the following PCR cycling parameters: 10 min at 95 °C, followed by 40 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. Results were analyzed by normalizing to the housekeeping genes ribosomal protein S18, peptidylprolyl isomerase A, and hypoxanthine phosphoribosyltransferase 1, using the 2−ΔΔCT method (28).
Homology Modeling and Molecular Dynamics (MD) Simulations
The human Bok protein sequence (NCBI accession number NP_115904.1) was modeled using human Bax (PDB entry 1F16) and human Bak (PDB entry 2YV6) as templates and the Modeler v9.12 program (29) with the very slow refinement setting for MD optimization. The N-terminal 21 residues and C-terminal 10 residues of Bok were intentionally not modeled, as the N-terminal is disordered or absent in the Bax and Bak structures and the C terminus does not align with Bax and Bak. The best of 50 models identified by the GA341 and DOPE score functions was then further refined using the loop-refine script to create a set of 20 models with modified loop conformations. The model with the lowest MOLPDF score was then selected as the final Bok model.
To assess the structural effects of mutating Leu34 to Gly, RosettaBackrub (30) was first used to create a model of BokL34G, which revealed little effect on the overall structure of the residue 24–42 α-helical region, but significant shifts in the position of some amino acid side chains. MD simulations of this region of BokWT and BokL34G were then performed using the Gromacs 4.5.5 package (31) and Amber99SB force field (32). Each peptide was solvated in an octahedral box filled with TIP3P water and the net charges were neutralized with 0.15 m NaCl. The PME method was used to treat long-range electrostatic interactions, a cutoff of 10Å was used for the Coulomb energy and Van Der Waals interactions, the system was energy minimized until reaching 1000 kJ/mol/nm, and then equilibrated for 100 ps using the v-rescale method at 300 K, followed by pressure equilibration for 100 ps using the Parrinello-Rahman method at 300 K, and the LINCS algorithm was applied to constrain all bond lengths. In the final 10-ns MD simulation, the time step was 2 fs, and structures were saved every 2 ps.
Data Analysis
All experiments were repeated at least once (n = the number of independent experiments) and representative images of gels and traces are shown. Quantitated data are graphed as mean ± S.E. when n ≥ 3 or mean ± range when n = 2.
Results
Bok-IP3R1 Binding in Vivo and the Origin of Bok Variants
We have shown previously, via IP, that Bok binds tightly to IP3Rs in various cultured cell lines (14). Examination of mouse brain shows that such binding also occurs in vivo (Fig. 1), since IP of IP3R1 from brain cerebral cortex and hippocampus lysates specifically isolates Bok (lanes 2 and 3). Further, anti-IP3R1 depleted the vast majority of both IP3R1 and Bok from the lysates (compare lanes 4 and 5 with 6 and 7), indicating that essentially all Bok is IP3R1-associated.
FIGURE 1.
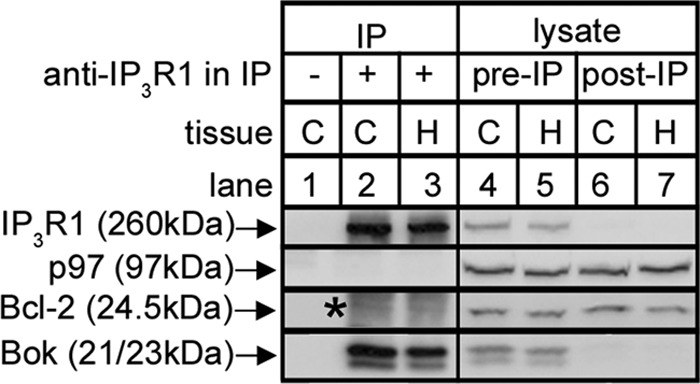
Co-IP of Bok with IP3R1 in mouse brain tissues. Mouse brain cerebral cortex (C) and hippocampus (H) lysates were incubated without or with anti-IP3R1, and IPs (lanes 1–3) and lysates (either pre- or post-IP; lanes 4–7) were subjected to SDS-PAGE and probed for the proteins indicated. p97 and Bcl-2 served as negative controls that did not co-IP and show that Bok co-IP is specific. Co-migrating IgG light chain seen in the Bcl-2 probe is indicated by the asterisk.
It is noteworthy that in mouse tissues and cell lines (Figs. 1 and 2, lanes 1–2) (14), anti-Bok, which is raised against amino acids 19–32 of mouse Bok (Fig. 3A), recognizes two species (at 21 and 23 kDa). Bands at 21 and 23 kDa were also seen after expression of exogenous Bok from mouse Bok cDNA in HEK cells (Fig. 2, lane 4). The existence of 21- and 23-kDa bands appears to be due to alternative translation initiation (33, 34), since mutation of the second AUG codon in the Bok coding region (that encodes Met15) blocks formation of the 21-kDa band (lane 5). Interestingly, the 3F-Bok construct (Fig. 3A) used previously to map the Bok-IP3R interaction (14) generated anti-Bok-immunoreactive bands at 27, 23, and 21 kDa (lane 6), with the anti-FLAG immunoreactive band at 27 kDa corresponding to full-length 3F-Bok, and the bands at 23 and 21 kDa corresponding to untagged Bok starting at Met1 or Met15. Again, evidence for alternative translation initiation comes from the observation that 3F-BokM15A does not generate the 21kDa band (lane 7) and that 3F-BokΔ1–14 (Fig. 3A) does not generate the 23kDa band. Thus, Bok mRNA appears to be translated using either Met1 or Met15 as the initiation codon, perhaps because of “leaky scanning” (33, 34) during the initiation of translation.
FIGURE 2.
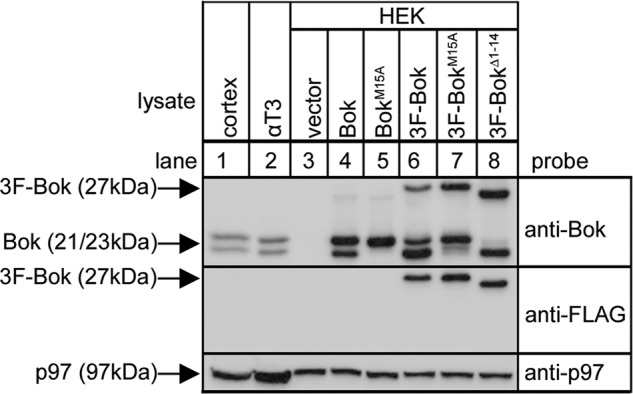
Analysis of the origin of Bok variants. Lysates from mouse brain cerebral cortex and αT3 cells (lanes 1 and 2) and HEK cells transfected to express the constructs indicated (lanes 3–8) were subjected to SDS-PAGE and probed with anti-Bok, anti-FLAG and anti-p97 as a loading control.
FIGURE 3.

Analysis of the determinants of Bok binding and molecular modeling. A, 3F-Bok constructs used to define the region that binds to IP3Rs. Shown are the N-terminal triple FLAG tag (gray box), the position of the BH1–4 domains (1–3, 14), the TM region, and the sequence of the mouse Bok BH4 domain (LGREYV), together with the mutants used to resolve the binding site. The anti-Bok epitope, residues 19–32 (4, 7), is indicated by the black bar. B, Bok homology model, based on Bax and Bak, showing the α-helical region (bright green, residues 24–42) that contains the BH4 domain (bright pink), and also the locations of the BH3 (yellow), BH1 (cyan), BH2 (blue) and TM (light pink) domains. The model starts at Pro22 and ends at Lys202 of Bok. C, models of the residue 24–45 region of BokWT and BokL34G after MD simulation for 10 ns. The positions of the BH4 domain (bright pink) and truncation points used in deletion mutants (arrows) are indicated. D, HEK 293T cells were transfected with cDNAs encoding 3F-Bok constructs and IP3R1HA, were harvested 24 h later, lysates were incubated without or with rabbit anti-HA, and IPs and input lysates were subjected to SDS-PAGE and probed for IP3R1HA and the 3F-Bok constructs.
Resolution of the Determinants of Bok Binding
To resolve the determinants of Bok binding to IP3R1 we made a series of truncations and point mutations in the vicinity of the BH4 domain (Fig. 3A), analyzed the ability of these constructs to interact with IP3R1HA (Fig. 3D), and generated molecular models to gain insight into the effects of these modifications on the structure of Bok (Fig. 3, B and C). The Bok model (Fig. 3B), which is based on Bax and Bak, indicates that the BH4 domain (bright pink) is located within the first α-helical region of Bok (residues 24–42), which lies in an accessible cleft. Sequential truncations within this α-helical region (Fig. 3, A and C) confirmed the role for the BH4 domain in mediating binding, since 3F-BokΔ1–27 and 3F-BokΔ1–32 bound well to IP3R1HA, but 3F-BokΔ1–39 did not (Fig. 3D, lanes 2–5). Surprisingly, it does not appear that it is specific residues in the BH4 domain that mediate binding, but rather the overall structure of the domain, as 3F-BokA34–36 and 3F-BokA37–38 bind well, but 3F-BokA34–38 does not (Fig. 3D, lanes 6–8). To confirm this idea, we mutated Leu34 to Gly, since adjacent Gly residues destabilize α-helices (35) and a previous study has shown that a di-Gly mutation in the BH4 domain of Bcl-2 blocks binding to IP3Rs (36). This mutation distorts the BH4 domain of Bok, as indicated by MD simulation (Fig. 3C), and indeed, the 3F-BokL34G mutant does not bind to IP3R1HA (Fig. 3D, lane 9).
Exogenous Bok Expression Is Dependent upon Binding to IP3R1
We noted in certain experiments using the co-transfection approach shown in Fig. 3D, that binding-deficient Bok mutants did not express as well as constructs that bound strongly to IP3R1HA. To examine this more carefully we monitored the effect of IP3R1HA on 3F-Bok expressed from a range of cDNA amounts (Fig. 4A). This revealed that IP3R1HA dramatically enhanced 3F-Bok expression, particularly when the lowest amounts of 3F-Bok cDNA were used (lanes 1–6). This was truly because of binding to IP3R1HA, because IP3R1HAΔ1–1903, which does not bind Bok (14), did not affect 3F-Bok expression, while IP3R1HAΔ1–1884, which does bind to Bok (14), enhanced 3F-Bok expression (Fig. 4A, lanes 7–12). The effect of binding to IP3R1 on Bok expression was confirmed by the fact that the expression level of binding-deficient mutants (3F-BokL34G and 3F-BokA34–38) was essentially unaffected by co-expression of IP3R1HA (Fig. 4B, lanes 4 and 8), while 3F-BokWT and 3F-BokA34–36 expression was greatly enhanced (lanes 2 and 6).
FIGURE 4.

Exogenous Bok expression is enhanced by IP3R1 and is degraded by the ubiquitin-proteasome pathway when not bound to IP3R1. HEK cells were transfected to express various 3F-Bok and/or IP3R1HA constructs, were incubated without or with 20 μg/ml CHX or 10 μm MG132 for the times indicated, were lysed, and were subjected to SDS-PAGE and probed as indicated. Erlin2 and p97 served as loading controls. The histograms show combined quantitated anti-FLAG immunoreactivity from multiple independent experiments. The asterisk in panel A marks the position in lanes 7–12 of high molecular mass (ubiquitinated) species derived from the truncated IP3R1HA constructs. The asterisked image in panel C was obtained by loading 15 μg and 3 μg of protein from vector- and IP3R1HA-transfected cells, respectively, to demonstrate that the signal from IP3R1HA-transfected cells is not saturated.
CHX-chase experiments showed that 3F-Bok was turned over more rapidly in the absence of IP3R1HA than in the presence of IP3R1HA (Fig. 4C), indicating that Bok is unstable in the absence of IP3R1, which in turn accounts for the low expression level. To probe the apparent degradation mechanism, cells were incubated with MG132, an inhibitor of the proteasome (Fig. 4D). This markedly increased 3F-Bok levels in the absence of IP3R1HA (lanes 3–6), but had no effect on 3F-Bok levels when IP3R1HA was present (lanes 1 and 2), indicating that “free” 3F-Bok is degraded by the proteasome. Conversely, the levels of 3F-Bak were unaffected by MG132 (lanes 7 and 8), showing that the increase seen for 3F-Bok is protein specific and not due to a generic effect on expression from the plasmid. Finally, the relatively rapid decline in 3F-Bok levels in the presence of CHX was partially blocked by MG132 (lanes 9–13), again implicating the proteasome in the turnover of free 3F-Bok.
To establish whether Bok is ubiquitinated, 3F-Bok-HA was immunoprecipitated from control and MG132-treated HEK cells and probed for ubiquitin. This revealed the presence of high molecular mass ubiquitinated species after MG132 treatment (Fig. 5, lane 3), that was not seen in control samples (lanes 1 and 5). Thus, ubiquitination of exogenous Bok appears to mediate its degradation.
FIGURE 5.
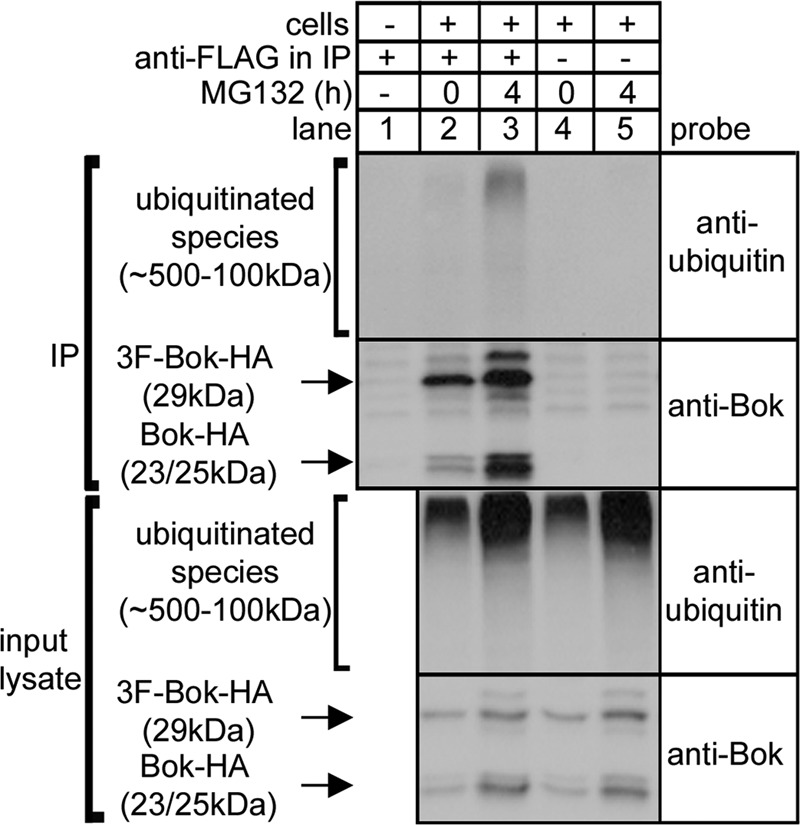
Bok ubiquitination. HEK cells transfected to express 3F-Bok-HA were incubated without or with 10 μm MG132, were lysed, were incubated without or with anti-FLAG, and IPs and input lysates were subjected to SDS-PAGE and probed as indicated.
Endogenous Bok Levels Are Dramatically Reduced by IP3R1 Deletion
To determine whether endogenous Bok levels are also IP3R-dependent, we examined αT3 cells, the cell type in which we originally discovered that endogenous Bok and IP3R1 interact strongly (14), and in which IP3R1 constitutes ∼99% of total IP3R content (22). Remarkably, CRISPR/Cas9-mediated deletion of IP3R1 caused a dramatic decline in Bok levels (Fig. 6A), to ∼2% of that seen in control cells (Fig. 6B), since the Bok immunoreactivity seen with 30 μg of IP3R1KO cell lysate (lane 1) was equivalent to that seen with 0.5–1 μg of control cell lysate (lanes 2 and 3). The decline in Bok expression was totally specific, as the levels of other pertinent proteins were unaffected (Fig. 6A), and was not due to loss of Bok mRNA, the levels of which were not substantially different in control αT3 and αT3IP3R1KO cells (Fig. 6C). Further, reintroduction of IP3R1HA and IP3R1HAΔ1–1884, which bind Bok (14), into αT3IP3R1KO cells caused a partial recovery of Bok expression, not seen with the binding-deficient construct IP3R1HAΔ1–1903 (Fig. 6D); it is likely that the recovery is only partial because of the relatively low transfection efficiency seen in αT3 cells (24, 25). This confirms that the Bok gene and mRNA are normal in αT3IP3R1KO cells and indicates that Bok is degraded in the absence of IP3R1. The degradation mechanism seems identical to that seen for exogenous 3F-Bok, since in αT3IP3R1KO cells, endogenous Bok levels were enhanced by MG132 (Fig. 6E, lanes 1–4) and CHX-chase showed that endogenous Bok is rapidly degraded (lanes 5–7) in a MG132-sensitive manner (lanes 8 and 9); none of these changes were seen in control αT3 cells (Fig. 6E, lanes 10–18). It is perhaps surprising that treatment with MG132 only doubled the level of Bok in αT3IP3R1KO cells (Fig. 6E, lanes 1–4) and that Bok levels did not approach the amount seen in control αT3 cells. This may be because MG132, in addition to inhibiting the proteasome, also causes ER stress and inhibits protein synthesis (12, 13). So, even though Bok degradation by the proteasome is blocked by MG132, so is the synthesis of new Bok, and only a modest increase in Bok levels are seen.
FIGURE 6.

Endogenous Bok is stabilized by IP3R1. A, levels of IP3R1 and Bok and other pertinent proteins in lysates from αT3 control and IP3R1KO cells obtained by targeting exon 5 (lane 2) and exon 13 (lane 4). B, comparison of Bok immunoreactivity in αT3 control and IP3R1KO cells obtained by loading different amounts of cell lysate. C, Bok mRNA levels (normalized to peptidylprolyl isomerase A) in exon 5 and exon 13 targeted αT3IP3R1KO cells, relative to the amount present in αT3 control cells. D, αT3 IP3R1KO cells were transfected with cDNAs encoding the IP3R1HA constructs indicated and cell lysates were probed for Bok. The asterisk marks the same species described in Fig. 4. E, αT3 control and IP3R1KO cells were treated as indicated with 10 μm MG132 and 20 μg/ml CHX, and 1 μg and 30 μg, respectively, of cell lysates were probed for Bok. The histogram shows combined quantitated Bok immunoreactivity normalized to levels in untreated cells from multiple independent experiments. An exon 5-targeted clone was used for the experiments shown in panels B, D, and E, and erlin2, and p97 served as loading controls.
Binding to IP3Rs Limits the Pro-apoptotic Effect of Overexpressed Bok
Since previous studies have shown that Bok overexpression triggers apoptosis (7–10), we examined whether binding to IP3Rs is important for this effect by comparing the pro-apoptotic effects of overexpressed BokWT and BokL34G (which does not bind to IP3Rs). Interestingly, we found that both BokWT and BokL34G trigger apoptosis, as indicated by enhanced caspase-3 and PARP cleavage, and remarkably, that BokL34G is more effective (Fig. 7, lanes 2 and 3). Under these conditions, some BokWT co-immunoprecipitates with endogenous IP3Rs, while BokL34G remains completely free (lower panels; lanes 2 and 3). This indicates that it is not necessary for exogenous Bok constructs to bind IP3Rs to be pro-apoptotic and, in fact, that the binding of Bok to IP3Rs limits apoptosis, apparently by limiting the amount of free Bok. Thus, free Bok, rather than IP3R-bound Bok, seems to be the trigger for apoptosis.
FIGURE 7.
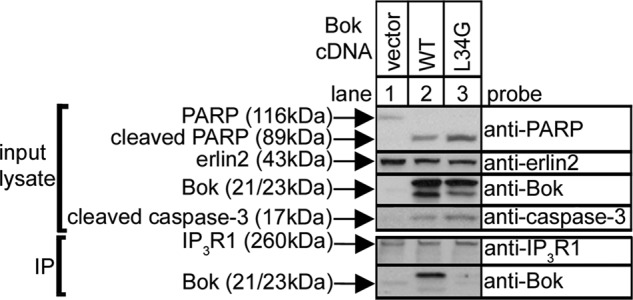
Enhanced pro-apoptotic effects of BokL34G. HeLa cells were transfected with pCAG-based vectors encoding BokWT and BokL34G, were harvested 24 h later, lysates were incubated with anti-IP3R1, and input lysates and IPs were subjected to SDS-PAGE and probed for the proteins indicated.
Stimuli Do Not Cause Release of Bok from IP3R1
Various agents were added to αT3 cells to determine whether Bok release from IP3R1 can be triggered, since that might cause Bok degradation and a decrease in cellular Bok levels. However, Fig. 8 (lanes 5–13) shows that neither the amount of Bok that co-IPs with IP3R1, nor cellular Bok levels, were substantially altered by staurosporine, a kinase inhibitor that triggers apoptosis and causes caspase-3-mediated IP3R1 cleavage (14–16), by forskolin, which triggers cAMP-dependent protein kinase-mediated phosphorylation of IP3R1 (15) and Bok (37), or by thapsigargin, which inhibits ER Ca2+-ATPase, depletes Ca2+ stores and leads to ER stress (12, 13). A slight but consistent decrease in Bok levels and co-IP was seen in response to GnRH (lanes 1–4), which triggers a robust increase in IP3 levels, IP3R1 activation, and proteasome-mediated IP3R1 degradation (14, 24, 25). However, that decrease is most likely because Bok bound to IP3R1 is degraded in unison with IP3R1 (14), rather than because it is released from IP3R1.
FIGURE 8.
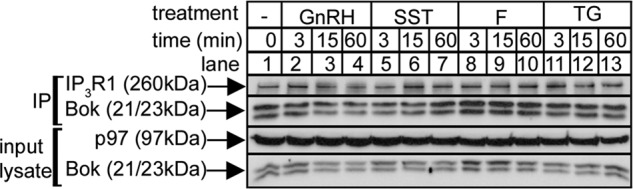
Lack of effect of cell stimulation on the Bok-IP3R1 interaction. αT3 cells were incubated as indicated with 1 μm GnRH, 1 μm staurosporine (SST), 10 μm forskolin (F), and 1 μm thapsigargin (TG), and cell lysates and anti-IP3R1 IPs were subjected to SDS-PAGE and probed in immunoblots for the proteins indicated.
Discussion
Our data indicate that the stability and thus the expression level of Bok is highly dependent upon binding to IP3Rs. This was demonstrated by focusing on the interaction of both exogenous and endogenous Bok with IP3R1, the most widely expressed and best understood IP3R type (15, 16). When IP3R1 is absent and, thus, the predominant binding site for Bok is also absent, “free” Bok appears to be degraded by the ubiquitin-proteasome pathway (UPP) (38).
The extent to which Bok levels are dependent upon IP3R1 is quite remarkable, with the steady-state level of Bok in αT3IP3R1KO cells being ∼2% of that seen in control αT3 cells. Using pharmacological agents and GnRH, we were unable to trigger dissociation of the Bok-IP3R1 complex and reduce endogenous Bok levels, suggesting that the extreme sensitivity of free Bok to the UPP is not a mechanism to control the levels of mature Bok that was once IP3R-associated. Rather, it likely represents a cellular mechanism to disallow newly synthesized Bok from existing in a free form. It is important to note that Bok, like several other Bcl-2 protein family members, has a TM domain at the very C terminus (1–3, 7) and is thus a “tail anchored protein” (39–42). It is currently unclear how newly synthesized TA proteins are inserted into membranes, but for at least some it appears that the BAG6/Ubl4A/TRC35 complex acts to chaperone the proteins from the ribosome to the ER membrane insertion machinery (42, 43). It seems plausible that when IP3R1 is absent, the lack of an appropriate docking site for newly synthesized Bok at the ER membrane means that its insertion into the ER membrane is impaired and that it is diverted to the UPP. Such a mechanism that causes the destruction of free Bok is entirely consistent with the observation that essentially all cellular Bok is bound to IP3Rs. Other Bcl-2 protein family members are also targeted by the UPP (44–47), and it will be interesting to see if they too are subject to a similar kind of “quality control.”
It is intriguing that the 23- and 21-kDa Bok variants appear to originate from alternative translational initiation at Met1 and Met15. It remains possible that proteolysis could account for the 21-kDa variant, but that seems unlikely because mutation of Met15 to Ala completely blocks formation of the 21-kDa band and highly Met-specific endoproteases are not known. Alternative translational initiation could result from “leaky scanning” (33, 34), although why this should occur for Bok mRNA is unclear, since the Kozak consensus sequence around Met1 (GCCAUGG) is ideal (33, 34). It is equally puzzling that exogenous 3F-Bok generates the 21- and 23-kDa variants, as well as the full-length construct at 27 kDa, since this indicates that “leaky scanning” also occurs at the initiation codon at the start of the 3× FLAG region, again despite an ideal Kozak sequence (ACCAUGG). Perhaps secondary structure of Bok mRNA disrupts the scanning process (33, 34). Whatever their origin, both forms of Bok bind to IP3R1 (e.g. Fig. 1), which is to be expected given that the binding is mediated by the BH4 domain (residues 34–39). However, it is likely that the two forms will have different properties, since full-length (23 kDa) Bok can be phosphorylated at Ser8 (37), which is absent from the 21-kDa form.
The region to which Bok binds appears to be a flexible, unstructured, surface-exposed loop within the ARM3 domain of IP3Rs (14, 16). The ARM3 domain is composed of an ensemble of 6 armadillo repeats, and together with the ARM 1 and 2 domains, is thought to form a flexible, solenoid-like structure that facilitates propagation of ligand-evoked signals to the channel pore (16). The ARM3 domain is also a regulatory hotspot, containing sites for phosphorylation, ATP binding, Ca2+ binding, caspase-3 cleavage, and ubiquitination (15, 16, 48). Proteolysis within the ARM3 domain has recently been suggested to provide a novel mode of IP3R regulation (49) and it is intriguing that such proteolysis is inhibited by Bok, most likely because of steric hindrance (14). In view of the ability of Bcl-2 to regulate IP3R channel activity (17–20), and the recently described findings that Bcl-xL activates channel gating by interacting with BH3 domain-like helices in the IP3R C-terminal tail (50), it is somewhat surprising that initial experiments with control and Bok−/− mouse embryonic fibroblasts did not reveal a major effect of Bok on the Ca2+-mobilizing function of IP3Rs (14). However, a caveat with this study is that the Bok−/− cells have adapted in terms of IP3R1–3 expression (14) and thus, whether Bok binding to the ARM3 domain directly regulates the Ca2+ mobilizing function of IP3Rs remains to be determined. Another possibility, given the strength of the Bok-IP3R interaction and that Bok has a TM domain, is that Bok contributes to the structural integrity or stability of IP3R tetramers. Interestingly, Bok is well expressed in the brain (4, 7, 12) and is enriched in the CA3 region of the hippocampus (51, 52). Our data indicate that in cerebral cortex and hippocampus, Bok is bound to IP3R1, and in this context, Bok binding should, at minimum, serve to protect IP3R1 from caspase 3-mediated proteolysis (14).
Bok is widely and highly expressed in mammalian tissues and is well conserved across mammalian species, yet attempts to define its biological role, and in particular the role it is suspected to play in apoptosis, have not led to a clear consensus view (4, 7, 11, 12, 53, 54). This may, in part, be because of the tight binding between Bok and IP3Rs and the degradation of free Bok by the UPP; in experiments in which exogenous Bok is introduced into cells, the amount of Bok expressed will be limited by the UPP and will vary between cell types because of differences in IP3R expression (14). Further, it is tempting to speculate that the existence of a cellular mechanism that efficiently destroys free Bok indicates that free Bok is harmful to the cell, and that in experiments in which Bok is overexpressed, apoptosis occurs because UPP-mediated Bok degradation is overwhelmed and free Bok is formed (7–10). This view is supported by our finding that BokL34G is more pro-apoptotic than BokWT. Certainly, if the apoptosis seen in Bok overexpression experiments (7–10) is considered to be “non-physiological,” it would help to rationalize data showing that there are no major apoptotic deficiencies attributable to Bok in Bok−/− mice (4, 11, 12), or in mice in which Bok is deleted in combination with Bak and Bax (11, 55).
Author Contributions
R. J. H. W. conceived and coordinated the study and wrote the paper with substantial editorial input from J. J. S. J. J. S. performed and analyzed all of the experiments shown, with input from E. J. Z. (Fig. 1) and L. M. S. (Fig. 6). X. H. performed the molecular modeling and MD simulations. F. A. W. created the cell lines used in Fig. 6. J. J. S. and F. A. W. both helped guide the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Richard Kopp and Michael Roe, SUNY Upstate Medical University, for help with the mRNA measurements, Dr Thomas Kaufmann, University of Bern, Switzerland, for providing anti-Bok and the cDNAs for mouse BokWT and human Bak, Dr. Jan Parys, KU Leuven, Belgium, for providing anti-IP3R1–3, Dr. Danielle Sliter and Elizabeth Snyder for helpful suggestions.
This work was supported by National Institutes of Health Grant DK049194 and the Carol M. Baldwin Breast Cancer Research Fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors-and does not necessarily represent the official views of the National Institutes of Health.
- BH
- Bcl-2 homology
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- inositol 1,4,5-trisphosphate receptor
- ER
- endoplasmic reticulum
- PARP
- poly (ADP-ribose) polymerase
- UPP
- ubiquitin-proteasome pathway
- TM
- transmembrane
- CHX
- cycloheximide
- GnRH
- gonadotropin-releasing hormone
- IP
- immunoprecipitation
- PEI
- polyethylenimine
- MD
- molecular dynamics.
References
- 1. Youle R. J., and Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 2. Kelly P. N., and Strasser A. (2011) The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 18, 1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moldoveanu T., Follis A. V., Kriwacki R. W., and Green D. R. (2014) Many players in BCL-2 family affairs. Trends Biochem. Sci. 39, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ke F., Voss A., Kerr J. B., O'Reilly L. A., Tai L., Echeverry N., Bouillet P., Strasser A., and Kaufmann T. (2012) BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 19, 915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chi X., Kale J., Leber B., and Andrews D. W. (2014) Regulating cell death at, on and in membranes. Biochim. Biophys. Acta 1843, 2100–2113 [DOI] [PubMed] [Google Scholar]
- 6. Chen H-C., Kanai M., Inoue-Yamauchi A., Tu H-C., Huang Y., Ren D., Kim H., Takeda S., Reyna D. E., Chan P. M., Ganesan Y. T., Liao C-P., Gavathiotis E., Hsieh J. J., and Cheng E. H. (2015) An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nature Cell Biol. 17, 1270–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Echeverry N., Bachmann D., Ke F., Strasser A., Simon H-U., and Kaufmann T. (2013) Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 20, 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu S. Y., Kaipia A., McGee E., Lomeli M., and Hsueh A. J. W. (1997) Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl. Acad. Sci. U.S.A. 94, 12401–12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsu S. Y., and Hsueh A. J. W. (1998) A splicing variant of the Bcl-2 member Bok with a truncated BH3 domain induces apoptosis but does not dimerize with antiapoptotic Bcl-2 proteins in vitro. J. Biol. Chem. 273, 30139–30146 [DOI] [PubMed] [Google Scholar]
- 10. Inohara N., Ekhterae D., Garcia I., Carrio R., Merino J., Merry A., Chen S., and Núñez G. (1998) Mtd, a novel Bcl-2 family member activates apoptosis in the absence of heterodimerization with Bcl-2 and Bcl-xL. J. Biol. Chem. 273, 8705–8710 [DOI] [PubMed] [Google Scholar]
- 11. Ke F., Bouillet P., Kaufmann T., Strasser A., Kerr J., and Voss A. K. (2013) Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 4, e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carpio M. A., Michaud M., Zhou W., Fisher J. K., Walensky L. D., and Katz S. G. (2015) BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc. Natl. Acad. Sci. U.S.A. 112, 7201–7206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao L., and Ackerman S. L. (2006) Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 18, 444–452 [DOI] [PubMed] [Google Scholar]
- 14. Schulman J. J., Wright F. A., Kaufmann T., and Wojcikiewicz R. J. H. (2013) The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J. Biol. Chem. 288, 25340–25349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foskett J. K., White C., Cheung K. H., and Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan G., Baker M. L., Wang Z., Baker M. R., Sinyagovskiy P. A., Chiu W., Ludtke S. J., and Serysheva I. I. (2015) Gating machinery of InsP3R channels revealed by electron microscopy. Nature 527, 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rong Y-P., Aromolaran A. S., Bultynck G., Zhong F., Li X., McColl K. S., Matsuyama S., Herlitze S., Roderick H. L., Bootman M. D., Mignery G. A., Parys J. B., De Smedt H., and Distelhorst C. W. (2008) Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol. Cell 31, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Distelhorst C. W., and Bootman M. D. (2011) Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: Role in Ca2+ signaling and disease. Cell Calcium 50, 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Monaco G., Decrock E., Akl H., Ponsaerts R., Vervliet T., Luyten T., De Maeyer M., Missiaen L., Distelhorst C. W., De Smedt H., Parys J. B., Leybaert L., and Bultynck G. (2012) Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-xL. Cell Death Differ. 19, 295–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lavik A. R., Zhong F., Chang M. J., Greenberg E., Choudhary Y., Smith M. R., McColl K. S., Pink J., Reu F. J., Matsuyama S., and Distelhorst C. W. (2015) A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 6, 27388–27402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wojcikiewicz R. J. H. (1995) Type I, II and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 270, 11678–11683 [DOI] [PubMed] [Google Scholar]
- 22. Pearce M. M. P., Wang Y., Kelley G. G., and Wojcikiewicz R. J. H. (2007) SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J. Biol. Chem. 282, 20104–20115 [DOI] [PubMed] [Google Scholar]
- 23. Bultynck G., Szlufcik K., Kasri N. N., Assefa Z., Callewaert G., Missiaen L., Parys J. B., and De Smedt H. (2004) Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem. J. 381, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu J. P., Wang Y., Sliter D. A., Pearce M. M. P., and Wojcikiewicz R. J. H. (2011) RNF170, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem. 286, 24426–24433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wright F. A., Lu J. P., Sliter D. A., Dupré N., Rouleau G. A., and Wojcikiewicz R. J. H. (2015) A point mutation in the ubiquitin ligase RNF170 that causes autosomal dominant sensory Ataxia destabilizes the protein and impairs inositol 1,4,5-trisphosphate receptor-mediated Ca2+ signaling. J. Biol. Chem. 290, 13948–13957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuda T., and Cepko C. L. (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 29. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 30. Smith C. A., and Kortemme T. (2008) Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 380, 742–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Computat. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 32. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 15, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kozak M. (1991) An analysis of vertebrate mRNA sequences: Intimations of translational control. J. Cell Biol. 115, 887–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kochetov A. V. (2008) Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. BioEssays 30, 683–691 [DOI] [PubMed] [Google Scholar]
- 35. Pace C. N., and Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Monaco G., Decrock E., Nuyts K., Wagner L. E. II., Luyten T., Strelkov S. V., Missiaen L., De Borggraeve W. M., Leybaert L., Yule D. I., De Smedt H., Parys J. B., and Bultynck G. (2013) α-Helical destabilization of the Bcl-2-BH4-domain peptide abolishes its ability to inhibit the IP3 receptor. PLOS One. 8, e73386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller R. L., Sandoval P. C., Pisitkun T., Knepper M. A., and Hoffert J. D. (2013) Vasopressin inhibits apoptosis in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 304, F177–F188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kleiger G., and Mayor T. (2014) Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 24, 352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Q., Liu Y., Soetandyo N., Baek K., Hegde R., and Ye Y. (2011) A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 42, 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalbfleisch T., Cambon A., and Wattenberg B. W. (2007) A bioinformatics approach to identifying tail-anchored proteins in the human genome. Traffic 8, 1687–1694 [DOI] [PubMed] [Google Scholar]
- 41. Wilfling F., Weber A., Potthoff S., Vögtle F-N., Meisinger C., Paschen S. A., and Häcker G. (2012) BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mariappan M., Li X., Stefanovic S., Sharma A., Mateja A., Keenan R. J., and Hegde R. S. (2010) A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 466, 1120–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawahara H., Minami R., and Yokota N. (2013) BAG6/BAT3: emerging roles in quality control of nascent polypeptides. J. Biochem. 153, 147–160 [DOI] [PubMed] [Google Scholar]
- 44. Breitschopf K., Haendeler J., Malchow P., Zeiher A. M., and Dimmeler S. (2000) Post-translational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol. Cell. Biol. 20, 1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang P., Wu Y., Li Y., Zheng J., and Tang J. (2013) A novel RING finger E3 ligase RNF186 regulate ER stress-mediated apoptosis through interaction with BNip1. Cell. Signal. 25, 2320–2333 [DOI] [PubMed] [Google Scholar]
- 46. Rooswinkel R. W., van de Kooij B., de Vries E., Paauwe M., Braster R., Verheij M., and Borst J. (2014) Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 123, 2806–2815 [DOI] [PubMed] [Google Scholar]
- 47. Carroll R. G., Hollville E., and Martin S. J. (2014) Parkin sensitizes towards apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Reports 9, 1538–1553 [DOI] [PubMed] [Google Scholar]
- 48. Wojcikiewicz R. J. H., Pearce M. M. P., Sliter D. A., and Wang Y. (2009) When worlds collide: IP3 receptors and the ERAD pathway. Cell Calcium 46, 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L., Alzayady K. J., and Yule D. I. (2015) Proteolytic fragmentation of inositol 1,4,5-trisphosphate receptors: a novel mechanism regulating channel activity? J. Physiol. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang J., Vais H., Gu W., and Foskett J. K. (2016) Biphasic regulation of IP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc. Natl. Acad. Sci. U.S.A. 113, E1953–E1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lein E. S., Zhao X., and Gage F. H. (2004) Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J. Neurosci. 24, 3879–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lein E. S., Callaway E. M., Albright T. D., and Gage F. H. (2005) Redefining the boundaries of the hippocampal CA2 subfield in the mouse using gene expression and 3-dimensional reconstruction. J. Comp. Neurol. 485, 1–10 [DOI] [PubMed] [Google Scholar]
- 53. Fernandez-Marrero Y., Ke F., Echeverry N., Bouillet P., Bachmann D., Strasser A., and Kaufmann T. (2016) Is Bok required for apoptosis induced by endoplasmic reticulum stress? Proc. Natl. Acad. Sci. U.S.A. 113, E492–E493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carpio M. A., Michaud M., Zhou W., Fisher J. K., Walensky L. D., and Katz S. G. (2016) Role of Bok at the intersection of endoplasmic reticulum stress and apoptosis regulation. Proc. Natl. Acad. Sci. U.S.A. 113, E494–E495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ke F., Grabow S., Kelly G. L., Lin A., O'Reilly L. A., and Strasser A. (2015) Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis. 6, e1938. [DOI] [PMC free article] [PubMed] [Google Scholar]