

Abstract
Cytochrome P450 46A1 (CYP46A1) is a microsomal enzyme and cholesterol 24-hydroxylase that controls cholesterol elimination from the brain. This P450 is also a potential target for Alzheimer disease because it can be activated pharmacologically by some marketed drugs, as exemplified by efavirenz, the anti-HIV medication. Previously, we suggested that pharmaceuticals activate CYP46A1 allosterically through binding to a site on the cytosolic protein surface, which is different from the enzyme active site facing the membrane. Here we identified this allosteric site for efavirenz on CYP46A1 by using a combination of hydrogen-deuterium exchange coupled to MS, computational modeling, site-directed mutagenesis, and analysis of the CYP46A1 crystal structure. We also mapped the binding region for the CYP46A1 redox partner oxidoreductase and found that the allosteric and redox partner binding sites share a common border. On the basis of the data obtained, we propose the mechanism of CYP46A1 allostery and the pathway for the signal transmission from the P450 allosteric site to the active site.
Keywords: allosteric regulation, Alzheimer disease, brain, cholesterol, cholesterol metabolism, cytochrome P450, drug design, hydrogen-deuterium exchange
Introduction
Cytochrome P450 46A1 (CYP46A1)2 is a heme-containing, microsomal monooxygenase that catalyzes regio- and stereoselective hydroxylation of cholesterol to 24(S)-hydroxycholesterol (24HC) (1, 2). Cholesterol 24-hydroxylation represents the major mechanism for cholesterol elimination from the mammalian brain and accounts for ∼75–85% of cerebral cholesterol removal in humans and ∼40–50% of cholesterol disposal in mice (3–5). Studies in vitro and in vivo revealed that CYP46A1 can bind compounds other than cholesterol, including sterols and some marketed drugs (6–13), with compound binding leading to enzyme inhibition (8, 10). Unexpectedly, several inhibitory pharmaceuticals were also found to act, at specific concentrations, as enzyme activators enhancing CYP46A1-mediated 24HC production (7, 13). Both increase and decrease of CYP46A1 activity have therapeutic potential (14); therefore, we began to ascertain the molecular basis of CYP46A1-drug interactions. Spectroscopic and crystallographic characterizations of substrate-free, substrate-bound, and seven CYP46A1-drug complexes established that inhibitory drugs bind to the enzyme active site because it is plastic and can accommodate compounds of different sizes and shapes because of a ligand-induced conformational fit (7, 9–12). In contrast, binding of the activating drugs is not yet fully understood but began to be elucidated for efavirenz (EFV), a Food and Drug Administration-approved anti-HIV medication. We established that, both in vitro and in vivo (in mice), the dependence of CYP46A1 activity on EFV concentration represents a bell-shaped curve with CYP46A1 activation at low EFV concentrations and inhibition at higher drug concentrations (13). We also generated a model of CYP46A1 activation by EFV (13) according to which CYP46A1 has two separate sites: the allosteric site located on the cytosolic surface of the molecule and the active site embedded in the lipid bilayer. At low concentrations, EFV is suggested to activate CYP46A1 because of preferential binding to the allosteric site which is on the CYP46A1 surface and is more accessible to EFV than the active site interacting with the membrane. At high concentrations, however, EFV begins to also bind to the CYP46A1 active site and thereby either prevents cholesterol from binding to the active site or displaces cholesterol from this site, thus causing CYP46A1 inhibition. To gain insight into a possible location of the allosteric site, we conducted in silico EFV dockings that identified several regions on the CYP46A1 surface that have potential to bind EFV (13).
CYP46A1 activation by EFV is of high clinical relevance because mouse models of Alzheimer disease with genetically increased CYP46A1 activity have improvements in memory and learning and reduction in amyloid β pathology (15, 16). Even in the absence of amyloid deposits, CYP46A1 up-regulation could be beneficial for cognition, as demonstrated by the memory improvement in aged normal mice (17). Accordingly, pharmacologic activation of CYP46A1 by EFV, a drug of good (85%) central nervous system penetration (18), has a strong potential as a new anti-Alzheimer disease therapy.
Conversely, CYP46A1 ablation or down-regulation is associated with negative effects. Cyp46a1−/− mice have severe deficiencies in spatial, associative, and motor learning and in hippocampal long-term potentiation (5). Normal mice with suppressed hippocampal CYP46A1 expression have increased neuronal cholesterol in the hippocampus, cognitive deficits, hippocampal atrophy, aberrant electrical activities, and, ultimately, neuronal death (19, 20). Thus, CYP46A1 activity is important for normal brain function.
Here we mapped the EFV-binding site experimentally by utilizing hydrogen-deuterium exchange (HDX) MS. Specifically, we employed differential HDX, in which CYP46A1 was subjected to HDX in the presence and absence of cholesterol and EFV, and the HDX kinetics for the liganded and unliganded protein states were monitored and compared (21, 22). Reduced HDX is usually observed at the site(s) of ligand interaction but sometimes reflects allosteric effects (22). Therefore, to better interpret our HDX data, we revisited our previous computational dockings (13) and conducted site-directed mutagenesis based on one of our previous in silico models of the EFV-CYP46A1 allosteric binding. The data obtained significantly enhance our understanding of the CYP46A1 allostery and structure-function relationships in the P450 superfamily and provide important structural information for the search of additional CYP46A1 activators among marketed drugs and development of novel, more potent CYP46A1-activating compounds.
Experimental Procedures
Materials
The S isomer of EFV (brand name Sustiva) was purchased from Toronto Research Chemicals Inc. (Toronto, ON, Canada) and used for preparation of stock solutions (0.5–10 mm) in methanol. Cholesterol was obtained from Steraloids (Newport, RI) and dissolved in 4.5% aqueous 2-hydroxypropyl-β-cyclodextrin for the 0.2–5 mm stocks. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Full-length and two truncated forms, Δ(3–27) and Δ(2–50), of CYP46A1 with a four-histidine tag on the C terminus were used (6, 23, 24). The Δ(3–27) or Δ(2–50) truncations do not abolish CYP46A1 activation by EFV or alter the shape of the enzyme activity curves versus EFV concentrations (Fig. 1). Also, these truncations do not significantly affect the CYP46A1 catalytic properties in the absence of EFV because they remove the N-terminal transmembrane region that is not a part of the enzyme active site. This transmembrane region anchors the CYP46A1 catalytic domain to the endoplasmic reticulum because the catalytic domain is associated with the membrane only peripherally (7, 23, 25). Full-length and truncated forms of CYP46A1 were expressed in Escherichia coli and purified as described previously (23, 24). Rat NADPH cytochrome P450 oxidoreductase (OR) was also expressed in E. coli and purified as described previously (26). Rat OR shares >90% of sequence identity with human OR, has a very similar crystal structure, and is frequently used instead of human OR in studies of human microsomal P450 enzymes (27, 28). The expression plasmid for rat OR was provided by Dr. F. Guengerich (Vanderbilt University).
FIGURE 1.

Dependence of enzyme activity on EFV concentrations in the in vitro enzyme assay utilizing purified CYP46A1. The assay conditions are described under “Experimental Procedures.” The results are mean ± S.D. of the measurements in three independent experiments. FL, full-length CYP46A1; Δ(3–27)CYP46A1 and Δ(2–50)CYP46A1, two CYP46A1 truncated forms.
Sample Preparation for HDX
Four different samples of 4 μm Δ(2–50)CYP46A1 in 50 mm potassium phosphate buffer (KPi, pH 7.2) containing 100 mm NaCl were prepared: CYP46A1 only, CYP46A1 with 20 μm cholesterol, CYP46A1 with 20 μm EFV, and CYP46A1 with both 20 μm EFV and 20 μm cholesterol. The final sample volume was 1 ml, and reagents were mixed in the following order. First, chilled 50 mm KPi (pH 7.2) containing 100 mm NaCl was added to the tube placed on ice. Then 20 μm EFV, when included, was added and mixed with the buffer by gently tapping the tube. Next, 4 μm CYP46A1 was added and gently mixed. Lastly, 20 μm cholesterol, when included, was added and gently mixed. Sample mixtures were incubated for 15 min at room temperature to establish binding equilibrium. After incubation, protein samples were stored prior to analysis in the autosampler, which was maintained at 1 °C.
HDX-LC-MS
These studies were conducted on a fully automated HDX PAL robot (LEAP Technologies, Carrboro, NC) carrying out sample labeling, quenching, in-line proteolysis, desalting, and analytical separation coupled to a Thermo LTQ Velos Orbitrap Elite mass spectrometer (Thermo Fisher, San Jose, CA). Protein samples (4 μm, a total of 5 μl) were diluted by the PAL robot into 21 μl of D2O buffer (50 mm KPi (pH 7.2) containing 100 mm NaCl), and HDX was performed at 25 °C in triplicate for 30-s, 1-min, 5-min, 15-min, and 1-h exchange times. CYP46A1 samples in undeuterated buffer served as 0-s controls. Exchange reactions were quenched by addition of 35 μl of 3 m urea and 100 mm sodium phosphate (pH 2.5) at 1 °C, followed by either digestion for 3 min in-line with an immobilized pepsin column (Enzymate BEH, 2.1 × 30 mm, 5 μm, Waters, Milford, MA) or digestion for 2 min with an immobilized protease XIII column (2.1 × 30 mm, NovaBioAssays, Woburn, MA). Peptide digests were loaded onto a C18 guard column (1.0 mm, 5 μm, Grace Discovery Sciences) for desalting, followed by separation on a Dionex Ultimate 3000 ultra performance liquid chromatograph (UPLC) with a C18 Hypersil Gold analytical column (1.0 mm × 5 cm, 1.9 μm, Thermo Scientific) at a flow rate of 50 μl/min. A 9.5-min gradient between solvent A (0.1% aqueous formic acid) and solvent B (80% aqueous acetonitrile containing 0.1% formic acid) was used: from 5% to 35% of solvent B for 3 min, from 35% to 70% of solvent B for 5 min, from 70% to 100% solvent B for 0.5 min, 100% solvent B for 0.5 min, and from 100% to 5% solvent B for 0.5 min. To minimize back exchange, the temperature of all LC connection lines and valves in the HDX PAL compartment was 1 °C. Peptide analysis was performed on the Orbitrap of a Thermo LTQ Velos Orbitrap Elite using the following settings: spray voltage, 3.7 kV; sheath gas flow rate, 25; capillary temperature, 270 °C; and resolution, 60,000.
To reduce carryover between protein samples, several washing steps were performed. First, after each sample injection, the protease column was washed with 50 μl of 1.5 m guanidine-HCl in water containing 0.1% formic acid. Second, after each chromatographic separation, a short blank was run to wash the C18 Hypersil Gold analytical column. Additionally, after each protein sample, a full PAL-Orbitrap system blank was run twice using H2O in place of protein. These washing steps removed all carryovers, as determined by MS/MS analyses of the blank runs.
Peptide Mapping and Sequence Coverage of CYP46A1
Pepsin and protease XIII digests of substrate-free CYP46A1 under the conditions of 0-s HDX were subjected to LC-MS/MS followed by MASCOT (Matrix Science, Oxford, UK) analysis for peptide identification. MS data were acquired by the Orbitrap to accurately measure peptide precursor masses. Fragment peptides and product ions were then obtained by collision-induced dissociation and measured in the LTQ ion trap. Data-dependent acquisition was performed for the six most abundant ions per full scan. To increase the number of identifications, dynamic exclusion was used by excluding an ion for 40 s after the ion was detected twice within a 30-s interval. Subsequent data analysis by MASCOT used the following settings: enzyme, none; MS tolerance, 20 ppm; MS/MS tolerance, 0.5 Da; variable modification, oxidation; peptide charge of 1+, 2+, 3+, and 4+. Peptides with an ion score below 20 were not considered. Other peptides were manually confirmed by fragment ion assignment. Identified peptide sequences and their associated retention times were imported into HDX WorkBench (Scripps Research Institute, Jupiter, FL) (29) to serve as a reference for HDX-MS data.
Data Acquisition and HDX-MS Analyses
Full-scan LC-MS data without fragmentation were acquired in triplicate for four CYP46A1 samples under HDX conditions of 30-s, 1-min, 5-min, 15-min, and 1-h exchange times. Peptides matching the LC-MS/MS reference set and having suitable signal intensity were then identified and extracted from all HDX LC-MS data files. These data were saved as the peptide set. The centroid mass of each isotopic envelope (m) and the percentage of deuterium (D%) were calculated by HDX WorkBench using the following equation (30): D% = {[m(partially deuterated) − m(undeuterated)]/[m(fully deuterated) − m(undeuterated)]} × 100%. The resulting data were manually interrogated to adjust the width of spectra included around the isotopic envelope to exclude background ions outside of the measured isotopic envelope. Chromatography was optimized to minimize co-elution during LC-MS. In instances of co-elution, isotopic envelopes were distant in m/z values, avoiding any measurement errors. In rare cases of overlapping isotopic envelopes, individual isotope peaks were matched to expected values calculated by the software, thereby omitting ions from an overlapping isotopic envelope. The three levels of separation (chromatography, MS, and HDX WorkBench) ensured confidence in the data quality and measurements. To reduce redundancy in peptides with multiple charge states, the charge state with the greatest signal intensity for each peptide was selected to report the data because the charge state had no effect on deuterium measurements (supplemental Fig. S1). Correction factors for back exchange and deuterium content were not necessary for relative quantification in two-sample comparisons. HDX WorkBench also generated kinetic plots of deuterium uptake, with each plotted data point representing an average of triplicate measurements.
Differential HDX
These analyses were carried out by HDX WorkBench. Differential D% values were determined (ΔD%) for each peptide pair by calculating the difference between their mean D% values representing the average of all time points. The significances in ΔD% (p < 0.05) were then established by paired t tests (HDX Workbench), and differential HDX maps were generated by consolidating peptide-level information into a single, linear amino acid sequence and showing only the regions with the statistically significant differences. Consolidation of differential HDX peptide data were performed by initially using data from the shortest non-overlapping peptides, which yielded the highest resolution, and then by using successively longer peptides to complete gaps in coverage areas.
Site-directed Mutagenesis, CYP46A1 Expression, and Purification
The mutations were introduced in the Δ(3–27)CYP46A1 expression construct (23) using an in vitro QuikChange site-directed mutagenesis kit (Stratagene) according to the instructions. The correct generation of desired mutations and the absence of undesired mutations were confirmed by DNA sequencing of the entire CYP46A1 coding region as well as the restriction analysis. The mutant P450s were heterologously expressed in E. coli and purified as described previously (24).
Enzyme Assay for CYP46A1 Activation by EFV
The conditions of the assay were as described previously (13) with the following reagent concentrations: 50 mm KPi (pH 7.2), 100 mm NaCl, 40 μg/ml of dilauroylglycerol-3-phosphtidylcholine, 0.02% Cymal 6, 0.5 μm purified P450, 1.0 μm OR, 10 nm [3H]cholesterol, 20 μm cold cholesterol, 5–100 μm or 20 μm EFV, and 1 mm NADPH. Enzymatic reactions were carried out for 30 min. The total product formation was linear with time and CYP46A1 concentration, and the formation of 24HC (the first CYP46A1 product) was maximal.
Kinetic Parameters for the OR-CYP46A1 Complex
These were determined by measuring the rates of cholesterol 24 hydroxylation by Δ(3–27)CYP46A1 at constant P450 (0.5 μm) and cholesterol (15 μm) concentrations and varying redox partner concentrations (0–10 μm). The assay was performed in 1 ml of 50 mm KPi (pH 7.2) and 100 mm NaCl. Enzymatic reactions proceeded for 5 min, a time when only one product, 24HC, was formed. The reaction rates for 24HC formation were linear with time and CYP46A1 concentration. The Km and kcat of OR for CYP46A1 were calculated with GraphPad Prism software (GraphPad Software, San Diego, CA) using the Michaelis-Menten equation.
Spectral Binding Assay
This assay and subsequent Kd determination were carried out as described previously (11, 13) in 1 ml of 50 mm KPi (pH 7.2), containing 100 mm NaCl and 0.4 μm Δ(3–27)CYP46A1. The spectral data were fit using the hyperbolic equation (when apparent spectral Kd was higher than the enzyme concentration), quadratic equation (when apparent spectral Kd was lower than the enzyme concentration assuming 1:1 stoichiometry), or the Hill equation (when cooperativity of binding was observed). No corrections were done for the affinity of cholesterol for 2-hydroxypropyl-β-cyclodextrin and other components in the assay buffer. Hence, the determined Kd values represent the apparent values.
Results
Sequence Coverage of CYP46A1 in the Presence of Different Ligands
Pepsin is traditionally the protease of choice for HDX-MS analysis because of its nonspecific proteolysis and production of short, overlapping peptides that improve sequence coverage. However, we found that some of the CYP46A1 regions were overdigested by pepsin, yielding peptides too small for MS analysis. This overdigestion created gaps in the enzyme sequence coverage and could not be overcome by a short, 3-min treatment time (data not shown). Therefore, we employed another protease, protease XIII, which is operational under the same conditions as pepsin and generates a different sequence coverage map. This strategy led to a more complete sequence coverage than using a single protease: 85% for substrate-free and EFV-bound CYP46A1 and 88% for CYP46A1 either in complex with cholesterol alone or in complex with both cholesterol and EFV (supplemental Figs. S2–S4). The high sequence coverage from the combinatorial protease approach and the data collection (triplicate measurements at each of the five exchange time points) produced high-quality deuterium uptake results that enabled subsequent differential HDX analyses.
Differential HDX
Three CYP46A1 pairs were compared: complex with cholesterol versus substrate-free enzyme to test the validity of differential HDX, double complex with EFV and cholesterol versus complex with cholesterol to map the EFV binding site and gain insight into the mechanism of CYP46A1 allostery, and complex with EFV versus substrate-free enzyme because EFV-bound CYP46A1 has not yet been crystallized. Differential HDX for the first CYP46A1 pair showed only a decrease in HDX (Fig. 2, A and C, and supplemental Fig. S3), consistent with our previous crystallographic studies establishing that substrate (cholesterol sulfate) binding makes the P450 active site more compact and less flexible (7). Indeed, the affected regions in this differential HDX were mostly those involved in the interactions with cholesterol, either directly (the A, B', F, and I (C terminus) helices along with the β1-1, β1-2, β1-4, β4-1, and β4-2 sheets) or indirectly (the B and I (N terminus) helices and the β3-3-β4-1 loop, Fig. 2, A and B). Only the D helix and adjacent D-E loop were away from the active site.
FIGURE 2.

Differential HDX analysis for the CYP46A1 complex with cholesterol versus substrate-free CYP46A1. A, colored bar mapping of the consolidated regions with altered deuterium uptake in the CYP46A1 primary sequence. Only negative ΔD% values were found, as indicated by the black, dark olive, and light olive bars below the primary CYP46A1 sequence according to the color code shown at the bottom. The dashed lines above the primary P450 sequence indicate the CYP46A1 secondary structural elements. B, mapping of the regions with altered deuterium uptake on the CYP46A1 crystal structure (PDB code 2Q9F). The heme is shown in red and cholesterol sulfate in green. The nitrogen, oxygen, and sulfate atoms are shown in blue, red, and yellow, respectively. The black horizontal line separates the cytosolic (above) and membrane-associated (below) portions of CYP46A1. C, kinetics of deuterium incorporation in eight representative peptides from the CYP46A1 regions that showed significant differences between cholesterol-bound CYP46A1 (blue) versus substrate-free CYP46A1 (red). The results are mean ± S.D. of triplicate measurements.
Differential HDX comparing cholesterol-bound CYP46A1 versus CYP46A1 double complex with EFV and cholesterol revealed a good candidate region for the EFV allosteric site. This candidate allosteric site was the only region with decreased deuterium uptake (the K” helix and adjacent K”-L loop; Fig. 3, A and C, and supplemental Fig. S4) and was located on the surface of the P450 molecule facing the cytosol (Fig. 3B). An increase in deuterium uptake was found in this analysis as well (Fig. 3, A and C) and was associated with the peptides involved in substrate binding (Fig. 3B). This result further supports the validity of our differential HDX analysis and the proposed model because it demonstrates that binding of EFV to the allosteric site does affect the CYP46A1 active site.
FIGURE 3.

Differential HDX analysis for the CYP46A1 double complex with EFV and cholesterol versus the CYP46A1 complex with cholesterol. A, colored bar mapping of the consolidated regions with altered deuterium uptake in the CYP46A1 primary sequence. The color code is shown at the bottom. The dashed lines above the CYP46A1 primary sequence indicate the protein secondary structural elements. B, mapping of the regions with altered deuterium uptake on the CYP46A1 crystal structure (PDB code 2Q9F). Coloring of the atoms is as in Fig. 2. The black horizontal line separates the cytosolic (above) and membrane-associated (below) portions of CYP46A1. C, kinetics of deuterium incorporation in eight representative peptides from the CYP46A1 regions that showed significant differences between the CYP46A1 double complex with EFV and cholesterol (blue) versus the CYP46A1 complex with cholesterol (red). The results are mean ± S.D. of triplicate measurements.
Finally, a comparison of EFV-bound CYP46A1 versus substrate-free enzyme did not find any peptides with statistically significant differences in the kinetics of HDX exchange because the observed differences were very small (<5%, supplemental Fig. S2).
Site-directed Mutagenesis
Mapping of the putative allosteric site to the K” helix region prompted us to investigate whether EFV interacts with this region in our previous computational dockings (13). One such model was found, in which EFV was sandwiched between the K” helix and the K”-L loop with the Lys-422 carbonyl and Arg-424 side chain from the latter restricting the position of the cyclopropyl ring of the drug (Fig. 4A). Lys-422 and Arg-424 could also play other functional roles because their side chains are positively charged and form (along with Lys-94, Arg-138, Arg-139, and R147A) the borders of the concave surface depression, which, in microsomal P450s, serves as a site for binding of OR, a common redox partner (31, 32). Hence, we generated the K422A and R424A CYP46A1 mutants to evaluate whether they affect EFV and OR binding and the K94A, R138A, R139A, and R147A mutants to map the OR binding site. The K422A and R424A CYP46A1 replacements were assessed for activation by EFV, spectral responses to the addition of cholesterol and EFV, as well as apparent OR binding, as evaluated by the Km and kcat of the OR-CYP46A1 complex. The R138A, R139A, and R147A mutants were studied for the apparent OR binding only. The K94A replacement produced inactive P420 protein and therefore was not evaluated. The K422A mutant retained the ability to be activated by EFV, although to a slightly lower extent than the wild-type CYP46A1, whereas the R424A mutant had a total loss of this ability (Fig. 5). Similar to cholesterol-bound wild-type CYP46A1, the cholesterol-bound K422A mutant showed a cooperativity (sigmoidal curve) of EFV binding in the spectral assay and had essentially the same spectral parameters (the apparent Kd and Hill coefficient) of this binding (Fig. 6A). In contrast, no cooperatively was observed in EFV binding to the cholesterol-bound R424A mutant whose binding curve was of a hyperbolic shape. The spectral Kd of EFV in this assay was altered as well and was >10 times lower than that for the wild type. Cholesterol binding to the R424A mutant was tighter as indicated by its apparent Kd, which was lower than those for the wild-type and K422A mutant (Fig. 6B). However, in the presence of EFV, cholesterol elicited only a very weak spectral response (ΔAmax = 0.009/nmol of P450) in the R424A mutant, precluding the determination of its spectral Kd (Fig. 6C). The cholesterol Kd values for the wild type and K422A mutant were similar in this assay. Thus, the R424A replacement affected EFV binding to the allosteric site and cholesterol binding to the CYP46A1 active site.
FIGURE 4.

Views of surface representation of the CYP46A1 structure (PDB code 2Q9F) with the docked EFV. EFV is shown in cyan, and the side chains and surface of the positively charged resides with potential for EFV and/or OR binding are shown in blue. The nitrogen, oxygen, fluorine, and chlorine atoms are shown in blue, red, light cyan, and green, respectively. A, close proximity in CYP46A1 of the putative EFV- and cytochrome P450 oxidoreductase-binding sites. The peptide coloring is the same as in Fig. 3. B and C, spatial positioning of the putative EFV- and oxidoreductase-binding sites relative to the regions with altered deuterium uptake in the CYP46A1 complex with cholesterol versus substrate-free CYP46A1 and the CYP46A1 double complex with EFV and cholesterol versus the CYP46A1 complex with cholesterol, respectively. The CYP46A1 orientation and peptide coloring are the same as in Figs. 2B and 3B.
FIGURE 5.
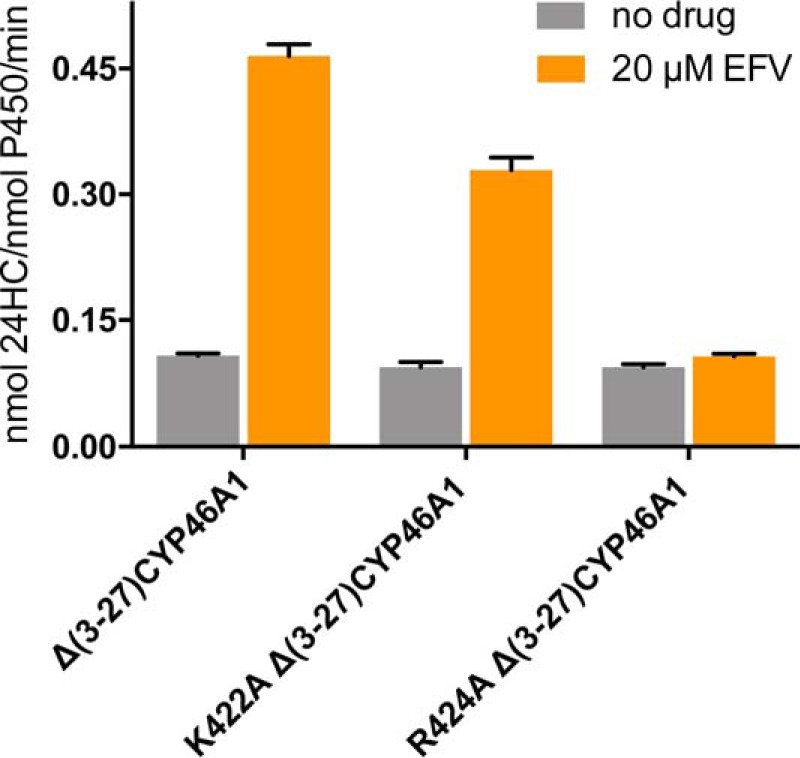
Effects of mutations on CYP46A1 activation by EFV in the in vitro enzyme assay. Both Δ(3–27)CYP46A1 and the K422A mutant are activated by EFV. However, the R424A mutation abolishes P450 activation by EFV. The assay conditions are described under “Experimental Procedures.” The results are mean ± S.D. of the measurements in three independent experiments.
FIGURE 6.

Effect of CYP46A1 mutations on EFV and cholesterol binding as assessed by spectral assays. The top of each vertical panel shows a schematic of the assay conditions: circles for CYP46A1, green rectangles for cholesterol, and magenta triangles for EFV. Each panel also shows the spectral parameters of binding (mean of triplicate experiments, S.D. ≤ 20% of a mean value and not shown), representative binding curve for the titrating ligand, and ligand-induced P450 difference spectra (insets) indicating the positions of the spectral peaks and troughs. A, EFV titrations of CYP46A1 in the presence of 20 μm cholesterol. B, cholesterol titrations of substrate-free CYP46A1. C, cholesterol titrations of CYP46A1 in the presence of 20 μm EFV. The assay conditions are described under “Experimental Procedures.” The spectral Kd values in B and C are estimates only because cholesterol and EFV bind very tightly, with the apparent Kd values being much lower than the concentration of CYP46A1 (0.4 μm). The Kd of cholesterol for the R424A mutant in the presence of EFV was not determined because of the weak P450 spectral response.
Both the K422A and R424A mutants had impaired interactions with OR, as indicated by the increased redox partner Km values for these mutants relative to that for wild-type CYP46A1 (5.5- and 3.8-fold, respectively) (Fig. 7). A weakened OR binding to the K422A mutant could be a reason for its slightly lower activation by EFV. The OR Km values for the R138A and R139A mutants were increased as well, and that for the R147A mutant remained unchanged. Thus, the concave depression on the proximal CYP46A1 surface defined by Lys-422, Arg-424, Arg-138, and Arg-139 seems to indeed represent the site of the P450 OR binding. The effect of the K422A, R424A, R138A, and R139A replacements on catalysis was different. The former three did not significantly alter the CYP46A1 kcat, whereas the later increased the kcat almost 2-fold.
FIGURE 7.

Effect of CYP46A1 mutations on kinetic parameters of the CYP46A1-cytochrome P450 OR complex. Each panel shows a representative kinetic curve and mean ± S.D. of the kinetic parameters, which represent the measurements in three independent experiments. The assay conditions are described under “Experimental Procedures.”
Discussion
In this work, we combined differential HDX with computational docking and site-directed mutagenesis and mapped the allosteric site for EFV responsible for positive modulation of CYP46A1-mediated cholesterol 24 hydroxylation. The identified site turned out to be very close to one of the three sites (near Pro-429) that had the lowest free energy of EFV binding to CYP46A in our computational docking (13). However, this site had a different EFV binding pose and was among the other potential 35 EFV binding sites suggested by our in silico predictions based on their negative free energies of EFV binding. Thus, our previous computational studies provided valuable information about potential sites and modes of EFV binding and demonstrated that all predicted sites should be considered until additional experimental data become available.
We also outlined the binding region for the CYP46A1 redox partner OR and established that the CYP46A1 allosteric site and the redox partner-binding region are adjacent and share a common border formed by Lys-422 and Arg-424. The K422A and R424A substitutions had similar effects on CYP46A1 interactions with OR, namely impairing these interactions (Fig. 7), but differential effects on EFV-induced CYP46A1 activation as well as drug and cholesterol binding (Figs. 5 and 6). Overall, the studied properties of the K422A mutant were similar to those of the wild type (except impaired OR binding), whereas the properties of the R424A mutant were very different. The R424A mutant did not seem to bind EFV at the allosteric site and undergo EFV-induced activation. It also showed a tighter cholesterol binding at the active site in the absence of EFV and a very weak spectral response to cholesterol in the presence of EFV.
The differential HDX data as well as the properties of the K422A and R424A mutants are consistent with one of our previous models of allosteric EFV binding to CYP46A1 (13), which was generated based on a high-resolution, 1.9-Å crystal structure of cholesterol sulfate-bound CYP46A1 (PDB code 2Q9F). This is a very hydrated structure containing 125 water molecules that are bound to both the protein surface and protein interior (Fig. 8A) and stabilize the P450 structure by forming an extensive hydrogen bond network. For EFV docking, however, water molecules were removed from the CYP46A1 structure, thus enabling the positioning of EFV in a surface depression filled by five water molecules (Wat-725, 730, 748, 749, and 751; Fig. 8B). The location of one of these molecules, Wat-751, overlaps with that of the EFV cyclopropyl ring, suggesting that, upon binding, EFV displaces this and likely other water molecules in the CYP46A1 allosteric site and thereby alters the hydrogen bond network involving these molecules. Indeed, the hydrogen bond contacts of Wat-751 include Wat-748, which in turn is hydrogen-bonded to the side chain of Glu-91 through Wat-808, Wat-783, and Wat-793 (Fig. 8B). Glu-91 is located in helix B, a surface helix that is flanked on both sides by the secondary structural elements that define the sides of the CYP46A1 active site: the β1-1/β1-2 sheets and the A helix from the N terminus and the B' helix from the C terminus (Fig. 8B). The β1-1/β1-2 loop region, in turn, has contacts with the F-G loop and the F helix, which also form the sides of the CYP46A1 active site. All active site regions connected to the B helix showed an increase in the differential HDX for the double CYP46A1 complex with EFV and cholesterol versus complex with cholesterol (Fig. 3). Hence, we propose that the mechanism of CYP46A1 allostery could involve the alteration of the hydrogen bond network of the enzyme (either its disruption, reconfiguration, or weakening), leading to an increase in the HDX of the active site regions covered by this network. An increased rate of amide exchange could in turn affect the rate of CYP46A1 catalysis because water plays an important role in the catalytic cycle of P450 by providing protons. Substrate binding could be affected as well because of the altered hydration of the CYP46A1 active site and easier displacement of Wat-718 serving as the sixth heme iron ligand.
FIGURE 8.

Cartoon representation of the crystal structure of substrate-bound CYP46A1 (PDB code 2Q9F). A, the distribution of crystallographic water molecules (blue spheres). B, putative amino acid residues and water molecules involved in signal transmission from the allosteric site to the P450 active site. Water molecules in EFV-binding site are colored in wheat. The color code is the same as in Fig. 4. Black dashed lines indicate hydrogen bonds.
Water is often necessary for the formation of protein crystals and may not bind to the protein when it is in solution. Therefore, we analyzed the properties of the K422A and R424A mutants to obtain further support for our proposed mechanism of CYP46A1 allostery. If this mechanism is correct and water is bound to CYP46A1 in solution, the replacement of Lys-422, whose carbonyl is hydrogen bonded to Wat-751, should not affect the CYP46A1 hydrogen bond network because Wat-751 is displaced anyway by EFV binding. EFV should still activate the K422A mutant and did not significantly affect the enzyme active site. Indeed, the K422A mutant was not very different in this respect from the wild type (Figs. 6 and 7), consistent with its predicted properties. In contrast, the substitution of Arg-424 could have more significant effects on the CYP46A1 hydrogen bond network because the nitrogen and oxygen from the Arg-424 peptide bond interact with Wat-748 and 783, respectively, from the water network, linking the allosteric and active sites (Fig. 8B). Thus, if the R424A substitution alters the position of the 424 peptide bond atoms, then it may also abolish CYP46A1 allosteric activation and have distant effects on the active site. These predicted consequences are also in a good agreement with the observed properties of the R424A mutant (Figs. 5–7). Furthermore, the Arg-424 replacement revealed that the alterations in the CYP46A1 hydrogen bond network lead to the altered Kd values of cholesterol and EFV for the R424A mutant. These effects could be due to changes in the active site hydration and/or conformation. Thus, the results of the differential HDX and functional characterizations of the CYP46A1 mutants are in agreement with each other and could be interpreted based on hydration of an available CYP46A1 crystal structure. All these data support our proposed mechanism of CYP46A1 allostery.
Besides the mechanistic insight, mapping of the EFV binding site is of practical relevance because it will help us identify additional CYP46A1 activators among marketed drugs. Our in silico dockings will now be restricted to the mapped allosteric site, and our intuitive predictions (10) will be based on the knowledge of the size and shape of the CYP46A1 allosteric site. We also obtained an opportunity for a structure-based design of compounds more potent for CYP46A1 activation than EFV.
This investigation also seems to provide an explanation for several structure-function observations in the P450 field that are still not well understood. The first is the effect of substrate binding on the redox partner binding and vice versa when binding of the redox partner and substrate exhibits strong positive cooperativity. The phenomenon is exemplified by CYP11A1, which, like CYP46A1, uses cholesterol as a substrate (33). The second is the data from the directed evolution approach, as exemplified by the mutagenesis of CYP2B1. These studies revealed that substrate binding and metabolism are influenced not only by the active site residues but also by residues outside of the active site (34, 35). Our work suggests that, in both cases, this could be when a distant residue/region in question participates in the hydrogen bond network connected to the P450 active site. This explanation is illustrated by Arg-424 in CYP46A1, found to be involved in both EFV and redox partner binding, as well as Lys-139, whose mutation impairs OR binding but increases the P450 kcat almost 2-fold (Fig. 7). Thus, studies of the CYP46A1 allostery provided information of general significance for the P450 superfamily.
In conclusion, we mapped the allosteric site in P450 (CYP46A1), which is the principle cholesterol hydroxylase in the brain and a potential therapeutic target for Alzheimer disease. We suggest how changes in the P450 surface allosteric site could be transmitted to the distant active site and will test our proposed mechanism in the future by crystallizing the R424A mutant and performing additional site-directed mutagenesis. Our findings appear to be applicable to other members of the P450 superfamily and will hopefully advance our understanding of some of the structure-function relationships in this important family of enzymes.
Author Contributions
K. W. A. conducted the HDX experiments and analyzed the data obtained with J. W. H. and I. V. T. N. M. carried out all protein expression, purification, site-directed mutagenesis, and enzyme activity assays. J. B. L. conducted spectral titrations. I. A. P. conceived the project, analyzed the data obtained, and wrote the paper with K. W. A. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported in part by Public Health Service Grant GM062882 (to I. A. P). The authors declare that they have no conflicts of interest with the contents of this article. Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment identified are necessarily the best available for the purpose.

This article contains supplemental Figs. S1–S4.
- CYP
- cytochrome P450
- 24HC
- 24(S)-hydroxycholesterol
- EFV
- efavirenz
- HDX
- hydrogen-deuterium exchange
- OR
- cytochrome P450 oxidoreductase.
References
- 1. Lütjohann D., Breuer O., Ahlborg G., Nennesmo I., Sidén A., Diczfalusy U., and Björkhem I. (1996) Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24s-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U.S.A. 93, 9799–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lund E. G., Guileyardo J. M., and Russell D. W. (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Björkhem I., Lütjohann D., Diczfalusy U., Ståhle L., Ahlborg G., and Wahren J. (1998) cholesterol homeostasis in human brain: turnover of 24s-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600 [PubMed] [Google Scholar]
- 4. Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., and Russell D. W. (2003) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278, 22980–22988 [DOI] [PubMed] [Google Scholar]
- 5. Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., and Russell D. W. (2006) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3869–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Bjorkhem I., and Pikuleva I. A. (2003) Broad substrate specificity of human cytochrome P450 46a1 which initiates cholesterol degradation in the brain. Biochemistry 42, 14284–14292 [DOI] [PubMed] [Google Scholar]
- 7. Mast N., White M. A., Bjorkhem I., Johnson E. F., Stout C. D., and Pikuleva I. A. (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46a1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U.S.A. 105, 9546–9551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shafaati M., Mast N., Beck O., Nayef R., Heo G. Y., Björkhem-Bergman L., Lütjohann D., Björkhem I., and Pikuleva I. A. (2010) The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24s-hydroxylase (Cyp46a1) in vitro and in vivo. J. Lipid Res. 51, 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mast N., Charvet C., Pikuleva I. A., and Stout C. D. (2010) Structural basis of drug binding to Cyp46a1, an enzyme that controls cholesterol turnover in the brain. J. Biol. Chem. 285, 31783–31795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mast N., Linger M., Clark M., Wiseman J., Stout C. D., and Pikuleva I. A. (2012) In silico and intuitive predictions of Cyp46a1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol. Pharmacol. 82, 824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mast N., Zheng W., Stout C. D., and Pikuleva I. A. (2013) Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46a1: unusual features and spectral response. J. Biol. Chem. 288, 4613–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mast N., Zheng W., Stout C. D., and Pikuleva I. A. (2013) Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing Cyp46a1. Mol. Pharmacol. 84, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mast N., Li Y., Linger M., Clark M., Wiseman J., and Pikuleva I. A. (2014) Pharmacologic stimulation of cytochrome P450 46a1 and cerebral cholesterol turnover in mice. J. Biol. Chem. 289, 3529–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun M. Y., Linsenbardt A. J., Emnett C. M., Eisenman L. N., Izumi Y., Zorumski C. F., and Mennerick S. (2016) 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 22, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hudry E., Van Dam D., Kulik W., De Deyn P. P., Stet F. S., Ahouansou O., Benraiss A., Delacourte A., Bougnères P., Aubourg P., and Cartier N. (2010) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol. Ther. 18, 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bryleva E. Y., Rogers M. A., Chang C. C., Buen F., Harris B. T., Rousselet E., Seidah N. G., Oddo S., LaFerla F. M., Spencer T. A., Hickey W. F., and Chang T. Y. (2010) Acat1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. U.S.A. 107, 3081–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maioli S., Båvner A., Ali Z., Heverin M., Ismail M. A., Puerta E., Olin M., Saeed A., Shafaati M., Parini P., Cedazo-Minguez A., and Björkhem I. (2013) Is it possible to improve memory function by upregulation of the cholesterol 24s-hydroxylase (Cyp46a1) in the brain? PLoS ONE 8, e68534–e68534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Decloedt E. H., Rosenkranz B., Maartens G., and Joska J. (2015) Central nervous system penetration of antiretroviral drugs: pharmacokinetic, pharmacodynamic and pharmacogenomic considerations. Clin. Pharmacokinet. 54, 581–598 [DOI] [PubMed] [Google Scholar]
- 19. Chali F., Djelti F., Eugene E., Valderrama M., Marquer C., Aubourg P., Duykaerts C., Miles R., Cartier N., and Navarro V. (2015) Inhibiting cholesterol degradation induces neuronal sclerosis and epileptic activity in mouse hippocampus. Eur. J. Neurosci. 41, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Djelti F., Braudeau J., Hudry E., Dhenain M., Varin J., Bièche I., Marquer C., Chali F., Ayciriex S., Auzeil N., Alves S., Langui D., Potier M. C., Laprevote O., Vidaud M., Duyckaerts C., Miles R., Aubourg P., and Cartier N. (2015) Cyp46a1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain 138, 2383–2398 [DOI] [PubMed] [Google Scholar]
- 21. Chalmers M. J., Pascal B. D., Willis S., Zhang J., Iturria S. J., Dodge J. A., and Griffin P. R. (2011) Methods for the analysis of high precision differential hydrogen deuterium exchange data. Int. J. Mass Spectrom. 302, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konermann L., Rodriguez A. D., and Sowole M. A. (2014) Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein-ligand complexes. Analyst 139, 6078–6087 [DOI] [PubMed] [Google Scholar]
- 23. Mast N., Andersson U., Nakayama K., Bjorkhem I., and Pikuleva I. A. (2004) Expression of human cytochrome P450 46a1 in Escherichia coli: effects of N- and C-terminal modifications. Arch. Biochem. Biophys. 428, 99–108 [DOI] [PubMed] [Google Scholar]
- 24. White M. A., Mast N., Bjorkhem I., Johnson E. F., Stout C. D., and Pikuleva I. A. (2008) Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46a1. Acta Crystallogr. D. Biol. Crystallogr. 64, 487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mast N., Liao W. L., Pikuleva I. A., and Turko I. V. (2009) Combined use of mass spectrometry and heterologous expression for identification of membrane-interacting peptides in cytochrome P450 46a1 and NADPH-cytochrome P450 oxidoreductase. Arch. Biochem. Biophys. 483, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2b2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 [DOI] [PubMed] [Google Scholar]
- 27. Xia C., Panda S. P., Marohnic C. C., Martásek P., Masters B. S., and Kim J. J. (2011) Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc. Natl. Acad. Sci. U.S.A. 108, 13486–13491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shinkyo R., and Guengerich F. P. (2011) Cytochrome P450 7a1 cholesterol 7α-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J. Biol. Chem. 286, 4632–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pascal B. D., Willis S., Lauer J. L., Landgraf R. R., West G. M., Marciano D., Novick S., Goswami D., Chalmers M. J., and Griffin P. R. (2012) HDX Workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass. Spectrom. 23, 1512–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hasemann C. A., Kurumbail R. G., Boddupalli S. S., Peterson J. A., and Deisenhofer J. (1995) Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure 3, 41–62 [DOI] [PubMed] [Google Scholar]
- 32. Poulos T. L., and Johnson E. F. (2005) in Cytochrome P450: Structure, Mechanism, and Biochemistry (Montellano P. R. O., ed.), 3rd Ed., pp 87–114, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 33. Lambeth J. D., Seybert D. W., and Kamin H. (1980) Phospholipid vesicle-reconstituted cytochrome P-450scc: Mutually facilitated binding of cholesterol and adrenodoxin. J. Biol. Chem. 255, 138–143 [PubMed] [Google Scholar]
- 34. Kumar S., Chen C. S., Waxman D. J., and Halpert J. R. (2005) Directed Evolution of mammalian cytochrome P450 2b1: mutations outside of the active site enhance the metabolism of several substrates, including the anticancer prodrugs cyclophosphamide and ifosfamide. J. Biol. Chem. 280, 19569–19575 [DOI] [PubMed] [Google Scholar]
- 35. Kumar S., and Halpert J. R. (2005) Use of directed evolution of mammalian cytochromes P450 for investigating the molecular basis of enzyme function and generating novel biocatalysts. Biochem. Biophys. Res. Commun. 338, 456–464 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.