

Abstract
Background and Purpose
Kinins are vasoactive and pro‐inflammatory peptides whose biological effects are mediated by two GPCRs, named B1 and B2 receptors. While the B2 receptor plays a protective role in the cardiovascular system via the activation of endothelial NOS, the B1 receptor is associated with vascular inflammation, insulin resistance and diabetic complications. Because the B1 receptor is a potent activator of the inducible form of NOS (iNOS), this study has addressed the role of iNOS in the deleterious effects of B1 receptors in insulin resistance.
Experimental Approach
Male Sprague–Dawley rats (50–75 g) had free access to a drinking solution containing 10% d‐glucose or tap water (control) for 9 weeks. During the last week, a selective iNOS inhibitor (1400W, 1 mg·kg−1 twice daily) or its vehicle was administered s.c.
Key Results
Prolonged glucose treatment caused insulin resistance and several hallmarks of type 2 diabetes. Whereas the treatment with 1400W had no impact on the elevated systolic blood pressure and leptin levels in glucose‐fed rats, it significantly reversed or attenuated hyperglycaemia, hyperinsulinaemia, insulin resistance (HOMA index), body weight gain, peroxynitrite formation (nitrotyrosine expression) and the up‐regulation of biomarkers of inflammation (B1 receptor, carboxypeptidase M, iNOS and IL‐1β) in renal cortex and aorta and to some extent in the liver.
Conclusions and Implications
Pharmacological blockade of iNOS prevents the formation of peroxynitrite, which amplifies the pro‐inflammatory effects of B1 receptors through a positive feedback mechanism. Hence, targeting iNOS can prevent the deleterious effects of B1 receptors in insulin resistance and peripheral inflammation.
Abbreviations
- 1400W
N‐(3‐(aminomethyl)benzyl)acetamidine
- BK
bradykinin
- CPM
carboxypeptidase M
- HOMA
homeostasis model assessment index
- iNOS
inducible NOS
- O2●¯
superoxide anion
- ONOO¯
peroxynitrite
- qRT‐PCR
real‐time quantitative RT‐PCR
- SSR240612
[(2R)‐2‐[((3R)‐3‐(1,3‐benzodioxol‐5‐yl)‐3‐[[(6‐methoxy‐2‐naphthyl)sulfonyl]amino]propanoyl)amino]‐3‐(4‐[[2R,6S)‐2,6‐dimethylpiperidinyl]methyl]phenyl)‐N‐isopropyl‐N‐methylpropanamide hydrochloride]
Tables of Links
| TARGETS | ||
|---|---|---|
| GPCRs a | Catalytic receptors b | Enzymes c |
| B1 receptor | IRS‐1 | Carboxypeptidase M |
| B2 receptor | ERK | |
| Inducible NOS |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Insulin resistance is considered as an early event in the development of type 2 diabetes mellitus in which mild vascular inflammation is a predictive factor. Vascular inflammation can be initiated by sustained hyperglycaemia‐induced oxidative stress, which activates a series of receptors and transcriptional factors leading to increased levels of cellular adhesion molecules, macrophages and leukocyte infiltration in the vasculature and to endothelial dysfunction (He and King, 2004; de Vries et al., 2015; Gleissner, 2015). Recently, we reported that the kinin B1 receptor is involved in low‐grade vascular inflammation and contributes to amplify and perpetuate the vascular oxidative stress in a rat model of insulin resistance induced by prolonged glucose feeding (Dias et al., 2010; Dias and Couture, 2012b; Couture et al., 2014). In this model, oral treatment with the non‐peptide B1 receptor antagonist SSR240612 for 1 week reversed the enhanced expression of B1 receptors, inducible nitric oxide synthase (iNOS) and other inflammatory key biomarkers (IL‐1β, macrophage migration inhibitory factor, adhesion molecules and macrophages) in the vascular bed (Dias and Couture, 2012b). The same prolonged blockade of B1 receptors reversed hypertension, metabolic abnormalities (hyperglycaemia, hyperinsulinaemia and insulin resistance), the enhanced NADPH oxidase activity and the basal production of vascular superoxide anion (O2 ●¯) (Dias et al., 2010). Moreover, B1 receptor activation was shown to enhance the production of O2 ●¯ through NADPH oxidase in the aorta of glucose‐fed rats and in human epithelial cells (Dias et al., 2010; Talbot et al., 2011). NADPH oxidase is known to produce the predominant source of reactive oxygen species in the vasculature that leads to diabetic complications and cardiovascular diseases (Cai et al., 2003; Taniyama and Griendling, 2003; Griendling and FitzGerald, 2003a; Griendling and FitzGerald, 2003b).
B1 receptors can also generate high output and prolonged NO production from endothelial cells following the activation of iNOS via Gαi and the Src‐dependent activation of the ERK/MAPK pathway (Kuhr et al., 2010; Brovkovych et al., 2011). In an oxidative stress environment, the high concentration of NO can rapidly react with O2 ●¯ to form the highly reactive peroxynitrite (ONOO¯), which causes DNA damage (altering gene expression), inflammation and oxidative stress, notably lipid peroxidation (membranes) and nitration of proteins (enzymes, transporters and ion channels) (Johansen et al., 2005). Importantly, the genetic deletion of iNOS prevents the development of insulin resistance in mice exposed to a high‐fat diet (Perreault and Marette, 2001) and that of age‐associated insulin resistance (Ropelle et al., 2013). Mice lacking iNOS were protected against endotoxaemia‐induced skeletal muscle insulin resistance and from lipid‐induced hepatic insulin resistance through the iNOS/NO/ONOO¯ pathway that causes tyrosine nitration (the covalent addition of NO2 to the tyrosine residues) of insulin receptor substrate 1 (IRS‐1) and thereby reduces its subsequent insulin‐dependent tyrosine phosphorylation (Charbonneau and Marette, 2010; Pilon et al., 2010).
Hence, B1 receptor‐mediated iNOS activation and enhanced expression can be detrimental in cardiometabolic diseases as highlighted in a model of insulin resistance associated with hypertension (Dias and Couture, 2012b). The latter hypothesis was directly tested in the present study by determining whether a 1 week pharmacological treatment with the selective inhibitor of iNOS 1400W (Garvey et al., 1997) could reproduce the beneficial effects of the B1 receptor antagonist on insulin resistance, vascular and non‐vascular inflammation and oxidative stress in the model of glucose‐fed rat. The impact of 1400W was also determined on the expression of B1 receptors and carboxypeptidase M (CPM), a key enzyme involved in the generation of B1 receptor ligands. This study highlights a partnership between B1 receptors and iNOS in insulin resistance.
Methods
Animal care and treatments
Male Sprague–Dawley rats (24–30 days old), weighing 50‐75 g, were purchased from Charles River Laboratories (Saint‐Constant, QC, Canada). Rats were housed two per cage, under standard conditions (22.5°C and 42.5% humidity, on a 12 h/12 h light–dark cycle, using heated wood chip litter as bedding material) in a pathogen‐free environment, and allowed free access to a standard chow diet (Teklad Global 18% Protein Rodent Diet) and to a drinking solution containing 10% d‐glucose (querySigma‐Aldrich, Oakville, ON, Canada) or tap water (control) for a period of 9 weeks.
A total of 24 rats were used in the experiments described here. During the last week (week 9), rats fed with glucose (G) or tap water (C) were randomly divided into four groups of six rats: group 1 (G + 1400W), group 2 (G + Vehicle), group 3 (C + 1400W) and group 4 (C + Vehicle). 1400W (Focus Biomolecules, Plymouth Meeting, PA, USA), a selective inhibitor of iNOS (Garvey et al., 1997), was given s.c. twice daily at 12 h intervals at the dose of 1 mg·kg−1 in a volume of 1 mL·kg−1 for 7 days. The vehicle (saline) was given in groups 2 and 4. Rats were subjected to several measurements, including systolic blood pressure, blood glucose and body weight during the treatment with 1400W. The dose of 1400W was selected on the basis of studies showing complete inhibition of endotoxin‐induced vascular leakage in various rat tissues at the dose of 1 mg·kg−1. When given i.v., rats tolerated a dose of 120 mg·kg−1 of 1400W for a 7 day period, and no apparent haemodynamic effect was seen up to 25 mg·kg−1 (Garvey et al., 1997).
All animal care and experimental procedures complied with the Use of Laboratory Animals and were approved by the Université de Montréal's Committee on Ethics in the Care and Use of Laboratory Animals (protocol 11–140) http://www.cdea.umontreal.ca in accordance with the guiding principles as enunciated by the Canadian Council on Animal Care. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Experimental procedures
At the end of the ninth week, the blood was collected by cardiac puncture in overnight‐fasted rats under isoflurane anesthesia. Blood samples were collected in tubes containing anticoagulant (Heparin; Sandoz, Boucherville, QC, Canada), and the plasma was separated by centrifugation at 600× g for 15 min at 4°C and stored at −20°C.
Three peripheral tissues (aorta, renal cortex and liver) were removed and kept frozen at −80°C. The thoracic aorta was the sentinel tissue to assess the anti‐inflammatory and anti‐oxidative effects of B1 receptor blockade in our previous studies (Dias et al., 2010; Dias and Couture, 2012b). The renal cortex and the liver were selected because diabetes induces glomerulosclerosis and nephropathy and the liver is the organ of glucose metabolism. Proteins, DNA and RNA of these organs were extracted to measure the expression of several inflammatory biomarkers [western blotting and quantitative RT‐PCR (qRT‐PCR)]. O2 ●¯ was measured in the thoracic aorta by a chemiluminescence technique using lucigenin.
Measurement of systolic blood pressure
Systolic blood pressure was measured by tail‐cuff plethysmography (ADI Instruments Inc., Colorado, CO, USA) using a pad heated at 37°C, and registered with the ADI Instruments software (Lab Chart Pro7.Ink). Each measurement corresponds to the average of 5–7 blood pressure readings taken at 1–2 min intervals.
Biochemical analysis
Blood glucose concentration was determined with a glucometer (Accu‐Chek Aviva; Roche Diagnostics, Laval, QC, Canada). Plasma insulin and leptin concentrations were determined by RIA using the double antibody/polyethylene glycol technique (rat insulin RIA kit and rat leptin RIA kit) from Millipore (St. Charles, MO, USA). The homeostasis model assessment (HOMA) index was used as an index of insulin resistance and calculated with the following formula: [insulin (μU·mL−1) × glucose (mmol·L−1)/22.5] (Matthews et al., 1985).
Measurement of O2 ●¯
The production of O2 ●¯ was measured in the thoracic aorta according to the lucigenin‐enhanced chemiluminescence method (Munzel et al., 1995). The frozen aortic slices (2–5 mg) were pre‐incubated in a Krebs‐HEPES buffer (saturated with 95% O2 and 5% CO2 at room temperature) for 30 min and transferred to a glass scintillation vial containing 5 μM of lucigenin (200 μL in 2 mL Krebs‐HEPES) to determine the O2 ●¯ basal level. The chemiluminescence was recorded every minute for 10 min in a dark room using a liquid scintillation counter (Wallac 1409, Turku, Finland). To ascertain the contribution of NADPH oxidase in the basal production of O2 ●¯ in the aorta of control and glucose‐fed rats treated or not with 1400W, diphenyleneiodonium (DPI, 10 μmol·L−1), a selective inhibitor of NADPH oxidase, was added to the medium 30 min prior to O2 ●¯ measurement in two additional slices of aortas in each group. The background was counted using a vial with no tissue solution. The final value expressed as cpm·mg−1 of dry weight tissue was calculated as follows: (tissue value − background value) ÷ tissue dry weight.
Western blot analysis
The western blot was performed according to a previous method (Dias et al., 2010; Pouliot et al., 2011). Total proteins were loaded (20–30 μg) in each well of 10% SDS‐PAGE. After completion of the migration by electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Bio‐Rad, Montreal, QC, Canada) at 100 V for 1 h. The membrane was then washed with PBS‐Tween 20 and incubated for 1 h at room temperature in a blocking solution of 5% skimmed milk dissolved in PBS‐Tween 20. The membrane was then cut in pieces according to the molecular weight of protein and incubated with specific primary antibodies in PBS‐Tween 20 overnight at 4°C. Dynein and β‐actin were used as standard proteins. After three sets of 10 min washing with PBS‐Tween 20, the membrane was incubated for 1 h at room temperature in PBS‐Tween 20 containing 5% skimmed milk with secondary antibodies. The membrane was washed three times (10 min per wash) with PBS‐Tween 20 before being subjected to a reaction with a chemiluminescent detection reagent improved for the western blot (Super‐Signal®; Thermo Scientific, Rockford, IL, USA). A quantitative analysis of proteins was provided by scanning densitometry using the MCID‐M1 system (Imaging Research, St. Catharines, ON, Canada).
Detection of bradykinin (BK) receptor proteins was made with selective anti‐B1 receptor and anti‐B2 receptor antibodies (1:1000) rose in rabbits (Biotechnology Research Institute, Montreal, QC, Canada) against a conserved amino acid sequence from B1 and B2 receptor proteins of mouse and rat. The epitopes used contained 15 amino acids localized in the C‐terminal part of the B1 receptor (VFAGRLLKTRVLGTL) and 15 amino acids localized in the second extracellular domain of the B2 receptor (TIANNFDWVFGEVLC). Care was taken to avoid sequences with similarity to related mammalian proteins, including the opposite receptor. One negative control was run for each antibody using the pre‐immune serum. Specificity of anti‐B1 receptor and anti‐B2 receptor antibodies was further determined using mouse kidney extracts from wild‐type, B1 and B2 receptor knockout (KO) mice (Lin et al., 2010; Lacoste et al., 2013).
The other primary antibodies were as follows: dynein (1:4000 mouse, SC‐13524; Santa Cruz Biotechnology, CA, USA), β‐actin (1:5000 mouse, SC‐47778; Santa Cruz), IL‐1β (1:500 rabbit, SC‐7884; Santa Cruz), iNOS (NOS2) (1:1000 rabbit, SC‐650; Santa Cruz), nitrotyrosine (1:1500 mouse, 1A6‐05233; Millipores, Billerica, MA, USA) and carboxypeptidase M (1:500 rabbit, SC‐98698; Santa Cruz). Secondary antibodies were horseradish peroxidase (HRP)‐linked goat anti‐rabbit SC‐2004 and HRP‐linked goat anti‐mouse SC‐2005 (Santa Cruz) used at dilution of 1:25000 (for B1 and B2 receptors), 1:5000 (for Dynein, β‐actin, IL‐1β, iNOS and CPM) and 1:3000 (nitrotyrosine).
qRT‐PCR
After the animals had been killed the various organs were removed and around 10 mg of tissue (renal cortex, liver and thoracic aorta) were put in a RNAlater stabilization reagent (QIAGEN, Toronto, ON, Canada) and frozen at −56°C. Total RNA was extracted from the tissue using Qiazol according to the manufacturer's instructions. The single‐stranded cDNA was synthesized according to the procedure in the manual supplied by Bio‐Rad. qRT‐PCR was performed in the SYBR Green Master Mix (QIAGEN) by adding 300 nM of each primer, and the signal was detected by Stratagene Mx3000p device (Thermo Fisher Scientific, Waltham, MA, USA) and using rat 18S as standard. The primer pairs were designed by Vector NTI software (Table 1). The PCR conditions were as follows: 95°C for 15 min followed by 46 cycles at 94°C for 15 s, 60°C for 30 s and 72°C for 30 s. The cycle threshold value represents the number of cycles during which a fluorescent signal increases above the background noise. The relative quantification of gene expression was analysed by the method of 2‐ΔΔCt (Livak and Schmittgen, 2001).
Table 1.
List of primer pairs designed by Vector NTI software and used in qRT‐PCR analysis
| Gene | Sequence | Position | GenBank accession no. | |||
|---|---|---|---|---|---|---|
| 18S | Forward | 5′ | TCA ACT TTC GAT GGT AGT CGC CGT | 3′ | 363–386 | X01117 |
| 18S | Reverse | 5′ | TCC TTG GAT GTG GTA GCC GTT TCT | 3′ | 470–447 | X01117 |
| B1R | Forward | 5′ | GCA GCG CTT AAC CAT AGC GGA AAT | 3′ | 367–391 | NM_030851 |
| B1R | Reverse | 5′ | CCA GTT GAA ACG GTT CCC GAT GTT | 3′ | 478–454 | NM_030851 |
| IL‐1β | Forward | 5′ | TGT CAC TCA TTG TGG CTG TGG AGA | 3′ | 247–270 | NM_031512 |
| IL‐1β | Reverse | 5′ | TGG GAA CAT CAC ACA CTA GCA GGT | 3′ | 411–388 | NM_031513 |
Statistical analysis
Data are presented as mean ± SEM, and n represents the number of rats. Statistical analysis was performed using PrismTM version 5.0 (GraphPad Software Inc., La Jolla, CA, USA); data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical significance was determined with Student's t‐test for unpaired samples or with the one‐way ANOVA followed by the Bonferroni test for multiple comparisons when F achieved P < 0.05 and there was no significant variance in homogeneity. Only the value of P ≤ 0.05 was considered to be statistically significant.
Results
Effect of 1400W on clinical parameters
Blood glucose level was increased by twofold (P < 0.05) in overnight‐fasted glucose‐fed rats compared with control rats, yet glycaemia was no longer significantly different from control values after 1 week treatment with 1400W (1 mg·kg−1 twice daily). Plasma insulin level was significantly increased by fourfold (P < 0.05) in glucose‐fed rats and was blunted by the treatment with 1400W. The HOMA index of insulin resistance was also significantly enhanced (P < 0.05) in glucose‐fed rats, yet this value was markedly reduced (P < 0.05) but not completely normalized by 1400W. In contrast, the same treatment with 1400W failed to affect glycaemia, insulinaemia and the HOMA index in control rats (Figure 1).
Figure 1.
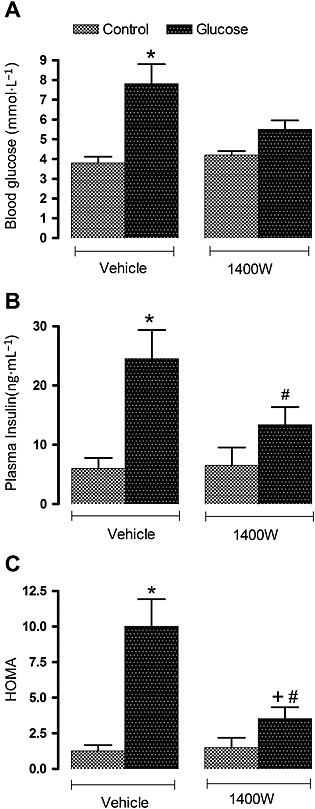
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on (A) blood glucose, (B) plasma insulin and (C) insulin resistance assessed by the HOMA index. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle; #P < 0.05 compared with control + 1400W.
The body weight was not significantly different between glucose‐fed and control rats before (8 weeks) and after (9 weeks) treatment with 1400W (Table 2). Nevertheless, the gain in body weight was significantly higher (P < 0.05) in glucose‐fed rats after 9 weeks when compared with age‐matched controls. The 1 week treatment with 1400W had no impact on the gain in body weight in control rats, but a significant loss in body weight (P < 0.05) was measured after treatment with 1400W in glucose‐fed rats. As indicated in Table 2, the leptin levels were significantly increased (P < 0.05) in the plasma of glucose‐fed rats in comparison with control. The 1 week treatment with 1400W had no significant effect on plasma leptin levels in both control and glucose‐fed rats, which remained significantly enhanced in the latter group. Systolic blood pressure was significantly enhanced (P < 0.05) in 9 week glucose‐fed rats compared with control rats, and the high systolic blood pressure value was not significantly affected by the 1 week treatment with 1400W (Table 2).
Table 2.
Body weight, plasma leptin levels and systolic blood pressure
| Parameters | Control + Vehicle | Glucose + Vehicle | Control + 1400W | Glucose + 1400W |
|---|---|---|---|---|
| Body weight at the initiation of protocol (g) | 114 ± 3 | 110 ± 3 | 107 ± 3 | 110 ± 3 |
| Body weight (g) at 8 weeks | 587 ± 25 | 592 ± 29 | 566 ± 21 | 571 ± 37 |
| Body weight (g) at 9 weeks | 603 ± 26 | 635 ± 18 | 573 ± 20 | 537 ± 15 |
| Body weight gain/loss (g) | 16 ± 2 | 43 ± 2 * | 13 ± 3 | (−) 34 ± 2 + , # |
| Plasma leptin (ng·mL−1) | 3.8 ± 0.3 | 10.4 ± 2.2 * | 4.6 ± 0.8 | 12.7 ± 2.5 # |
| Systolic blood pressure (mm Hg) at 9 weeks | 109 ± 3 | 127 ± 3 * | 119 ± 3 | 127 ± 3 |
Values represent the mean ± SEM of six rats per group. Statistical comparison (*) to control + vehicle rats, (+) to glucose + vehicle and (#) to control + 1400W is indicated by
P ≤ 0.05,
P ≤ 0.05,
P ≤ 0.05.
Effect of 1400W on oxidative stress
The impact of 1400W on oxidative stress was evaluated on the production of O2 ●¯ and the expression of nitrotyrosine, a marker of ONOO¯. ONOO¯ can modify tyrosine residue in various proteins to form nitrotyrosine, which could alter protein function and stability. As shown in Figure 2, the basal production of O2 ●¯ was significantly enhanced (P < 0.05) in the aorta of glucose‐fed rats when compared with control rats. The 1 week treatment with 1400W increased significantly (P < 0.05) the basal production of O2 ●¯ in control rats but failed to affect significantly the increased basal production of O2 ●¯ in glucose‐fed rats. The basal production of O2 ●¯ in the aorta of control and glucose‐fed rats treated or not with 1400W was completely abolished when the tissue was treated for 30 min with DPI (10 μmol·L−1), confirming the involvement of NADPH oxidase in the production of O2 ●¯ (data not shown).
Figure 2.
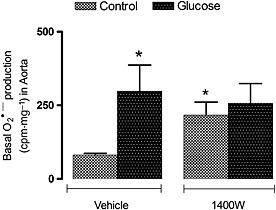
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on basal O2 ●¯ production in the thoracic aorta. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle.
In control renal cortex, nitrotyrosine epitopes were detected in proteins with molecular masses of about 55 and 70–85 kDa. In glucose renal cortex, the intensity of those original nitrotyrosine epitopes increased, and other nitrosylated proteins occurred at about 50 and 60 kDa. Treatment with 1400W significantly reduced the intensity of nitrotyrosine‐containing proteins in glucose renal cortex to levels not significantly different from control. Likewise, nitrosylated proteins with molecular masses of about 70–80 and 90–95 kDa were detected in control aorta, and their intensity was higher in glucose aorta, which also displayed additional nitrosylated proteins at molecular masses of 110 and 150 kDa. Treatment with 1400W significantly reduced the expression of these nitrosylated proteins in both control and glucose aortas (Figure 3).
Figure 3.
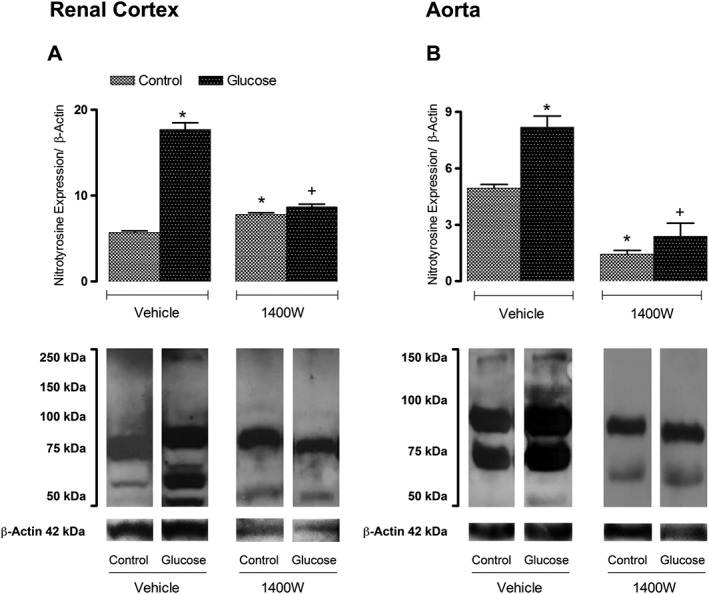
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on nitrotyrosine expression in (A) renal cortex and (B) thoracic aorta. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle.
Effect of 1400W on B1 receptors and carboxypeptidase M
Levels of B1 receptor mRNA and protein were markedly and significantly enhanced in the three tissues (renal cortex, aorta and the liver) of glucose‐fed rats (Figure 4). The 1 week treatment with 1400W reduced B1 receptor expression, particularly at the mRNA level in renal cortex, aorta and liver. While B1 receptor mRNA levels were completely abolished by 1400W, B1 receptor protein levels remained significantly increased when compared with control values in aorta and liver. The lack of correlation between B1 receptor gene and protein expression is unexplained but may indicate that the B1 receptor remains expressed at the cell membrane beyond the inhibition of its synthesis by 1400W. The treatment with 1400W failed, however, to affect significantly B1 receptor mRNA and protein expression in the three tissues in control rats (Figure 4).
Figure 4.
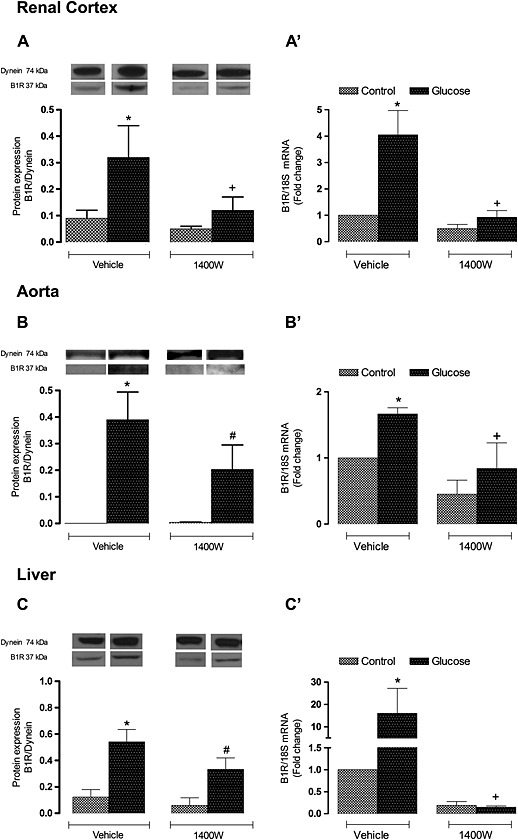
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on B1 receptor expression in (A, A′) renal cortex, (B, B′) thoracic aorta and (C, C′) liver. The expression of B1 receptors was measured at the protein level by western blot (A, B, C) and at mRNA level by qRT‐PCR (A′, B′, C′). Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle; #P < 0.05 compared with control + 1400W.
Protein expression of CPM was also significantly increased (P < 0.05) in the renal cortex, aorta and liver of glucose‐fed rats. The 1 week treatment with 1400W abolished the overexpression of CPM in the renal cortex and aorta of glucose‐fed rats but failed to affect that in the liver. 1400W had no significant impact in the three control tissues (Figure 5).
Figure 5.
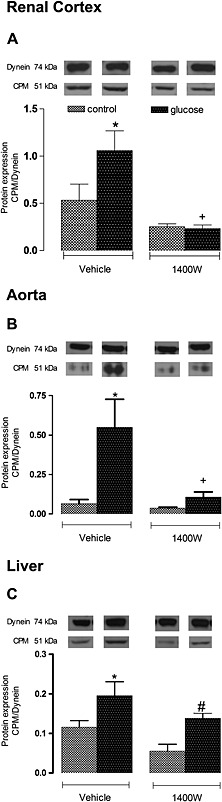
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on membrane CPM expression in (A) renal cortex, (B) thoracic aorta and (C) liver. The expression of CPM was measured at protein level by western blot. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle; #P < 0.05 compared with control + 1400W.
Effect of 1400W on iNOS and IL‐1β
Protein expression of iNOS was found to be significantly increased (P < 0.05) in the renal cortex, aorta and liver of glucose‐fed rats. The 1 week treatment with 1400W abolished the enhanced expression of iNOS in the two former tissues but had no significant effect on iNOS in the liver of glucose‐fed rats. Treatment with 1400W had no significant impact in the three control tissues (Figure 6). In glucose‐fed rats, the expression of IL‐1β was also significantly enhanced at mRNA and protein levels in the renal cortex, aorta and liver (Figure 7). The 1 week treatment with 1400W abolished the increased mRNA expression in the three tissues in glucose‐fed rats without affecting basal expression of IL‐1β in control rats. The impact of 1400W on IL‐1β protein expression was less striking. For instance, it had a reducing effect in the renal cortex and aorta but not in the liver where IL‐1β protein expression remained significantly elevated compared with its control (Figure 7).
Figure 6.
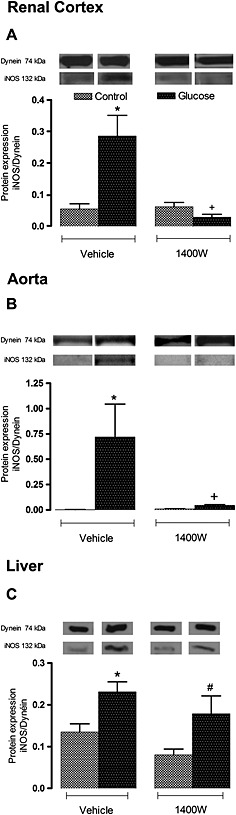
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on the iNOS expression in (A) renal cortex, (B) thoracic aorta and (C) liver. The expression of iNOS was measured at protein level by western blot. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle; #P < 0.05 compared with control + 1400W.
Figure 7.
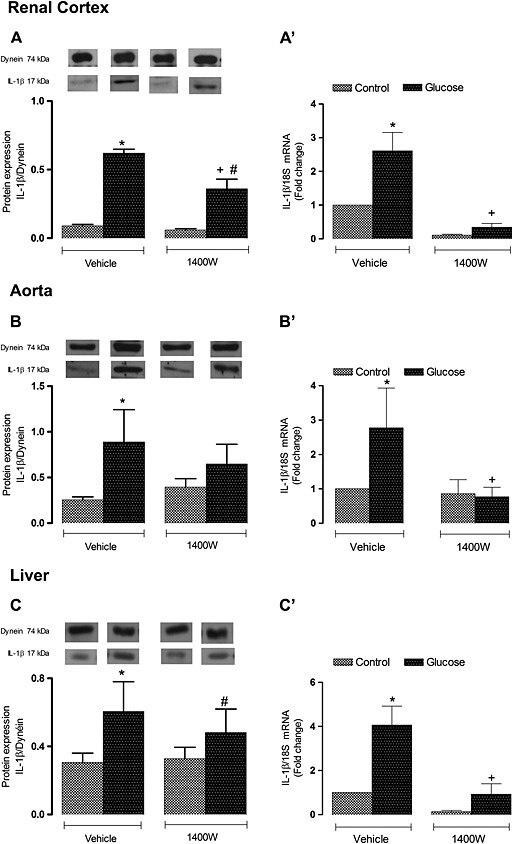
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on IL‐1β expression in (A, A′) renal cortex, (B, B′) thoracic aorta and (C, C′) liver. The expression of IL‐1β was measured at the protein level by western blot (A, B, C) and at mRNA level by qRT‐PCR (A′, B′, C′). Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle; #P < 0.05 compared with control + 1400W.
Effect of 1400W on B2 receptor expression
Protein expression of B2 receptors was not affected in the renal cortex and aorta but was significantly increased (P < 0.05) in the liver of glucose‐fed rats. Although B2 receptor expression was not altered by the 1 week treatment with 1400W in the aorta, it was significantly decreased in the control and glucose renal cortex (P < 0.05). In contrast, treatment with 1400W increased the expression of B2 receptors in the control liver (P < 0.05) to values measured in glucose‐fed rats, showing an absence of inhibition of the upregulated B2 receptors in the liver of glucose‐fed rats (Figure 8).
Figure 8.
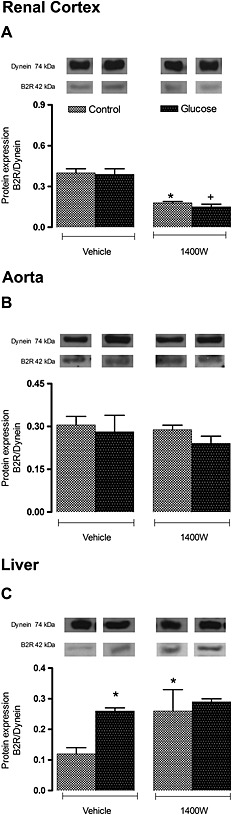
Effect of s.c. administered 1400W (1 mg·kg−1 twice daily) for 7 days on B2R expression in (A) renal cortex, (B) thoracic aorta and (C) liver. The expression of B2R was measured at protein level by western blot. Data are mean ± SEM obtained from six rats per group. *P < 0.05 compared with control + vehicle; +P < 0.05 compared with glucose + vehicle.
Discussion
In this study, we demonstrated that 1 week treatment with a selective inhibitor of iNOS (1400W) improved insulin resistance, hyperglycaemia, hyperinsulinaemia and reversed body weight gain without affecting hypertension and hyperleptinaemia in glucose‐fed rats. The beneficial effects of 1400W on those metabolic abnormalities were associated with a correction of ONOO¯ formation and of upregulated biomarkers of inflammation (B1 receptor, iNOS and IL‐1β) in peripheral tissues. In addition, the up‐regulation of CPM (kininase I), a key enzyme involved in the biotransformation of BK and Lys‐BK into their active C‐terminal metabolites (des‐Arg9‐BK and Lys‐des‐Arg9‐BK), the preferred endogenous agonists at B1 receptors (Regoli and Gobeil, 2015), was reversed by 1400W in glucose‐fed rats. These findings strongly suggest that the deleterious effects of iNOS are linked to B1 receptor activation. The production of ONOO¯ upon iNOS activation by B1 receptors may contribute to insulin resistance by blocking tyrosine phosphorylation of IRS‐1 subsequently to tyrosine nitration (Pilon et al., 2010). Because the B1 receptor is virtually absent in physiological situations, this study is the first to suggest that the reactive nitrogen derivative ONOO¯ is more important than O2 ●¯ in hyperglycaemia‐induced up‐regulation of B1 receptors and may act as a positive feedback mechanism (Figure 9).
Figure 9.
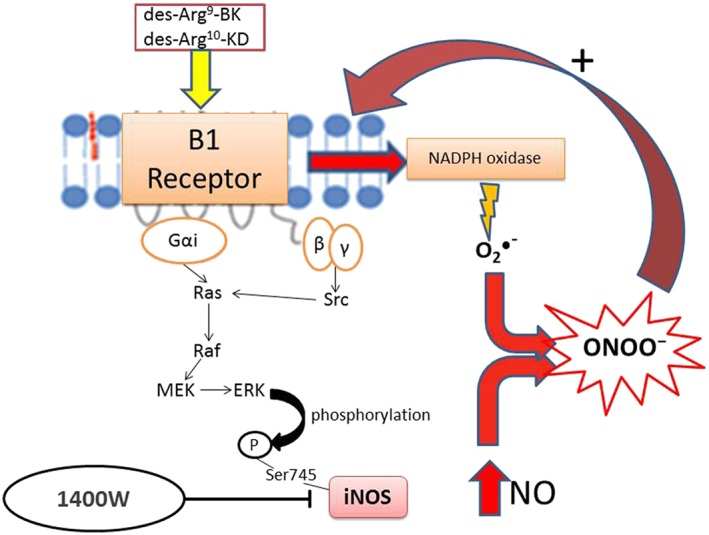
Schematic diagram of the signalling pathway activated by B1 receptor agonists leading to the formation of ONOO¯ upon post‐translational activation of iNOS and NADPH oxidase. Activation of the B1 receptor through Gαi and βγ‐dependent activation of Src, Ras, Raf and MEK leads to ERK‐dependent phosphorylation of iNOS on Ser745 that causes high NO output (Brovkovych et al., 2011). Simultaneously, B1 receptor agonists can activate NADPH oxidase to generate O2 ●¯ (Dias et al., 2010) that can rapidly react with NO to form ONOO¯. The latter exerts a positive feedback loop to enhance the expression of B1 receptors. Blockade of iNOS with 1400W prevents this vicious cycle and the pro‐inflammatory effects of B1 receptors.
Impact of 1400W on peripheral cardiometabolic targets and oxidative stress
Levels of O2 ●¯, a marker of oxidative stress, were significantly enhanced in the aorta of glucose‐fed rats and remained unaffected by 1400W. In this model of insulin resistance, basal production of O2 ●¯ derived mainly from NADPH oxidase (sensitive to DPI), which was also found to be activated by a B1 receptor agonist to raise the formation of O2 ●¯ in the aorta of glucose‐fed rats (Dias et al., 2010). Thus, the failure of 1400W to prevent the enhanced generation of O2 ●¯ is logical. Indeed, the inhibition of iNOS prevents the formation of NO and thereby is likely to compromise its binding with O2 ●¯ to form ONOO¯. This can afford an explanation for the inhibition of ONOO¯ in aorta and renal cortex with the consequent accumulation of O2 ●¯ in the tissue as observed in control aorta under iNOS inhibition.
Previous studies have reported that the B1 receptor can be upregulated by its own agonist in human fibroblasts (Schanstra et al., 1998) and in vivo on blood vessels as the blockade of B1 receptors with SSR240612 reversed the up‐regulation of B1 receptors in glucose‐fed rats (Dias et al., 2010; Dias and Couture, 2012a, 2012b). The enhanced formation of B1 receptor agonists (des‐Arg9‐BK and Lys‐des‐Arg9‐BK) following the overexpressed CPM appears therefore a feasible mechanism for B1 receptor up‐regulation. By reversing the up‐regulation of CPM with 1400W, the over activation of the B1 receptor by its endogenous agonists is suppressed, leading to a normalisation of B1 receptor expression at mRNA and protein levels. By this mechanism, iNOS inhibition targets both B1 receptor ligands and B1 receptor expression.
In a recent study, we have shown that a 1 week treatment with the B1 receptor antagonist SSR240612 reversed body weight gain without affecting hyperleptinaemia in glucose‐fed rats (Dias and Couture, 2012a), which reinforces a role for the B1 receptor in obesity that could be mediated by iNOS as the up‐regulation of iNOS in retroperitoneal adipose tissue was also reversed by SSR240612 in glucose‐fed rats. Obesity is associated with increased iNOS expression in insulin‐sensitive tissues in rodents and humans, and inhibition of iNOS ameliorates obesity‐induced insulin resistance (Kaneki et al., 2007). However, a primary role for leptin in body weight gain associated with B1 receptors and iNOS is not supported by our studies using either SSR240612 (Dias and Couture, 2012a) or 1400W. Thus, the B1 receptor contributes to insulin resistance through a mechanism that is iNOS‐dependent and leptin‐independent.
Although 1400W prevented insulin resistance, it failed to affect the associated hypertension in chronic glucose‐fed rats. This may indicate that hypertension is not related to peripheral iNOS or to the overproduction of vascular oxidative stress (O2 ●¯ and ONOO¯) or to peripheral B1 receptor expression and activation. Indeed, it was found that only non‐peptide antagonists of B1 receptors that could pass the blood–brain barrier to inhibit B1 receptors in the central nervous system can reverse high systolic blood pressure in glucose‐fed rats, spontaneously hypertensive rats and angiotensin II‐induced hypertension (Lungu et al., 2007; De Brito Gariepy et al., 2010; Dias et al., 2010).
Interestingly, increased expression and activity of iNOS were reported in superior mesenteric arteries from 12 to 14 week streptozotocin‐induced diabetic rats (Bardell and MacLeod, 2001). In this model of type 1 diabetes, rats are not hypertensive and display a phenotype of cardiovascular depression (depressed mean arterial blood pressure and heart rate resistant to 1400W) associated with enhanced expression of iNOS and nitrotyrosine in cardiovascular tissues and with endothelial dysfunction (Nagareddy et al., 2005). Consistently, iNOS knockout mice are resistant to endothelial dysfunction during diabetes (Gunnett et al., 2003). Hence, iNOS induction in cardiovascular tissues may not contribute directly to high systemic blood pressure in insulin resistance and to low systemic blood pressure in type 1 diabetes despite impairment of endothelial function.
Impact of 1400W on inflammatory markers
The suppression of the enhanced expression of B1 receptors and IL‐1β at mRNA levels by 1400W occurred in the three tissues studied (aorta, renal cortex and the liver) of chronic glucose‐fed rats, suggesting that the inflammatory process is not restricted to the vasculature. Nevertheless, protein expression of the B1 receptor, CPM, IL‐1β and iNOS was more resistant to 1400W in the liver, suggesting that the enhanced expression of these markers in the liver may be differently regulated at transcriptional and post‐transcriptional levels in this model of insulin resistance. Also, B2 receptor protein expression was increased in the liver but not in the two other tissues in glucose‐fed rats, and again, it was resistant to iNOS blockade. In agreement with this study, 1 week blockade of B1 receptors with SSR240612 reversed the overexpression of iNOS, B1 receptors and IL‐1β in the aorta of glucose‐fed rats (Dias et al., 2010; Dias and Couture, 2012b). Hence, in addition to being involved in the ERK/MAPK activation of iNOS, the B1 receptor may control the expression of iNOS through ONOO¯ formation in blood vessel. In this context, it is worth noting that B1 receptor‐induced activation of iNOS plays a primary role in lethal thrombosis in a rat model of septic shock in diabetes (Tidjane et al., 2015).
Conclusion
Pharmacological blockade of iNOS provides the same beneficial effects as B1 receptor blockade in insulin resistance and peripheral inflammation in chronic glucose‐fed rats. Inhibition of ONOO¯ formation by the selective iNOS blocker 1400W appears to be a possible downstream target mechanism that prevents the positive feedback up‐regulation and activation of the pro‐inflammatory B1 receptor by endogenous ligands under the control of carboxypeptidase M. These findings have clinical relevance in the treatment of insulin resistance and type 2 diabetes where iNOS and the B1 receptor represent promising therapeutic targets.
Author contributions
Y.H. and R.C. conceived and designed the experiments. Y.H. performed the experiments, analysed the data and drafted the paper. R.C. supervised the study and edited and wrote the final version of the manuscript. All authors approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Acknowledgements
This work was supported by a Grant‐in‐Aid from the Canadian Institutes of Health Research (MOP‐119329) to R.C. Y.H. received a PhD Studentship award from the Department of Molecular and Integrative Physiology of the Université de Montréal. The authors greatly appreciate the technical assistance of Jacques Sénécal for the pharmacological treatment and RIA studies.
Haddad, Y. , and Couture, R. (2016) Interplay between the kinin B1 receptor and inducible nitric oxide synthase in insulin resistance. British Journal of Pharmacology, 173: 1988–2000. doi: 10.1111/bph.13491.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardell AL, Macleod KM (2001). Evidence for inducible nitric‐oxide synthase expression and activity in vascular smooth muscle of streptozotocin‐diabetic rats. J Pharmacol Exp Ther 296: 252–259. [PubMed] [Google Scholar]
- Brovkovych V, Zhang Y, Brovkovych S, Minshall RD, Skidgel RA (2011). A novel pathway for receptor‐mediated post‐translational activation of inducible nitric oxide synthase. J Cell Mol Med 15: 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Griendling KK, Harrison DG (2003). The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24: 471–478. [DOI] [PubMed] [Google Scholar]
- Charbonneau A, Marette A (2010). Inducible nitric oxide synthase induction underlies lipid‐induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes 59: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture R, Blaes N, Girolami JP (2014). Kinin receptors in vascular biology and pathology. Curr Vasc Pharmacol 12: 223–248. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brito Gariepy H, Carayon P, Ferrari B, Couture R (2010). Contribution of the central dopaminergic system in the anti‐hypertensive effect of kinin B1 receptor antagonists in two rat models of hypertension. Neuropeptides 44: 191–198. [DOI] [PubMed] [Google Scholar]
- de Vries MA, Alipour A, Klop B, van de Geijn GJ, Janssen HW, Njo TL et al. (2015). Glucose‐dependent leukocyte activation in patients with type 2 diabetes mellitus, familial combined hyperlipidemia and healthy controls. Metabolism 64: 213–217. [DOI] [PubMed] [Google Scholar]
- Dias JP, Couture R (2012a). Blockade of kinin B(1) receptor reverses plasma fatty acids composition changes and body and tissue fat gain in a rat model of insulin resistance. Diabetes Obes Metab 14: 244–253. [DOI] [PubMed] [Google Scholar]
- Dias JP, Couture R (2012b). Suppression of vascular inflammation by kinin B1 receptor antagonism in a rat model of insulin resistance. J Cardiovasc Pharmacol 60: 61–69. [DOI] [PubMed] [Google Scholar]
- Dias JP, Talbot S, Senecal J, Carayon P, Couture R (2010). Kinin B1 receptor enhances the oxidative stress in a rat model of insulin resistance: outcome in hypertension, allodynia and metabolic complications. PLoS One 5: e12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ et al. (1997). 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric‐oxide synthase in vitro and in vivo. J Biol Chem 272: 4959–4963. [DOI] [PubMed] [Google Scholar]
- Gleissner CA (2015). The vulnerable vessel. Vascular disease in diabetes mellitus. Hamostaseologie 35: 267–271. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Fitzgerald GA (2003a). Oxidative stress and cardiovascular injury: part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108: 1912–1916. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Fitzgerald GA (2003b). Oxidative stress and cardiovascular injury: part II: animal and human studies. Circulation 108: 2034–2040. [DOI] [PubMed] [Google Scholar]
- Gunnett CA, Heistad DD, Faraci FM (2003). Gene‐targeted mice reveal a critical role for inducible nitric oxide synthase in vascular dysfunction during diabetes. Stroke 34: 2970–2974. [DOI] [PubMed] [Google Scholar]
- He Z, King GL (2004). Protein kinase Cbeta isoform inhibitors: a new treatment for diabetic cardiovascular diseases. Circulation 110: 7–9. [DOI] [PubMed] [Google Scholar]
- Johansen JS, Harris AK, Rychly DJ, Ergul A (2005). Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneki M, Shimizu N, Yamada D, Chang K (2007). Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal 9: 319–329. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhr F, Lowry J, Zhang Y, Brovkovych V, Skidgel RA (2010). Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 44: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste B, Tong XK, Lahjouji K, Couture R, Hamel E (2013). Cognitive and cerebrovascular improvements following kinin B1 receptor blockade in Alzheimer's disease mice. J Neuroinflammation 10: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Talbot S, Lahjouji K, Roy JP, Senecal J, Couture R et al. (2010). Mechanism of cigarette smoke‐induced kinin B(1) receptor expression in rat airways. Peptides 31: 1940–1945. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Lungu C, Dias JP, Franca CE, Ongali B, Regoli D, Moldovan F et al. (2007). Involvement of kinin B1 receptor and oxidative stress in sensory abnormalities and arterial hypertension in an experimental rat model of insulin resistance. Neuropeptides 41: 375–387. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985). Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412–419. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG (1995). Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross‐tolerance. J Clin Invest 95: 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Xia Z, McNeill JH, Macleod KM (2005). Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin‐induced diabetes. Am J Physiol Heart Circ Physiol 289: H2144–H2152. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault M, Marette A (2001). Targeted disruption of inducible nitric oxide synthase protects against obesity‐linked insulin resistance in muscle. Nat Med 7: 1138–1143. [DOI] [PubMed] [Google Scholar]
- Pilon G, Charbonneau A, White PJ, Dallaire P, Perreault M, Kapur S et al. (2010). Endotoxin mediated‐iNOS induction causes insulin resistance via ONOO¯ induced tyrosine nitration of IRS‐1 in skeletal muscle. PLoS One 5: e15912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot M, Hetu S, Lahjouji K, Couture R, Vaucher E (2011). Modulation of retinal blood flow by kinin B(1) receptor in Streptozotocin‐diabetic rats. Exp Eye Res 92: 482–489. [DOI] [PubMed] [Google Scholar]
- Regoli D, Gobeil F Jr (2015). Critical insights into the beneficial and protective actions of the kallikrein‐kinin system. Vascul Pharmacol 64: 1–10. [DOI] [PubMed] [Google Scholar]
- Ropelle ER, Pauli JR, Cintra DE, da Silva AS, De Souza CT, Guadagnini D et al. (2013). Targeted disruption of inducible nitric oxide synthase protects against aging, S‐nitrosation, and insulin resistance in muscle of male mice. Diabetes 62: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanstra JP, Bataille E, Marin Castano ME, Barascud Y, Hirtz C, Pesquero JB et al. (1998). The B1‐agonist [des‐Arg10]‐kallidin activates transcription factor NF‐kappaB and induces homologous upregulation of the bradykinin B1‐receptor in cultured human lung fibroblasts. J Clin Invest 101: 2080–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot S, Lin JC, Lahjouji K, Roy JP, Senecal J, Morin A et al. (2011). Cigarette smoke‐induced kinin B1 receptor promotes NADPH oxidase activity in cultured human alveolar epithelial cells. Peptides 32: 1447–1456. [DOI] [PubMed] [Google Scholar]
- Taniyama Y, Griendling KK (2003). Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42: 1075–1081. [DOI] [PubMed] [Google Scholar]
- Tidjane N, Hachem A, Zaid Y, Merhi Y, Gaboury L, Girolami JP et al. (2015). A primary role for kinin B1 receptor in inflammation, organ damage, and lethal thrombosis in a rat model of septic shock in diabetes. Eur J Inflamm 13: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]