

Abstract
Many conditions culminate in heart failure (HF), a multi‐organ systemic syndrome with an intrinsically poor prognosis. Pharmacotherapeutic agents that correct neurohormonal dysregulation and haemodynamic instability have occupied the forefront of developments within the treatment of HF in the past. Indeed, multiple trials aimed to validate these agents in the 1980s and early 1990s, resulting in a large and robust evidence‐base supporting their use clinically. An established treatment paradigm now exists for the treatment of HF with reduced ejection fraction (HFrEF), but there have been very few notable developments in recent years. HF remains a significant health concern with an increasing incidence as the population ages. We may indeed be entering the surgical era for HF treatment, but these therapies remain expensive and inaccessible to many. Newer pharmacotherapeutic agents are slowly emerging, many targeting alternative therapeutic pathways, but with mixed results. Metabolic modulation and manipulation of the nitrate/nitrite/nitric oxide pathway have shown promise and could provide the answers to fill the therapeutic gap between medical interventions and surgery, but further definitive trials are warranted. We review the significant evidence base behind the current medical treatments for HFrEF, the physiology of metabolic impairment in HF, and discuss two promising novel agents, perhexiline and nitrite.
Abbreviations
- ANT
adenine nucleotide translocase
- ARB
angiotensin receptor blocker
- BEAUTIFUL
morBidity‐mortality EvAlUaTion of the If inhibitor ivabradine in patients with coronary disease and left ventricULar dysfunction trial
- BNP
brain natriuretic peptide
- CHARM
Candesartan in Heart failure ‐ Assessment of moRtality and Morbidity
- CK
creatine kinase
- COPERNICUS
CarvedilOl ProspEctive RaNdomIzed CUmulative Survival Study
- CPT
carnitine palmitoyltransferase
- ESC
European Society of Cardiology
- FA
fatty acid
- FADH2
flavin adenine dinucleotide
- HF
heart failure
- HFpEF
heart failure and preserved ejection fraction
- HFrEF
heart failure and reduced ejection fraction
- ISDN
isosorbide dinitrate
- LVEF
left ventricular ejection fraction
- MERIT‐HF
MEtoprolol cr/xl Randomised Intervention Trial in congestive Heart Failure
- NYHA
New York Heart Association
- PDH
pyruvate dehydrogenase
- PCr
phosphocreatine
- Pi
inorganic phosphate
- PPP
pentose phosphate pathway
- RAAS
renin–angiotensin–aldosterone system
- RCT
randomized controlled trial
- SENIORS
Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with heart failure
- SHIFT
Systolic Heart failure treatment with the If inhibitor ivabradine Trial
- TCA
tricarboxylic acid
- UCP
uncoupling protein
- V‐HeFT
vasodilator heart failure trial
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| AT1 receptor |
| β‐adrenoceptors |
| Nuclear hormone receptors b |
| Mineralocorticoid receptor |
| PPARα |
| Enzymes c |
| ACE |
| Aldehyde dehydrogenase 2 |
| Neprilysin |
| NOS |
| PDE5 |
| Soluble guanylate cyclase |
| Transporters d |
| Adenine nucleotide translocase (ANT) |
| ATP synthase |
| GLUT1 |
| GLUT4 |
| Uncoupling protein (UCP) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a, 2015b, 2015c, 2015d).
Introduction
Chronic heart failure (HF) is a complex, systemic, multi‐organ syndrome, which represents a common endpoint for many cardiac conditions (Follath et al., 1998). Traditionally, it is characterized by haemodynamic impairment and progressive neurohormonal dysregulation, with increased sympathetic activation, elevated peripheral vascular resistance and cardiac remodelling (Braunwald and Bristow, 2000). More recently, metabolic impairment (Neubauer, 2007) and activation of inflammatory responses (Rauchhaus et al., 2000) have been increasingly recognized. At a population level, HF affects around 900 000 Britons, half of whom die within 5 years of diagnosis (Hobbs et al., 2007). At current hospitalization rates, 70% of which involve patients aged >65 years, HF costs the UK 2% of the total annual National Health Service budget (Basu et al., 2008), and this is set to increase further as the population ages.
Pharmacotherapeutic agents geared towards correcting neurohormonal dysregulation have occupied the forefront of developments within the treatment of HF in the past, resulting in a large and robust evidence‐base supporting their use clinically. This evidence underpins the European Society of Cardiology (ESC) Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure, most recently updated in 2012 (McMurray et al., 2012). The pace and success of development of novel pharmacotherapeutic agents over the last 20 years has been disappointing. However, new agents are slowly emerging. LCZ696 (a combination therapy of an angiotensin receptor blocker and a neprilysin inhibitor) demonstrated superiority over angiotensin converting enzyme (ACE) inhibitors in 8399 patients with HF and reduced ejection function (HFrEF), with a reduction in the risk of all‐cause mortality of 12.6% (95% CI 7–18%; P < 0.0001) (Packer et al., 2015). Inflammatory mediators such as the key cytokine TNF‐α were also identified as targets for novel therapies in patients with HF; however, human trials have shown disappointing results (Chung et al., 2003; Mann et al., 2004; Torre‐Amione et al., 2008). Despite this, a significant advancement in our understanding of inflammation in HF has been made and has generated further research in this field (Heymans et al., 2009). Investigation into cardiac myosin activators [such as omecamtiv mecarbil, which has been shown to improve systolic function and is well tolerated (Greenberg et al., 2015)], phosphodiesterase inhibition, manipulation of the nitrate/nitrite/NO pathway, metabolic modulation, stem cell therapy and gene therapy is currently underway (Shah and Mann, 2011; Tilemann et al., 2012).
Up to 50% of patients with HF have a preserved ejection fraction of ≥50% (HFpEF). The therapeutic paradigm that has been successful in HFrEF has failed in this syndrome (Yusuf et al., 2003; Cleland et al., 2006; Massie et al., 2008; Pitt et al., 2014). Therefore, effective therapies are urgently needed. With the rapid advancements in the development of implantable cardiac devices such as left ventricular assist devices, we may be entering the surgical era of HF treatment. These interventions, despite their clearly beneficial use in patients awaiting cardiac transplantation or as an alternative destination therapy, remain expensive and lead to device‐related complications. As such, surgery is reserved for patients with end‐stage HF (Mancini and Colombo, 2015).
We suggest that targeting alternative pathways other than those used by the well‐established HFrEF pharmacological agents will prove beneficial in the advancement of pharmacotherapy in HF (Figure 1). Metabolic modulation has shown a promising and favourable effect profile in Phase 2 randomized controlled trials (RCTs) performed by our group in patients with HFrEF (Lee et al., 2005) and hypertrophic cardiomyopathy (Abozguia et al., 2010). However, to date, there have been no Phase 3 trials. A meta‐analysis assessing the addition of trimetazidine to conventional therapy in patients with HFrEF demonstrated a reduction in hospitalization for cardiac causes (RR: 0.43, P = 0.03) and improved symptomatology and parameters of cardiac functioning, but had no effect on all‐cause mortality (Zhang et al., 2012). Our group and others have also investigated nitrite, which has been shown to be a hypoxic‐dependent vasodilator, significantly improving cardiac haemodynamics without causing symptomatic hypotension (Maher et al., 2008). Nitrite has also been shown to exhibit additional benefits of metabolic modulation within skeletal muscle (Bailey et al., 2009), which may translate to cardiac muscle. Herein, we will review the significant evidence base behind the current medical treatments for HFrEF, the physiology of metabolic impairment in HF and discuss two promising novel agents, perhexiline and nitrite.
Figure 1.
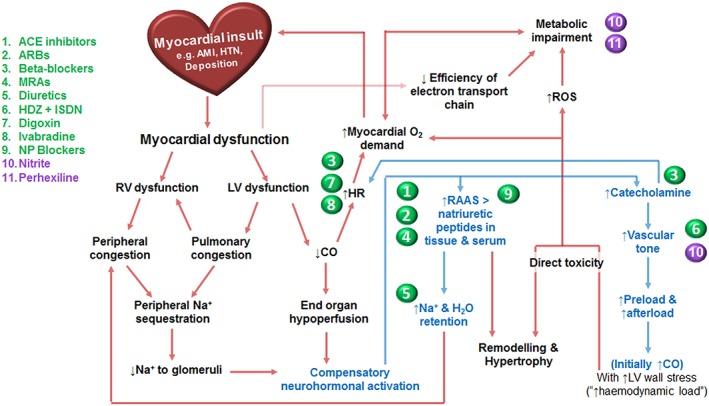
Overview of compensatory mechanisms in HF and complementary pharmacotherapeutic agents. Compensatory mechanisms and key pathophysiological changes take place in HF. Multiple pharmacotherapeutic agents have been developed to target these pathways and improve disease burden in HFrEF. ACE, angiotensin converting enzyme; AMI, acute myocardial infarction; ARBs, angiotensin receptor blockers; CO, cardiac output; H2O, water; HDZ, hydralazine; HR, heart rate; HTN, hypertension; ISDN, isosorbide dinitrate; LV, left ventricle; MRAs, mineralocorticoid receptor antagonists; Na, sodium; NP, neprilysin; RAAS, renin–angiotensin–aldosterone system; ROS, reactive oxygen species; RV, right ventricle.
ESC guidelines
The ESC guidelines recommend approved agents for use in HFrEF based on robust clinical evidence (Table 1). Pharmacotherapeutic agents with class I (strong evidence of benefit) recommendations include β‐blockers and ACE inhibitors [or angiotensin receptor blockers (ARBs) when ACE inhibitors are not tolerated] for all patients with HFrEF in the first instance, regardless of symptomatology. Additional agents including hydralazine and isosorbide dinitrate [in African‐American patients with New York Heart Association (NYHA) class III–IV on an ACE inhibitor and β‐blocker] and mineralocorticoid receptor antagonists [NYHA class II–IV with left ventricular ejection fraction (LVEF) ≤35%] are indicated as necessitated by ongoing NYHA class II–IV symptoms. These medications are supported by class A evidence (data from multiple RCTs). Recommendations in favour of treatment being beneficial (class IIb) have also been made by the ESC for the use of digoxin and ivabradine (if HR ≥70 bpm) in HFrEF with sinus rhythm, unless contraindicated, to decrease hospitalizations (McMurray et al., 2012).
Table 1.
Pivotal clinical trials for pharmacotherapeutic agents approved for the treatment of HFrEF
| HFrEF agent | Example | Relevant trials | Number of patients | Relative risk | Reference | |
|---|---|---|---|---|---|---|
| All‐cause mortality (95% CI) | Heart failure hospitalizations (95% CI) | |||||
| Angiotensin converting enzyme (ACE) inhibitors | Enalapril | CONSENSUS trial | 2289 | 0.73 (0.63–0.84, P < 0.00002) | 0.73 (N/A, P = 0.0001) | CONSENSUS Trial Study Group, 1987 |
| SOLVD trial | 2569 | 0.84 (0.74–0.95, P = 0.0036) | 0.74 (0.66–0.72, P < 0.0001) ǂ | The SOLVD Investigators, 1992 | ||
| Captopril | SAVE trial | 2231 | 0.81 (0.68–0.97, P = 0.019) | 0.78 (0.63–0.96, P = 0.019) | Pfeffer et al., 1992 | |
| Ramipril | AIRE trial | 2006 | 0.73 (0.60–0.89, P = 0.002) | N/A | The AIRE Study Group, 1993 | |
| Angiotensin receptor blockers (ARBs) | Candesartan | CHARM‐alternative trial | 2028 | 0·80 (0·66–0·96, P = 0.02) | 0·61 (0·51–0·73, P < 0.0001) | Granger et al., 2003 |
| Valsartan | Val‐HeFT trial (added to ACE‐Is) | 5010 | 1.02 (0.88–1.1, P = 0.80) § | 0.87 (0.77–0.97, P = 0.009) * , ǂ | Cohn and Togononi, 2001 | |
| VALIANT trial (non‐inferiority) | 14 703 | 1.00 (0.90–1.11, P = 0.98) * , ¥ | 0.97 (0.90–1.05, P = 0.51) ǂ , ¥ | Pfeffer et al., 2003 | ||
| Mineralocorticoid receptor antagonists | Spironolactone | RALES trial | 1663 | 0.70 (0.60–0.82, P < 0.001) | 0.65 (0.54–0.77, P < 0.001) | Pitt et al., 1999 |
| Eplerenone | EMPHASIS‐HF trial | 2737 | 0.76 (0.62–0.93, P = 0.008) | 0.58 (0.47–0.70, P < 0.001) | Zannad et al., 2011 | |
| β‐Blockers | Bisoprolol | CIBIS‐II trial | 2647 | 0.66 (0.54–0.81, P < 0.0001) | 0·64 (0·53–0·79, P = 0.001) | CIBIS‐II Study Group, 1999 |
| Metoprolol | MERIT‐HF trial | 3991 | 0.66 (0.53–0.81, P = 0.0062) | 0.65 (N/A) | MERIT‐HF Study Group, 1999 | |
| Carvedilol | COPERNICUS trial | 2289 | 0.65 (0.52–0.81, P = 0.00013) | 0.72 (N/A, P = 0.0001) | Packer et al., 2002 | |
| Nebivolol | SENIORS trial | 2128 | 0.88 (0.71–1.08, P = 0.21) | 0.86 (0.74–0.99, P = 0.039) ǂ | Flather et al., 2005 | |
| Hydralazine and isosorbide dinitrate | Hydralazine and ISDN | V‐HeFT I | 642 | 0.66 (0.46–0.96, P < 0.028) | N/A | Cohn et al., 1986 |
| A‐HeFT | 1050 | 0.57 (N/A, P = 0.01) | 0.77 (N/A, P = 0.001) | Taylor et al., 2004 | ||
| Cardiac glycosides | Digoxin | Digitalis Investigation Group trial | 6800 | 0.99 (0.91–1.07, P = 0.80) | 0.72 (0.66–0.79, P < 0.001) | The Digitalis Investigation Group, 1997 |
| Ivabradine | Ivabradine | The BEAUTIFUL trial | 10 917 | 1·04 (0·92–1·16, P = 0.55) | 0·99 (0·86–1·13, P = 0.85) | Fox et al., 2008 |
| The SHIFT trial | 6558 | 0·90 (0·80–1·02, P = 0.092) | 0.74 (0.66–0.83, P < 0.0001) | Swedberg et al., 2010 | ||
aire, the Acute Infarction Ramipril Efficacy; concensus, the Cooperative North Scandinavian Enalapril Survival Study; EMPHASIS‐HF, the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure; HFrEF, Heart Failure with Reduced Ejection Fraction; N/A, Not Available; RALES, Randomized Aldactone Evaluation Study; SAVE, Survival and Ventricular Enlargement; SOLVD, Studies of Left Ventricular Dysfunction; VALIANT, Valsartan in Acute Myocardial Infarction; Val‐HeFT, Valsartan Heart Failure Trial.
Includes death and hospitalization.
Confidence interval of 97.5%.
Confidence interval of 98%
Compared with the Captopril treatment group.
Evidence‐based pharmacological agents
ACE inhibitors
Diminishing sodium delivery and arterial pressure to the renal glomeruli in patients with HF activates the renin–angiotensin–aldosterone system (RAAS), which is further augmented by sympathetic activation. This compensatory mechanism causes potent vasoconstriction (with an increase in total peripheral resistance and therefore afterload) and salt and water retention to increase circulatory volume (Furberg and Yusuf, 1985). ACE is a common link between the RAAS and kallikrein–kinin pathways, converting angiotensin I to the salt‐retentive, vasoconstrictive and hypertrophic angiotensin II, and degrading the vasodilatory and salt‐wasting bradykinin (Brown and Vaughan, 1998). It is now recognized that RAAS activation incorporates a tissue component (particularly cardiac, vascular and renal) that is independent from the systemic endocrine effects (Dzau and Re, 1994). In the compensated state of HF, plasma renin and ACE levels have been shown to fall in a trend towards normal (Dzau and Hirsch, 1990). Despite this trend, elevations in tissue RAAS persist, indicating its critical importance in cardiovascular disease states. Components of the RAAS at a tissue level act in a paracrine or autocrine manner to produce many tissue‐specific effects, including increased vascular tone and cardiac remodelling and fibrosis (Paul et al., 2006). Indeed, amelioration of chronic tissue RAAS activation is probably more important in the long‐term benefits of neurohormonal blockade than the haemodynamic effects of systemic RAAS blockade (Dzau and Hirsch, 1990).
Several influential RCTs investigating ACE inhibitors in HFrEF demonstrated reductions in mortality in the enalapril treatment groups compared with placebo of up to 31% (P = 0.001) (The CONSENSUS Trial Study Group, 1987; The SOLVD Investigators, 1992). Similar mortality benefits were later demonstrated for captopril and ramipril in the Survival And Ventricular Enlargement Trial and The Acute Infarction Ramipril Efficacy Study trials in 1992 and 1993, respectively (Pfeffer et al., 1992; The AIRE Study Group, 1993). In 1992, the Studies of Left Ventricular Dysfunction Prevention trial further demonstrated reduced HF incidence and cardiovascular mortality with enalapril, in patients with asymptomatic HFrEF (Cleland et al., 2006).
Angiotensin receptor blockers
Angiotensin receptor blockers have similar therapeutic applications to ACE inhibitors in the setting of HF. They are useful as a monotherapy, provide an alternative option for individuals who are intolerant of ACE inhibitors (due to cough or angioedema) and may offer an augmented clinical effect in combination with ACE inhibitors (likely from a more extensive blockade of the RAAS axis). By competitively inhibiting angiotensin II at the angiotensin II type 1 (AT1) receptor directly, ARBs do not cause an elevation in bradykinin nor the attendant side effect profile. The most compelling data for ARBs versus placebo was presented by the Candesartan in Heart Failure ‐ Assessment of Mortality and Morbidity (CHARM) Program trials, the Valsartan Heart Failure Trial in 2001 and the Valsartan in Acute Myocardial Infarction trial in 2003. The CHARM Program trials demonstrated a significant reduction in cardiovascular death or hospital admission for HF in the candesartan group compared with placebo of 30% (95% CI 19–40%; P < 0.0001) in 2028 patients with symptomatic HFrEF who were not receiving ACE inhibitors due to previous intolerance (Granger et al., 2003; McMurray et al., 2003). The Valsartan Heart Failure Trial demonstrated a 24% (13.8% vs. 18.2%, P < 0.001) reduction in hospitalizations for worsening HF in the treatment group compared with placebo. However, 93% of study patients were also treated with ACE inhibitors, which is probably responsible for the lack of observed effect on mortality (Cohn and Tognoni, 2001). The Valsartan in Acute Myocardial Infarction trial was a three‐arm RCT of 14 808 patients comparing titrated valsartan, captopril and valsartan/captopril combination therapy in patients within 0.5–10 days of an acute myocardial infarction complicated by HFrEF and demonstrated non‐inferiority of valsartan to captopril (Pfeffer et al., 2003).
Mineralocorticoid receptor antagonists
Direct blockade of aldosterone at the distal nephron promotes fluid loss and retention of potassium ions (otherwise lost as a result of the action of loop diuretics) and further amelioration of systemic and chronic tissue RAAS activation. The two main trials to investigate the role of aldosterone antagonists in patients with HFrEF were the Randomized Aldactone Evaluation Study 1996 and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure 2011 studies. These RCTs compared spironolactone versus placebo in 1663 NYHA III–IV patients and eplerenone versus placebo in 2737 NYHA II patients, respectively on standard medical therapy. They demonstrated a 30% reduction in mortality in the spironolactone arm versus placebo at 24 months (95%CI 18–40%; P < 0.001) (Pitt et al., 1999) and a risk reduction of 37% in composite cardiovascular death and hospitalization for HF in the eplerenone group versus placebo at 21 months (95%CI 26–46%; P < 0.001) (Zannad et al., 2011) respectively. A further analysis of 261 patients from the Randomized Aldactone Evaluation Study revealed that high baseline levels of biomarkers of cardiac fibrosis (secondary to higher tissue RAAS activity) correlated with poorer prognosis, and the observed benefit from spironolactone was greater in such patients (Zannad et al., 2000).
β‐blockers
Sympathetic neural activation in cardiac failure is a key compensatory mechanism to counteract falling cardiac output and end‐organ hypoperfusion. Chronically, however, elevated catecholamines are deleterious, with pro‐arrhythmogenic effects [with the attendant decrease in coronary blood flow and increased myocardial O2 demand (Rona, 1985)] and direct cardiotoxic effects via cAMP‐mediated calcium overload, particularly influx through the verapamil‐sensitive calcium channel, which is pro‐apoptotic (Mann et al., 1992). These effects are ameliorated by β‐Adrenoceptors antagonists, a heterogeneous group of pharmacotherapeutic agents. These agents vary greatly in their selectivity for the adrenoceptor subtypes and may possess ancillary properties (e.g. the antioxidant and anti‐inflammatory properties of carvedilol that are theoretically of great importance in HF); however, their impact on mortality appears to be a class‐wide effect (Chatterjee et al., 2013).
The four cornerstone trials assessing β‐blockers in HF were the The Cardiac Insufficiency Bisoprolol Study II (CIBIS‐II; 1999), Metoprolol cr/xl Randomized Intervention Trial in Congestive Heart Failure (MERIT‐HF; 1999), Carvedilol Prospective Randomized Cumulative Survival Study (COPERNICUS; 2002) and the Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with heart failure (SENIORS; 2005) studies. The MERIT‐HF and CIBIS‐II trials in 1991 assessed controlled release metoprolol versus placebo in 3991 HFrEF patients and bisoprolol versus placebo in 2647 HFrEF patients, respectively (MERIT‐HF; The CIBIS‐II Study Group, 1999). Both trials demonstrated an impressive 34% reduction in mortality (95% CI 19–46%, P < 0.0001 and 95% CI 19–47%, P = 0.0062 respectively) in the treatment arm compared with placebo. The COPERNICUS trial assessed carvedilol versus placebo in 2289 patients with severe HF and LVEF ≤25% and demonstrated a 35% (95% CI 19–48%, P = 0.0014) reduction in mortality in the treatment group compared with placebo (Packer et al., 2002). A significant mortality benefit in 2128 patients >70 years of age was shown in the SENIORS trial in 2005, the first trial to specifically target HFrEF patients of advanced age. In the nebivolol treatment arm compared with placebo, there was a reduction in the composite primary outcome of all‐cause mortality and cardiovascular hospitalizations of 14% (95%CI 1–26%, P = 0.039) (Flather et al., 2005). This improvement in the composite outcome was driven mainly by a reduction in cardiovascular hospitalizations, with no effect on all‐cause mortality.
Diuretics
The careful balance between fluid retention following chronic neurohormonal activation and compensatory natriuretic mechanisms is precarious and may be affected by the slightest changes in homeostasis. Fluid overload causes pulmonary congestion (in left heart failure) and gastrointestinal, hepatic and peripheral congestion (in right heart failure). This leads to the often debilitating and unacceptable symptomatology of the HF syndrome. Indeed, the evidence for the use of diuretics in HF is geared for the treatment of these symptoms and is experience‐based, with a paucity of robust clinical data in relation to improvements in disease mortality (Faris et al., 2002). Loop diuretics are the most efficacious and potent in alleviating symptoms, both in acute decompensation and chronic disease, but as with all diuretics, they must be carefully titrated against hypokalaemia (excluding potassium sparing), symptomatic hypotension and renal decline (a significant cause of mortality in patients who die from worsening HF (Sarraf et al., 2009).
Hydralazine and isosorbide dinitrate
The effect of reducing both resistance vessel and venous tone on the failing heart via the simultaneous use of the arterial vasodilator hydralazine and the arteriolar and venodilator isosorbide dinitrate (ISDN) was assessed in the Vasodilator Heart Failure trials (V‐HeFT I/II) and the African‐American Heart Failure Trial in 1986, 1991 and 2004 respectively. The V‐HeFT I trial compared hydralazine and ISDN versus prazosin with placebo in 642 men with impaired systolic function and demonstrated a 34% relative risk reduction at 2 years (P < 0.028) (Cohn et al., 1986). V‐HeFT II was undertaken in 1991 to compare this combination with and without enalapril, with significant mortality benefit seen in the enalapril arm (Cohn et al., 1991). Hydralazine and ISDN in combination was also shown to reduce nitrate tolerance by previously unknown mechanisms. Organic nitrates activate NADPH oxidase, which results in deleterious ROS generation, partially contributing to nitrate tolerance via endothelial dysfunction (uncoupling of NOS) and inhibition of mitochondrial aldehyde dehydrogenase 2 (responsible for a large part of organic nitrate enzymatic bioactivation to NO) (Munzel et al., 2005). It seems fortuitous that hydralazine was used in addition to organic nitrates, as evidence later emerged that it prevents NADPH oxidase activation, reducing nitrate toxicity, tolerance and free radical production (Munzel et al., 1996). Given the mortality benefit, hydralazine and ISDN may provide a reasonable alternative to ACE inhibitors in patients who cannot tolerate ACE inhibitors or ARBs. More specifically, African‐American populations, in whom low‐renin, volume‐expansive hypertension is a common comorbid cardiovascular condition (Cook, 1988), the use of augmenters of ACE‐inhibitor action is encouraged to reduce the very high doses of ACE inhibitors otherwise required. The African‐American Heart Failure Trial randomly assigned 1050 Black patients with NYHA III/IV HF on standard medical therapy (including ACE inhibitors) to hydralazine and ISDN versus placebo. There was a significant decrease in all‐cause mortality in the treatment arm (P = 0.02) at 3 years (Taylor et al., 2004). Despite the compelling data for the use of organic nitrates in HFrEF, the use of nitrates in HFpEF has been relatively untested until recently, with many clinicians prescribing them to increase exercise tolerance and activity levels. The Nitrate's Effect on Activity Tolerance in Heart Failure trial, a multicentre, double‐blind crossover study of isosorbide mononitrate in HFpEF, demonstrated that, in fact, patients in the treatment arm trended towards being less active and had no beneficial increase in exercise capacity compared with placebo (Redfield et al., 2015).
Cardiac glycosides
Positive inotropic effects combined with vagally mediated negatively chronotropic effects present a favourable profile for the use of cardiac glycosides. Outside of their use in managing tachyarrhythmias however, there has been no convincing effect on cardiovascular mortality in HF demonstrated. The role of digoxin in HF remained heatedly controversial given the risks for toxicity, despite over 200 years of clinical experience, until more recently, and the argument has been left largely unresolved. The largest study to look at the use of digoxin in HF patients was the Digitalis Investigation Group trial in 1997. This multicentre double‐blind trial assessed digoxin versus placebo in 6800 patients with a LVEF ≤45% on conventional medical therapy including diuretics and ACE inhibitors. All‐cause mortality was not significantly affected; however, fewer patients were hospitalized for worsening HF with a risk reduction of 28% (95% CI 21–34%, P < 0.001) (The Digitalis Investigation Group, 1997). A post hoc analysis of the Digitalis Investigation Group trial demonstrated a correlation between serum digoxin concentrations and crude all‐cause mortality, with significantly higher mortality in the toxic range and no benefit outside of the target range of 0.5 to 0.8 ng⋅mL−1 (Rathore et al., 2003), stressing the importance of regular plasma monitoring. A randomized, placebo‐controlled trial investigating the addition of digoxin to conventional therapy is ongoing (EudraCT:2007‐006660‐30).
Ivabradine
A resting heart rate ≥70 bpm in patients with HFrEF has been shown to have a negative impact on cardiovascular outcomes (Fox et al., 2008). Ivabradine is a selective If current inhibitor in the sinoatrial node, causing exclusively negative chronotropic effects. Ivabradine has therefore also found a role in the clinical setting for patients in whom β‐blockade is not tolerated or is contraindicated. The morbidity‐mortality evaluation of the If inhibitor ivabradine in patients with coronary disease and left ventricular dysfunction trial (BEAUTIFUL; 2008) and systolic heart failure treatment with the If inhibitor ivabradine trial (SHIFT; 2010) studies were pivotal trials underpinning the evidence for ivabradine in HF. The BEAUTIFUL trial assessed 10 917 HFrEF patients with stable coronary artery disease and a resting heart rate ≥60 bpm in sinus rhythm despite maximally tolerated β‐blockade. This trial failed to demonstrate a reduction in mortality, but suggested a trend towards benefit in patients with resting HR ≥70 bpm (Fox et al., 2008). In 2010, The SHIFT trial randomized 6558 patients with HFrEF who were in sinus rhythm with HR ≥70 bpm and had been admitted to hospital for HF within the previous year to receive ivabradine or placebo. Ivabradine reduced both the risk of death due to HF compared with placebo and hospital admissions for worsening HF by 26% (P < 0.0001) (Swedberg et al., 2010). These promising data have been challenged however, as despite 90% of patients receiving β‐blockers, only 26% of patients were successfully titrated to full doses, and up to 40% of patients were not managed with a mineralocorticoid receptor antagonist, suggesting sub‐optimal concomitant conventional medical therapy (Teerlink, 2010). Indeed, in a recent and much larger study of 19 102 patients with stable coronary artery disease without clinical evidence of HF, targeted heart rate reduction to a mean of 61 bpm conferred no demonstrable benefit in the treatment arm, and in fact, a conflicting increase was observed in the composite primary endpoint of cardiovascular death and non‐fatal acute myocardial infarction in patients with severe activity‐limiting angina (Fox et al., 2014). The debate on the role of ivabradine in cardiovascular disease is therefore ongoing.
Neprilysin inhibition
Heart failure stimulates the compensatory natriuretic peptide system, which is composed of atrial natriuretic peptide, brain natriuretic peptide (BNP), C‐type natriuretic peptide and other vasoactive substances such as angiotensin II and bradykinin (Vardeny et al., 2014). Neprilysin degrades these vasoactive peptides, and so targeted inhibitors of neprilysin increase circulating levels of these substances and counteract the effects of neurohormonal over‐activation. Sole inhibitors of neprilysin, however, failed to demonstrate a significant impact on blood pressure in hypertensive cohorts and so were discontinued (McDowell and Nicholls, 1999). This may have been in part due to an attendant increase in angiotensin, necessitating the combination of neprilysin inhibitors with drugs that target the RAAS. Omapatrilat, the first dual neprilysin and ACE inhibitor, was shown to be superior to enalapril alone (Kostis et al., 2004). Unfortunately, there was a significant increase in the incidence of angioedema in the treatment arm (likely to be due to the substantial inhibition of bradykinin degradation), and the drug was discontinued. LCZ696, a combination of sacubitril (neprilysin inhibitor) and valsartan (ARB), as previously stated, showed a similar reduction in all‐cause mortality compared with an ACE inhibitor combination, with better safety data due to an attenuated effect on bradykinin breakdown (Packer et al., 2015).
Metabolic dysfunction in heart failure
Normal cardiac metabolism
The continuous and coordinated contractile activity of cardiac myocytes requires the consumption of vast amounts of energy, and this is reflected in the observation of tremendous mitochondrial density on histology (Goffart et al., 2004). Fatty acid (FA) metabolism predominates in adult healthy cardiac myocytes (around 70%), which is complemented by carbohydrate oxidation (20%) and, to a lesser extent, contribution from ketones and various amino acids (Stanley et al., 2005). However, the healthy heart is a metabolic ‘omnivore’, which is able to adjust substrate utilization according to substrate availability (Singh et al., 2014). Glucose enters the myocyte through glucose transporters (predominantly GLUT4). Glucose also undergoes a series of metabolic conversions in the cytosol known as glycolysis, resulting in the production of pyruvate, which is actively transported across the mitochondrial membrane where the pyruvate dehydrogenase (PDH) enzyme complex catalyses its conversion to acetyl CoA. PDH is the rate‐limiting step in carbohydrate metabolism and is negatively regulated by kinases and positively regulated by phosphatases (Doenst et al., 2013). Hypoxia potently inhibits PDH; therefore in these circumstances pyruvate is converted to lactate (anaerobic glycolysis). PDH is also subject to marked allosteric regulation, with acetyl CoA and NAD+ inhibiting PDH via kinase activation.
Long‐chain fatty acids enter the myocyte via fatty acid transporters and undergo esterification to long‐chain fatty acyl CoA. These are actively transported into the mitochondria via the ‘carnitine shuttle’ (van der Vusse et al., 2000). The enzyme carnitine palmitoyltransferase type 1 (CPT1) catalyses the addition of a carnitine group to the fatty acyl CoA molecule, facilitating transfer across the mitochondrial membrane where the enzyme CPT2 cleaves the carnitine (which is exported back across the mitochondrial membrane). In the mitochondria, the fatty acyl CoA undergoes β‐oxidation, a four‐step process generating acetyl CoA, NADH and flavin adenine dinucleotide (FADH2). Acetyl CoA arising from both fuel sources enters the tricarboxylic acid (TCA) cycle to produce NADH (Schwarz et al., 2014). High‐energy electrons donated from NADH and FADH2 (derived from succinate dehydrogenase, another component of the TCA cycle) are transferred from complex to complex of the electron transport chain to power extrusion of hydrogen ions across the membrane to generate an electrochemical gradient. This ultimately powers phosphorylation of ADP to ATP by ATP synthase within the mitochondrial inner membrane, with oxidation of the hydrogen ions to water (Figure 2). ATP within the mitochondria donates high‐energy phosphate to creatine catalysed by mitochondrial creatine kinase (CK), which is transported to the sites of energy utilization within the cytosol (Neubauer, 2007). Here, phosphocreatine (PCr) phosphorylates ADP to ATP (catalysed by cytosolic CK) to power the multitude of myocardial sarcomeres and various ion channels.
Figure 2.
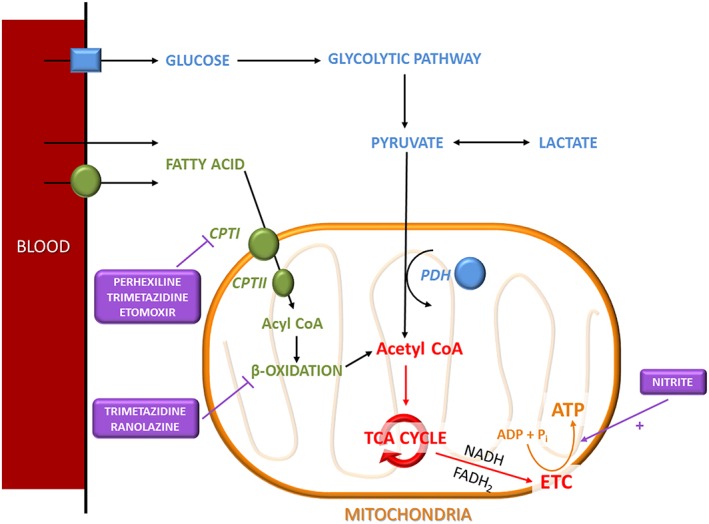
Schematic of the key metabolic pathways in cardiac myocytes. Fatty acid and glucose metabolism are key metabolic pathways within cardiac myocytes that are responsible for generating large amounts of ATP. Perhexiline and nitrite are therapeutic agents that have the ability to modulate and enhance cardiac metabolism. Acetyl CoA, acetyl coenzyme A; ADP, adenosine diphosphate; ATP, adenosine triphosphate; CPT, carnitine palmitoyltransferase; ETC, electron transport chain; FADH2, flavin adenine dinucleotide; NADH, nicotine adenine dinucleotide; Pi, inorganic phosphate; TCA, tricarboxylic acid.
The high‐energy nature of the electrons utilized in oxidative phosphorylation results in the formation of harmful reactive oxygen species (ROS) (Schwarz et al., 2014), even in physiological states. Complex homeostatic mechanisms, including NADPH from the pentose phosphate pathway (PPP), exist to manage low levels of ROS and protect cardiac myocytes from detrimental mitochondrial DNA damage and subsequent activation of apoptotic pathways. Uncoupling proteins [both uncoupling protein (UCP) and adenine nucleotide translocase (ANT)] facilitate proton leak back into the mitochondria resulting in reduced ROS generation but also less ATP production. The activity of UCPs and ANT are increased by ROS (Schwarz et al., 2014).
Metabolic changes in heart failure
Given the high‐energy consumption within the normal heart, it is appropriate to define HF as a condition in which metabolic impairment is a predominant feature. A decrease in ATP production (with resultant energetic starvation) resulting from impaired generation from glucose and FA and activation of energy‐consuming biosynthetic and redox‐stress pathways are mechanisms by which this metabolic shift occurs (Doenst et al., 2013). Creatine delivery and CK activity and flux are also reduced in HF (impairing ATP delivery to sarcomeres and ion channels necessary for pump function and cardiac homeostasis), which causes a reduction in the energy charge within the cytosol, as reduced delivery of ATP reduces the phosphorylation potential of ADP (given by Gibbs Law [ATP]/[ADP] [Pi]) (Siddiqi et al., 2013). These complex changes in the HF population are inconsistent across the aetiologies of HF, however, they become more uniform across groups at the onset of systolic dysfunction. A mechanistic study of 14 patients with aortic stenosis demonstrated a reduced PCr to ATP ratio on magnetic resonance spectroscopy in patients with symptoms of HF compared with those without (1.1 vs. 1.56, P < 0.0035) (Conway et al., 1991). PCr to ATP ratio has been well validated as a marker of cardiac metabolic status and efficiency (Beadle and Frenneaux, 2010).
The reduction in ATP production in HF is complex. Catecholaminergic stimulation in HF results in activation of lipolytic pathways and an increase in plasma‐free FAs. As a result, FA accumulation occurs within cardiac myocytes and mitochondria. Down‐regulation of enzymes for β‐oxidation occurs due to a decrease in activity of the PPARα pathway, which is a transcriptional regulator of FA use (Doenst et al., 2013). Decreased FA uptake and oxidation (with preferential utilization of carbohydrates) have been observed in patients with dilated cardiomyopathy and symptoms of HF compared with healthy controls (Neglia et al., 2007). In a further molecular study analysing human left ventricular biopsy tissue, total expression and mRNA levels of medium‐chain and long‐chain acyl CoA dehydrogenase (involved in FA β‐oxidation) were down‐regulated by >40% (P < 0.05) in patients with HF compared with age‐matched controls (Sack et al., 1996). A ‘funnelling’ effect occurs due to reduced β‐oxidation and increased mitochondrial FA in HF, and FAs begin to react with ROS within mitochondria to form lipid peroxides. Lipid peroxides damage mitochondrial DNA (hence impairing their ATP‐production capacity) and other cellular structures and activate MAPK pathways, triggering adverse remodelling and fibrosis (Siddiqi et al., 2013). Lipid peroxides also increase the activation of UCPs and ANT on the mitochondrial membrane, a protective mechanism allowing the mitochondrion to transport them back into the cytosol (Schrauwen and Hesselink, 2004). Unfortunately, this allows for proton leak at the electron transport chain and reduces the efficiency of ATP production.
Glucose oxidation is also impaired in patients with HFrEF. There is profound cardiac insulin resistance in HF, with decreased insulin‐dependent glucose uptake, but normal or increased insulin‐independent glucose uptake. G6P (formed by phosphorylation of glucose on entry into the cardiac myocyte) is also shunted down other metabolic pathways that do not produce ATP (Doenst et al., 2013). The hexosamine biosynthesis pathway, a growth and protein synthesis pathway, is increased in HF and contributes to cardiac hypertrophy and remodelling. The PPP is also up‐regulated to produce increased levels of NADPH, which are required for anti‐oxidative defence at low levels of ROS (Burgoyne et al., 2012), but may contribute to oxidative stress when ROS levels are high. G6P will also enter the glycolytic pathway to produce pyruvate. In HF, there is impaired oxidation of pyruvate to acetyl CoA by the PDH complex, reducing the amount available for entry into the TCA cycle. A high‐salt diet‐induced HF model in rats demonstrated a reduction in GLUT1 and PDH expression (Kato et al., 2010), and this was replicated in an in vivo thoracic aortic constriction HF murine model (Dai et al., 2013). PDH activity also appears to be reduced in HF resulting in impaired carbohydrate oxidation despite the availability of pyruvate (Singh et al., 2014). Pyruvate levels available for oxidation are also reduced in HF, as it is shunted along anaplerotic pathways (without ATP production) to replenish amino acids within the TCA cycle that have been utilized in hypertrophic growth and remodelling. The anaplerosis of pyruvate is enhanced in the presence of NADPH (derived from the up‐regulated PPP) and may consume large amounts of NADPH and impair anti‐oxidative defence (Doenst et al., 2013). The changes in cardiac metabolism culminate in hypertrophy and fibrosis reduced availability of ATP and high ROS levels, which further worsen the structural changes and energetic starvation.
Metabolic modulators
Glucose metabolism is more efficient than FA metabolism (in terms of oxygen requirement), and therefore, shifting energy production towards this system (generally through direct or indirect PDH complex activation) in patients with cardiovascular disease has been suggested in order to correct metabolic impairment and reduce ROS production (Lopaschuk et al., 2002). Various metabolic modulators exist, including perhexiline, trimetazidine, ranolazine and etomoxir. Of these agents, perhexiline has shown great promise due to its high potency and well‐documented anti‐ischaemic properties (Horowitz and Chirkov, 2010).
Perhexiline was widely used throughout the 1970s for the treatment of refractory angina pectoris (Ashrafian et al., 2007). It was later revealed that perhexiline improved cardiac function and myocardial efficiency by down‐regulating CPT1 activity, the rate‐limiting enzyme required for transport of FAs across the mitochondrial membrane (Kennedy et al., 1996). This in turn prevents mitochondrial FA build up, limiting harmful ROS production and subsequent energetic impairment. Recently, it has also been proposed that perhexiline may directly increase activity of the PDH complex (Yin et al., 2013). This improved glucose metabolism at low oxygen tensions improves metabolic efficiency and, when used at earlier stages of cardiovascular disease, may slow progression. Despite these promising findings, the use of perhexiline drastically declined during the 1980s due to several adverse effects. These ranged from minor nausea and lethargy to debilitating peripheral neuropathy and hepatotoxicity (Shah et al., 1982). Severe toxicity, however, results from direct inhibition of liver and brain isoforms of the CPT1 enzyme, which may be avoided by tightly monitoring plasma concentrations within a target therapeutic range of 0.5–2.2 μM (Horowitz et al., 1986).
In a cohort study involving elderly patients aged >75 years with inoperable aortic stenosis, treatment with perhexiline improved NYHA functional class (P < 0.01), with an 80% 12‐month actuarial survival rate (Unger et al., 1997). In an RCT performed by our group involving 46 patients with hypertrophic cardiomyopathy treated with perhexiline or placebo for a mean of 4.6 months, improved cardiac energetics were demonstrated by improved PCr/ATP ratio (1.27 to 1.73, P = 0.03) and NYHA class (P < 0.001) in the perhexiline arm (Abozguia et al., 2010). A further RCT by us focusing on patients with HFrEF has also shown significant improvements in peak exercise oxygen consumption (16.1 ± 0.6 vs. 18.8 ± 1.1 mL⋅kg−1⋅min−1, P < 0.001) and LVEF (24% vs. 34%, P < 0.001) following treatment with perhexiline (Lee et al., 2005).
Trimetazidine is a weak CPT1 inhibitor (Kennedy and Horowitz, 1998) with potent inhibitory effects on the long‐chain 3‐ketoacyl‐CoA thiolase enzyme, which is involved in the final steps of FA β‐oxidation (Kantor et al., 2000). Similarly to perhexiline, trimetazidine is an effective antianginal and has been shown to increase time to exercise tolerance in combination with metoprolol (420 ± 108 s in the placebo group to 485 ± 122 s in the treatment arm, P < 0.05) in patients with chronic stable angina (Trimetazidine in Poland II study trial) (Szwed et al., 2001). Trimetazidine was further shown to improve left ventricular end‐systolic volume (98 ± 1.36 mL vs. 81 ± 27 mL, P = 0.04), with a corresponding increase in ejection fraction (36 ± 7% vs. 43 ± 10%, P = 0.002) (Fragasso et al., 2006).
Ranolazine has also been found to have antianginal effects and holds a similar chemical structure to trimetazidine (Horowitz and Chirkov, 2010). The exact mechanism of action remains unknown; however, in a global ischaemia rat model, ranolazine was found to partially inhibit FA oxidation and stimulate glucose oxidation (McCormack et al., 1996). These metabolic changes were shown to have therapeutic effects in a three‐group (placebo vs. one of two doses of ranolazine), parallel, double‐blind placebo‐controlled trial involving 823 chronic stable angina sufferers on background antianginal therapy (Combination Assessment of Ranolazine in Stable Angina Study trial). The treatment groups experienced an increased time to angina on exercise and could exercise for longer compared with the placebo arm (Chaitman et al., 2004). Nitroglycerin use also decreased in the treatment arm by one use per week vs. placebo (P < 0.02). In a recent, small proof‐of‐concept RCT, ranolazine was further shown to improve haemodynamics [with decreases in left ventricular end‐diastolic pressure (P = 0.04) and pulmonary capillary wedge pressure (P = 0.04) in patients with HFpEF (Maier et al., 2013)].
Etomoxir also inhibits the CPT1 enzyme, although to a lesser extent than perhexiline (Luiken et al., 2009). In a failing rat‐heart model, perfusion with etomoxir improved left ventricular haemodynamics and indices of myocardial performance (P < 0.05) (Turcani and Rupp, 1997). A small first‐in‐human study of etomoxir in 10 HF patients of non‐ischaemic origin demonstrated haemodynamic improvement in HFrEF, with an increase in LVEF (21.5 ± 2.6% to 27.0 ± 2.3%, P < 0.01) and exercise cardiac output (from 9.72 ± 1.25 L⋅min−1 to 13.44 ± 1.50 L⋅min−1, P < 0.01) (Schmidt‐Schweda and Holubarsch, 2000). The long‐term safety of etomoxir, however, has been questioned due to a recent clinical trial involving 260 patients with moderate HF, which was halted early due to four patients developing significant hepatic transaminitis (Holubarsch et al., 2007).
A novel therapeutic pathway: nitrate–nitrite–NO
The nitrate–nitrite–NO pathway has been suggested as an alternative mechanism for systemic NO production. This is an alternative mechanism to the classical pathway in which NO is produced by oxidation of L‐arginine in a reaction catalysed by nitric oxide synthase (NOS) (Lundberg et al., 2008). NO generated through the nitrate–nitrite–NO pathway has been suggested to involve a series of nitric oxide synthase (NOS)‐independent and oxygen‐independent reactions. However, the precise mechanism(s) for nitrite conversion to NO remains to be fully elucidated because a number of enzymes have been implicated in the catalysis of nitrite to NO in various tissue compartments (Lundberg et al., 2008). Green leafy vegetables (such as spinach and beetroot) are the major source of dietary inorganic nitrate. Once consumed, the salivary inorganic nitrate is reduced to nitrite by commensal bacteria. Nitrite is then swallowed, absorbed from the stomach and reduced to NO, particularly under hypoxic conditions, by a number of nitrite reductases such as myoglobin, haemoglobin and xanthine oxidoreductase (Gladwin and Kim‐Shapiro, 2008; Webb et al., 2008). NO may then directly modify proteins [particularly via S‐nitrosylation of cysteine residues, which may enhance or dampen protein activity (Foster et al., 2003)] or, more classically, activate soluble guanylate cyclase within vascular smooth muscle cells and platelets to increase cGMP to exert its vasodilator and platelet‐inhibitory effects. Phosphodiesterase is pivotal in homeostatic control over cellular cGMP and cAMP activity; thus, targeted inhibition of cardiac and smooth muscle isoforms of phosphodiesterase have been shown to elicit vasodilatation and attenuate cardiac hypertrophy (Guazzi, 2008). A placebo‐controlled RCT involving 23 stable HF patients treated with the PDE5 inhibitor sildenafil for 6 months demonstrated an improvement in pulmonary artery systolic pressure (33.7 to 23.9 mmHg, P < 0.01) and aerobic efficiency and exercise ventilation (peak VO2 14.8 to 18.7 mL⋅min−1⋅kg−1, P < 0.01) (Guazzi et al., 2007). These promising data are yet to be explored in larger clinical trials.
Exploitation of the NO pathway has been further attempted with other stimulators and activators of soluble guanylate cyclase including riociguat, vericiguat and cinaciguat. Recently, the Soluble Guanylate Cyclase Stimulator in Heart Failure Study (in patients with LVEF <45%) trial, a randomized‐controlled, multicentre double‐blind trial to assess the tolerability and optimal dose of vericiguat in 456 patients with HFrEF, confirmed tolerability but failed to show a significant decrease in NT‐proBNP in the treatment groups (the primary endpoint). There was, however, a demonstrable dose–response relationship for the reduction in NT‐proBNP (P < 0.02), which was promising and requires further elucidation (Gheorghiade et al., 2015).
The nitrate–nitrite–NO pathway has been shown to regulate blood pressure and hypoxic vasodilatation and improve exercise performance and mitochondrial efficiency (Bailey et al., 2014). Our work and those of others have demonstrated that infusions of sodium nitrite in healthy subjects dilate forearm vasculature, with significant potentiation during hypoxia, thereby preferentially dilating capacitance vessels (Maher et al., 2008; van Faassen et al., 2009; Totzeck et al., 2012; Kapil et al., 2014). As such, these haemodynamic effects have sparked interest in inorganic nitrate (to increase serum nitrite) as a novel oral therapeutic intervention, particularly in cardiovascular diseases such as HFrEF. We have recently shown in a first‐in‐human HF efficacy/safety study that sodium nitrite infusion (50 μg⋅kg−1⋅min−1) in patients with severe HF resulted in an increase in ventricular stroke volume (43.22 ± 21.5 to 51.84 ± 23.6 mL, P = 0.003), with marked falls in pulmonary vascular resistance (29%; P = 0.03) and right atrial pressure (40%; P = 0.007), but only modest falls in mean arterial blood pressure (4 mm Hg; P = 0.004). The increase in stroke volume correlated (r = 0.67; P = 0.003) with an increase in estimated trans‐septal gradient (= pulmonary capillary wedge pressure − right atrial pressure), suggesting relief of diastolic ventricular interaction as a contributory mechanism (Ormerod et al., 2015).
Nitrite has also been shown to improve the metabolic efficiency of skeletal muscle. This may be partially due to its vasodilator properties, but it has also been shown to have direct effects on key metabolic components (Clerc et al., 2007; Basu et al., 2008). The Karolinska group showed in a double‐blind crossover trial in healthy volunteers that oral inorganic nitrate supplementation improves oxidative phosphorylation efficiency in skeletal muscle mitochondria. An analysis of mitochondrial function revealed decreased expression of ANT and decreased mitochondrial membrane uncoupling (Larsen et al., 2011). It has also been proposed that under hypoxic conditions, nitrite may act as the terminal electron acceptor in place of oxygen (Basu et al., 2008). Given the underlying metabolic impairment in HF and the potential benefit of metabolic modulation, determining whether changes observed in skeletal muscle translates to cardiac muscle would be of great benefit, but remains yet unclear. Zamani et al. (2015) reported in a double‐blind, placebo‐controlled crossover study that nitrate rich beetroot juice (12.9 mmol) in patients with HFpEF increased exercise cardiac output and improved peak VO2 compared with nitrate‐deplete placebo (Zamani et al., 2015). These new studies provide evidence of promising beneficial effects in patients with HF, and phase 2 studies for longer treatment regimens are underway (ISRCTN16356908; NCT02256345; ACTRN12615000906550; ACTRN12613000689774; NCT02401126).
Conclusion
Classic treatments for HF have resulted in significant improvements in disease morbidity and mortality over the last 20 years. Despite this, cohorts of patients remain who cannot tolerate maximal up‐titration of traditional treatment modalities or remain symptomatic despite them. With rapid improvements in cardiac devices, we may be entering the surgical age of HF treatment, but such interventions remain expensive and inaccessible to many. Novel pharmacotherapeutic agents, such as perhexiline and nitrite, may provide pharmacological alternatives to traditional treatments and fill the therapeutic gap, but further definitive trials are warranted.
Author contributions
B.L.L., H.N. and N.D.G. contributed to writing the article. M.P.F. and M.M. contributed to article drafting and revision. M.M. authorized the final version of the review.
Conflict of interest
M.P.F. is the inventor for a method‐of‐use patent for the use of perhexiline in heart failure and cardiomyopathies. B.L.L., H.N., N.D.G. and M.M. have no conflict of interest to report.
Acknowledgements
This study is supported by the UK Medical Research Council [G1001536].
Loudon, B. L. , Noordali, H. , Gollop, N. D. , Frenneaux, M. P. , and Madhani, M. (2016) Present and future pharmacotherapeutic agents in heart failure: an evolving paradigm. British Journal of Pharmacology, 173: 1911–1924. doi: 10.1111/bph.13480.
References
- Abozguia K, Elliott P, Mckenna W, Phan TT, Nallur‐Shivu G, Ahmed I et al. (2010). Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 122: 1562–1569. [DOI] [PubMed] [Google Scholar]
- AIRE Study Group (1993). Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Lancet 342: 821–828. [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafian H, Horowitz JD, Frenneaux MP (2007). Perhexiline. Cardiovasc Drug Rev 25: 76–97. [DOI] [PubMed] [Google Scholar]
- Bailey JC, Feelisch M, Horowitz JD, Frenneaux MP, Madhani M (2014). Pharmacology and therapeutic role of inorganic nitrite and nitrate in vasodilatation. Pharmacol Ther 144: 303–320. [DOI] [PubMed] [Google Scholar]
- Bailey SJ, Winyard P, Vanhatalo A, Blackwell JR, Dimenna FJ, Wilkerson DP et al. (2009). Dietary nitrate supplementation reduces the O2 cost of low‐intensity exercise and enhances tolerance to high‐intensity exercise in humans. J Appl Physiol 107: 1144–1155. [DOI] [PubMed] [Google Scholar]
- Basu S, Azarova NA, Font MD, King SB, Hogg N, Gladwin MT et al. (2008). Nitrite reductase activity of cytochrome c. J Biol Chem 283: 32590–32597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle R, Frenneaux M (2010). Magnetic resonance spectroscopy in myocardial disease. Expert Rev Cardiovasc Ther 8: 269–277. [DOI] [PubMed] [Google Scholar]
- Braunwald E, Bristow MR (2000). Congestive heart failure: fifty years of progress. Circulation 102: IV14–IV23. [DOI] [PubMed] [Google Scholar]
- Brown NJ, Vaughan DE (1998). Angiotensin‐converting enzyme inhibitors. Circulation 97: 1411–1420. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Mongue‐Din H, Eaton P, Shah AM (2012). Redox signaling in cardiac physiology and pathology. Circ Res 111: 1091–1106. [DOI] [PubMed] [Google Scholar]
- Chaitman BR, Pepine CJ, Parker JO, Skopal J, Chumakova G, Kuch J et al. (2004). Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA 291: 309–316. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Biondi‐Zoccai G, Abbate A, D'ascenzo F, Castagno D, Van Tassell B et al. (2013). Benefits of beta blockers in patients with heart failure and reduced ejection fraction: network meta‐analysis. BMJ 346: f55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT (2003). Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐alpha, in patients with moderate‐to‐severe heart failure: results of the anti‐TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 107: 3133–3140. [DOI] [PubMed] [Google Scholar]
- CIBIS‐II Study Group (1999). The Cardiac Insufficiency Bisoprolol Study II (CIBIS‐II): a randomised trial. Lancet 353: 9–13. [PubMed] [Google Scholar]
- Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J (2006). The Perindopril in Elderly People with Chronic Heart Failure (PEP‐CHF) study. Eur Heart J 27: 2338–2345. [DOI] [PubMed] [Google Scholar]
- Clerc P, Rigoulet M, Leverve X, Fontaine E (2007). Nitric oxide increases oxidative phosphorylation efficiency. J Bioenerg Biomembr 39: 158–166. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Archibald DG, Ziesche S, Franciosa JA, Harston WE, Tristani FE et al. (1986). Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med 314: 1547–1552. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F et al. (1991). A comparison of enalapril with hydralazine‐isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 325: 303–310. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Tognoni G (2001). A randomized trial of the angiotensin‐receptor blocker valsartan in chronic heart failure. N Engl J Med 345: 1667–1675. [DOI] [PubMed] [Google Scholar]
- CONSENSUS Trial Study Group (1987). Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 316: 1429–1435. [DOI] [PubMed] [Google Scholar]
- Conway MA, Allis J, Ouwerkerk R, Niioka T, Rajagopalan B, Radda GK (1991). Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 338: 973–976. [DOI] [PubMed] [Google Scholar]
- Cook CA (1988). Pathophysiologic and pharmacotherapy considerations in the management of the black hypertensive patient. Am Heart J 116: 288–295. [DOI] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L et al. (2013). Global proteomics and pathway analysis of pressure‐overload‐induced heart failure and its attenuation by mitochondrial‐targeted peptides. Circ Heart Fail 6: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digitalis Investigation Group (1997). The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 336: 525–533. [DOI] [PubMed] [Google Scholar]
- Doenst T, Nguyen TD, Abel ED (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113: 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzau VJ, Hirsch AT (1990). Emerging role of the tissue renin‐angiotensin systems in congestive heart failure. Eur Heart J 11 (Suppl B): 65–71. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Re R (1994). Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation 89: 493–498. [DOI] [PubMed] [Google Scholar]
- Faris R, Flather M, Purcell H, Henein M, Poole‐Wilson P, Coats A (2002). Current evidence supporting the role of diuretics in heart failure: a meta analysis of randomised controlled trials. Int J Cardiol 82: 149–158. [DOI] [PubMed] [Google Scholar]
- Flather MD, Shibata MC, Coats AJ, Van Veldhuisen DJ, Parkhomenko A, Borbola J et al. (2005). Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 26: 215–225. [DOI] [PubMed] [Google Scholar]
- Follath F, Cleland JG, Klein W, Murphy R (1998). Etiology and response to drug treatment in heart failure. J Am Coll Cardiol 32: 1167–1172. [DOI] [PubMed] [Google Scholar]
- Foster MW, Mcmahon TJ, Stamler JS (2003). S‐nitrosylation in health and disease. Trends Mol Med 9: 160–168. [DOI] [PubMed] [Google Scholar]
- Fox K, Ford I, Steg PG, Tardif JC, Tendera M, Ferrari R (2014). Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med 371: 1091–1099. [DOI] [PubMed] [Google Scholar]
- Fox K, Ford I, Steg PG, Tendera M, Ferrari R (2008). Ivabradine for patients with stable coronary artery disease and left‐ventricular systolic dysfunction (BEAUTIFUL): a randomised, double‐blind, placebo‐controlled trial. Lancet 372: 807–816. [DOI] [PubMed] [Google Scholar]
- Fragasso G, Palloshi A, Puccetti P, Silipigni C, Rossodivita A, Pala M et al. (2006). A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol 48: 992–998. [DOI] [PubMed] [Google Scholar]
- Furberg CD, Yusuf S (1985). Effect of vasodilators on survival in chronic congestive heart failure. Am J Cardiol 55: 1110–1113. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP et al. (2015). Effect of Vericiguat, a Soluble Guanylate Cyclase Stimulator, on Natriuretic Peptide Levels in Patients With Worsening Chronic Heart Failure and Reduced Ejection Fraction: The SOCRATES‐REDUCED Randomized Trial. JAMA 314: 2251–2262. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Kim‐Shapiro DB (2008). The functional nitrite reductase activity of the heme‐globins. Blood 112: 2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffart S, Von Kleist‐Retzow JC, Wiesner RJ (2004). Regulation of mitochondrial proliferation in the heart: power‐plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res 64: 198–207. [DOI] [PubMed] [Google Scholar]
- Granger CB, Mcmurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B et al. (2003). Effects of candesartan in patients with chronic heart failure and reduced left‐ventricular systolic function intolerant to angiotensin‐converting‐enzyme inhibitors: the CHARM‐Alternative trial. Lancet 362: 772–776. [DOI] [PubMed] [Google Scholar]
- Greenberg BH, Chou W, Saikali KG, Escandon R, Lee JH, Chen MM et al. (2015). Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail 3: 22–29. [DOI] [PubMed] [Google Scholar]
- Guazzi M (2008). Clinical use of phosphodiesterase‐5 inhibitors in chronic heart failure. Circ Heart Fail 1: 272–280. [DOI] [PubMed] [Google Scholar]
- Guazzi M, Samaja M, Arena R, Vicenzi M, Guazzi MD (2007). Long‐term use of sildenafil in the therapeutic management of heart failure. J Am Coll Cardiol 50: 2136–2144. [DOI] [PubMed] [Google Scholar]
- Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen‐Tervaert JW et al. (2009). Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 11: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs FD, Roalfe AK, Davis RC, Davies MK, Hare R (2007). Prognosis of all‐cause heart failure and borderline left ventricular systolic dysfunction: 5 year mortality follow‐up of the Echocardiographic Heart of England Screening Study (ECHOES). Eur Heart J 28: 1128–1134. [DOI] [PubMed] [Google Scholar]
- Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P et al. (2007). A double‐blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) 113: 205–212. [DOI] [PubMed] [Google Scholar]
- Horowitz JD, Chirkov YY (2010). Perhexiline and hypertrophic cardiomyopathy: a new horizon for metabolic modulation. Circulation 122: 1547–1549. [DOI] [PubMed] [Google Scholar]
- Horowitz JD, Sia ST, Macdonald PS, Goble AJ, Louis WJ (1986). Perhexiline maleate treatment for severe angina pectoris–correlations with pharmacokinetics. Int J Cardiol 13: 219–229. [DOI] [PubMed] [Google Scholar]
- Kantor PF, Lucien A, Kozak R, Lopaschuk GD (2000). The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long‐chain 3‐ketoacyl coenzyme A thiolase. Circ Res 86: 580–588. [DOI] [PubMed] [Google Scholar]
- Kapil V, Weitzberg E, Lundberg JO, Ahluwalia A (2014). Clinical evidence demonstrating the utility of inorganic nitrate in cardiovascular health. Nitric Oxide 38: 45–57. [DOI] [PubMed] [Google Scholar]
- Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y et al. (2010). Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail 3: 420–430. [DOI] [PubMed] [Google Scholar]
- Kennedy JA, Horowitz JD (1998). Effect of trimetazidine on carnitine palmitoyltransferase‐1 in the rat heart. Cardiovasc Drugs Ther 12: 359–363. [DOI] [PubMed] [Google Scholar]
- Kennedy JA, Unger SA, Horowitz JD (1996). Inhibition of carnitine palmitoyltransferase‐1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol 52: 273–280. [DOI] [PubMed] [Google Scholar]
- Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E (2004). Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens 17: 103–111. [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Schiffer TA, Borniquel S, Sahlin K, Ekblom B, Lundberg JO et al. (2011). Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab 13: 149–159. [DOI] [PubMed] [Google Scholar]
- Lee L, Campbell R, Scheuermann‐Freestone M, Taylor R, Gunaruwan P, Williams L et al. (2005). Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short‐term use of a novel treatment. Circulation 112: 3280–3288. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Rebeyka IM, Allard MF (2002). Metabolic modulation: a means to mend a broken heart. Circulation 105: 140–142. [PubMed] [Google Scholar]
- Luiken JJ, Niessen HE, Coort SL, Hoebers N, Coumans WA, Schwenk RW et al. (2009). Etomoxir‐induced partial carnitine palmitoyltransferase‐I (CPT‐I) inhibition in vivo does not alter cardiac long‐chain fatty acid uptake and oxidation rates. Biochem J 419: 447–455. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT (2008). The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167. [DOI] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA et al. (2008). Hypoxic modulation of exogenous nitrite‐induced vasodilation in humans. Circulation 117: 670–677. [DOI] [PubMed] [Google Scholar]
- Maier LS, Layug B, Karwatowska‐Prokopczuk E, Belardinelli L, Lee S, Sander J et al. (2013). RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI‐DHF proof‐of‐concept study. JACC Heart Fail 1: 115–122. [DOI] [PubMed] [Google Scholar]
- Mancini D, Colombo PC (2015). Left ventricular assist devices: a rapidly evolving alternative to transplant. J Am Coll Cardiol 65: 2542–2555. [DOI] [PubMed] [Google Scholar]
- Mann DL, Kent RL, Parsons B, Cooper GT (1992). Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 85: 790–804. [DOI] [PubMed] [Google Scholar]
- Mann DL, Mcmurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS et al. (2004). Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 109: 1594–1602. [DOI] [PubMed] [Google Scholar]
- Massie BM, Carson PE, Mcmurray JJ, Komajda M, Mckelvie R, Zile MR et al. (2008). Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 359: 2456–2467. [DOI] [PubMed] [Google Scholar]
- Mccormack JG, Barr RL, Wolff AA, Lopaschuk GD (1996). Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 93: 135–142. [DOI] [PubMed] [Google Scholar]
- Mcdowell G, Nicholls DP (1999). The endopeptidase inhibitor, candoxatril, and its therapeutic potential in the treatment of chronic cardiac failure in man. Expert Opin Investig Drugs 8: 79–84. [DOI] [PubMed] [Google Scholar]
- Mcmurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K et al. (2012). ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 33: 1787–1847. [DOI] [PubMed] [Google Scholar]
- Mcmurray JJ, Ostergren J, Swedberg K, Granger CB, Held P, Michelson EL et al. (2003). Effects of candesartan in patients with chronic heart failure and reduced left‐ventricular systolic function taking angiotensin‐converting‐enzyme inhibitors: the CHARM‐Added trial. Lancet 362: 767–771. [DOI] [PubMed] [Google Scholar]
- Merit‐Hf Study Group (1999). Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF). Lancet 353: 2001–2007. [PubMed] [Google Scholar]
- Munzel T, Daiber A, Mulsch A (2005). Explaining the phenomenon of nitrate tolerance. Circ Res 97: 618–628. [DOI] [PubMed] [Google Scholar]
- Munzel T, Kurz S, Rajagopalan S, Thoenes M, Berrington WR, Thompson JA et al. (1996). Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane‐bound NADH oxidase. A new action for an old drug. J Clin Invest 98: 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C et al. (2007). Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 293: H3270–H3278. [DOI] [PubMed] [Google Scholar]
- Neubauer S (2007). The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151. [DOI] [PubMed] [Google Scholar]
- Ormerod JO, Arif S, Mukadam M, Evans JD, Beadle R, Fernandez BO et al. (2015). Short‐term intravenous sodium nitrite infusion improves cardiac and pulmonary hemodynamics in heart failure patients. Circ Heart Fail 8: 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H et al. (2002). Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106: 2194–2199. [DOI] [PubMed] [Google Scholar]
- Packer M, Mcmurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR et al. (2015). Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 131: 54–61. [DOI] [PubMed] [Google Scholar]
- Paul M, Poyan Mehr A, Kreutz R (2006). Physiology of local renin‐angiotensin systems. Physiol Rev 86: 747–803. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ Jr, Cuddy TE et al. (1992). Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med 327: 669–677. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Mcmurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP et al. (2003). Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 349: 1893–1906. [DOI] [PubMed] [Google Scholar]
- Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B et al. (2014). Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 370: 1383–1392. [DOI] [PubMed] [Google Scholar]
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A et al. (1999). The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341: 709–717. [DOI] [PubMed] [Google Scholar]
- Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM (2003). Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 289: 871–878. [DOI] [PubMed] [Google Scholar]
- Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C et al. (2000). Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 102: 3060–3067. [DOI] [PubMed] [Google Scholar]
- Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH et al. (2015). Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N Engl J Med 373: 2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rona G (1985). Catecholamine cardiotoxicity. J Mol Cell Cardiol 17: 291–306. [DOI] [PubMed] [Google Scholar]
- Sack MN, Rader TA, Park S, Bastin J, Mccune SA, Kelly DP (1996). Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94: 2837–2842. [DOI] [PubMed] [Google Scholar]
- Sarraf M, Masoumi A, Schrier RW (2009). Cardiorenal syndrome in acute decompensated heart failure. Clin J Am Soc Nephrol 4: 2013–2026. [DOI] [PubMed] [Google Scholar]
- Schmidt‐Schweda S, Holubarsch C (2000). First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (Lond) 99: 27–35. [PubMed] [Google Scholar]
- Schrauwen P, Hesselink MK (2004). The role of uncoupling protein 3 in fatty acid metabolism: protection against lipotoxicity? Proc Nutr Soc 63: 287–292. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Siddiqi N, Singh S, Neil CJ, Dawson DK, Frenneaux MP (2014). The breathing heart – mitochondrial respiratory chain dysfunction in cardiac disease. Int J Cardiol 171: 134–143. [DOI] [PubMed] [Google Scholar]
- Shah AM, Mann DL (2011). In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JD (1982). Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. Br Med J (Clin Res Ed) 284: 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi N, Singh S, Beadle R, Dawson D, Frenneaux M (2013). Cardiac metabolism in hypertrophy and heart failure: implications for therapy. Heart Fail Rev 18: 595–606. [DOI] [PubMed] [Google Scholar]
- Singh S, Schwarz K, Horowitz J, Frenneaux M (2014). Cardiac energetic impairment in heart disease and the potential role of metabolic modulators: a review for clinicians. Circ Cardiovasc Genet 7: 720–728. [DOI] [PubMed] [Google Scholar]
- SOLVD Investigators (1992). Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 327: 685–691. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–1129. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost‐Brama A et al. (2010). Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 376: 875–885. [DOI] [PubMed] [Google Scholar]
- Szwed H, Sadowski Z, Elikowski W, Koronkiewicz A, Mamcarz A, Orszulak W et al. (2001). Combination treatment in stable effort angina using trimetazidine and metoprolol: results of a randomized, double‐blind, multicentre study (TRIMPOL II). TRIMetazidine in POLand. Eur Heart J 22: 2267–2274. [DOI] [PubMed] [Google Scholar]
- Taylor AL, Ziesche S, Yancy C, Carson P, D'agostino R Jr, Ferdinand K et al. (2004). Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 351: 2049–2057. [DOI] [PubMed] [Google Scholar]
- Teerlink JR (2010). Ivabradine in heart failure–no paradigm SHIFT…yet. Lancet 376: 847–849. [DOI] [PubMed] [Google Scholar]
- Tilemann L, Ishikawa K, Weber T, Hajjar RJ (2012). Gene therapy for heart failure. Circ Res 110: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre‐Amione G, Anker SD, Bourge RC, Colucci WS, Greenberg BH, Hildebrandt P et al. (2008). Results of a non‐specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): a placebo‐controlled randomised trial. Lancet 371: 228–236. [DOI] [PubMed] [Google Scholar]
- Totzeck M, Hendgen‐Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ et al. (2012). Nitrite regulates hypoxic vasodilation via myoglobin‐dependent nitric oxide generation. Circulation 126: 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcani M, Rupp H (1997). Etomoxir improves left ventricular performance of pressure‐overloaded rat heart. Circulation 96: 3681–3686. [DOI] [PubMed] [Google Scholar]
- Unger SA, Robinson MA, Horowitz JD (1997). Perhexiline improves symptomatic status in elderly patients with severe aortic stenosis. Aust N Z J Med 27: 24–28. [DOI] [PubMed] [Google Scholar]
- Van Der Vusse GJ, Van Bilsen M, Glatz JF (2000). Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 45: 279–293. [DOI] [PubMed] [Google Scholar]
- Van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim‐Shapiro DB et al. (2009). Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev 29: 683–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeny O, Miller R, Solomon SD (2014). Combined neprilysin and renin–angiotensin system inhibition for the treatment of heart failure. JACC Heart Fail 2: 663–670. [DOI] [PubMed] [Google Scholar]
- Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ et al. (2008). Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res 103: 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Dwyer J, Langley SR, Mayr U, Xing Q, Drozdov I et al. (2013). Effects of perhexiline‐induced fuel switch on the cardiac proteome and metabolome. J Mol Cell Cardiol 55: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, Mcmurray JJ et al. (2003). Effects of candesartan in patients with chronic heart failure and preserved left‐ventricular ejection fraction: the CHARM‐preserved trial. Lancet 362: 777–781. [DOI] [PubMed] [Google Scholar]
- Zamani P, Rawat D, Shiva‐Kumar P, Geraci S, Bhuva R, Konda P et al. (2015). Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation 131: 371–380 ; discussion 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannad F, Alla F, Dousset B, Perez A, Pitt B (2000). Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investig Circ 102: 2700–2706. [DOI] [PubMed] [Google Scholar]
- Zannad F, Mcmurray JJ, Krum H, Van Veldhuisen DJ, Swedberg K, Shi H et al. (2011). Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 364: 11–21. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lu Y, Jiang H, Zhang L, Sun A, Zou Y et al. (2012). Additional use of trimetazidine in patients with chronic heart failure: a meta‐analysis. J Am Coll Cardiol 59: 913–922. [DOI] [PubMed] [Google Scholar]