

Abstract
Background and purpose
The reliable assessment of proarrhythmic risk of compounds under development remains an elusive goal. Current safety guidelines focus on the effects of blocking the KCNH2/HERG ion channel‐in tissues and animals with intact repolarization. Novel models with better predictive value are needed that more closely reflect the conditions in patients with cardiac remodelling and reduced repolarization reserve.
Experimental Approach
We have developed a model for the long QT syndrome type‐5 in rabbits (LQT5 ) with cardiac‐specific overexpression of a mutant (G52R) KCNE1 β‐subunit of the channel that carries the slow delayed‐rectifier K+‐current (IKs). ECG parameters, including short‐term variability of the QT interval (STVQT), a biomarker for proarrhythmic risk, and arrhythmia development were recorded. In vivo, arrhythmia susceptibility was evaluated by i.v. administration of the IKr blocker dofetilide. K+ currents were measured with the patch‐clamp technique.
Key Results
Patch‐clamp studies in ventricular myocytes isolated from LQT5 rabbits revealed accelerated IKs and IKr deactivation kinetics. At baseline, LQT5 animals exhibited slightly but significantly prolonged heart‐rate corrected QT index (QTi) and increased STVQT. Dofetilide provoked Torsade‐de‐Pointes arrhythmia in a greater proportion of LQT5 rabbits, paralleled by a further increase in STVQT.
Conclusion and Implications
We have created a novel transgenic LQT5 rabbit model with increased susceptibility to drug‐induced arrhythmias that may represent a useful model for testing proarrhythmic potential and for investigations of the mechanisms underlying arrhythmias and sudden cardiac death due to repolarization disturbances.
Abbreviations
- HERG
human ether‐a‐go‐go gene
- ICa,L
L‐type Ca2 + current
- IK1
inward rectifier potassium current
- IKr
rapid delayed rectifier potassium current
- IKs
slow delayed rectifier potassium current
- Ito
transient outward potassium current
- KCNE1
potassium voltage‐gated channel subfamily E member 1
- LQT5
long QT syndrome type 5
- minK
minimum sequence required for a potassium current
- QTi
heart rate‐corrected QT index
- STVQT
short‐term variability of the QT interval
- STVRR
short‐term variability of the RR interval
- TdP
Torsade‐de‐Pointes
- TG
transgenic
- WT
wild type
Tables of Links
| TARGETS |
|---|
| Voltage‐gated ion channels |
| KCNQ1/KvLQT1, Kv7.1 channels |
| LIGANDS |
|---|
| Dofetilide |
| Nisoldipine |
| HMR 1556 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
In cardiac myocytes, the ion channel that carries the slow delayed‐rectifier K+‐current (IKs) is composed of a pore‐forming α(KCNQ1/KvLQT1) and a modulatory β‐subunit (KCNE1/minK) (Barhanin et al., 1996; Sanguinetti et al., 1996. Although KCNQ1 alone forms a voltage‐gated K+‐channel, KCNE1/minK is required to reproduce the kinetic properties of native IKs (Sanguinetti et al., 1996). IKs is an important determinant of myocardial repolarization, particularly with increased adrenergic stimulation (Bennett and Begenisich, 1987; Han et al., 2001), and is a key contributor to repolarization reserve in mammalian and human myocardium (Varro et al., 2000; Jost et al., 2005; Jost et al., 2007). Repolarization reserve refers to the redundant repolarizing capacity of the myocardium, that is, with loss of function of a single K+‐current, other repolarizing currents increase to compensate (Roden, 2006, 1998; Varro and Baczko, 2011). Loss of IKs function reduces this reserve and makes the heart more vulnerable to arrhythmias (Roden, 2006; Varro and Baczko, 2011).
KCNE1, also known as minK, was the first Kv‐channel accessory subunit cloned from human hearts (Murai et al., 1989). MinK is a small (129 amino acid) protein with a single transmembrane spanning domain (Nerbonne and Kass, 2005). Mutations in either KCNQ1 or KCNE1 can alter the biophysical properties of IKs and KCNE1 mutations are known to underlie congenital long QT syndrome (LQTS) type 5 (LQT5) (Panaghie et al., 2006).
LQTS, characterized by a prolonged QT‐interval, can cause sudden death by causing a characteristic ventricular tachycardia known as Torsade‐de‐Pointes (TdP) (Lehnart et al., 2007). LQTS can be acquired, following drug treatment, or be part of an inherited syndrome (Harmer et al., 2010). Mutations in genes encoding the pore‐forming subunits of the rapid delayed‐rectifier K+‐current (IKr) and IKs account for >90% of genotyped LQTS (Moss and Kass, 2005; Nerbonne and Kass, 2005). Mutations in the KCNE1 gene are rare and account for ~3% of all LQT mutations (Splawski et al., 2000).
A complete understanding of the mechanisms by which individual mutations may lead to arrhythmias and sudden death requires the study of experimental animal models. Transgenic mouse models of LQT syndromes have several limitations (Salama and London, 2007): including heart rates 10 times faster than humans and different repolarizing currents (Nerbonne and Kass, 2005). KCNE1 knockout (Drici et al., 1998; Kupershmidt et al., 1998), knock‐in (Rizzi et al., 2008; Nishio et al., 2009) and loss‐of‐function mutations (Demolombe et al., 2001) in transgenic mouse models only partly mimic the human LQT phenotype (Salama and London, 2007). The laboratory rabbit (Oryctolagus cuniculus) has several advantages. The heart rate is slower than in mice. The larger size of rabbit hearts enables tools developed for the evaluation of human cardiac function. Moreover, rabbits have similar repolarizing ion channels to humans (Nerbonne, 2000). To date, only two transgenic LQT rabbit models, overexpressing dominant‐negative pore mutants of KvLQT1 (LQT1) or HERG channels (LQT2), have been described (Brunner et al., 2008). A novel missense mutation, which leads to a substitution of arginine (R) for glycine (G) at position 52 of the minK protein, was described in a Chinese family (Ma et al., 2003). Among seven mutation carriers, five were clinically affected; however, two family members had normal ECG (Ma et al., 2003). In Xenopus oocytes, co‐expression of the mutant G52R‐KCNE1 with KCNQ1 had a dominant‐negative effect, reducing IKs by 50% and prolonging the cardiac action potential (Ma et al., 2003). The G52R mutation did not alter channel‐subunit assembly or trafficking to the cell membrane but rendered KCNE1 unable to modulate the gating properties of KCNQ1 in CHO‐K1 cells (Harmer et al., 2010).
The present study was designed to create and characterize an LQT5 rabbit model with reduced repolarization reserve in order to model impairments of repolarization that are silent until challenged by an exogenous K+‐channel blocking drug and then potentiate proarrhythmic risk.
Methods
Human heart samples
The experimental protocol was approved by the Scientific and Research Ethical Committee of the Medical Scientific Board at the Hungarian Ministry of Health (ETT‐TUKEB: 4991–0/2010‐1018EKU), and followed the principles outlined in the Declaration of Helsinki. Samples from the lateral left ventricular wall of a non‐diseased human heart, technically unusable for transplantation, were obtained from a general organ donor and were frozen immediately in liquid nitrogen until processing.
Animals
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals (USA NIH publication NO 85–23, revised 1996) and conformed to the Directive 2010/63/EU of the European Parliament; they were approved by the Animal Care and Ethics Committee of the Agricultural Biotechnology Center and the Ethical Committee for the Protection of Animals in Research of the University of Szeged (approval number: I‐74‐5‐2012) and by the Department of Animal Health and Food Control of the Ministry of Agriculture and Rural Development (authority approval numbers 22.1/433/003/2010 and XIII/1211/2012). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and the editorial on reporting animal studies (McGrath and Lilley, 2015).
New Zealand White rabbits were obtained from Tetrabbit Kft. (Hungary). The experimental animals were young adults (approximately 3.5 kg) of either sex, unless otherwise indicated. The animals were kept in stainless steel rabbit cages containing an elevated resting place and fulfilling the requirements of authorities in terms of size and environmental enrichment. Rabbits were individually housed and were kept at standard temperature, humidity and lighting. Food and water were provided ad libitum. The water was provided in separate drinking bowls to each animal individually and the water was regularly checked for quality and any pathogens.
Collection of zygotes and laparoscopic transfer of injected embryos to recipient rabbits was performed as described earlier (Besenfelder and Brem, 1993). Pseudopregnant animals were moved to a nesting cage 3–4 days before expected delivery date. For the in vivo ECG studies, rabbits were anaesthetized with ketamine‐S/xylazine i.m. (12.5 and 3.5 mg·kg−1) or thiopental (50 mg·kg−1) in the marginal ear‐vein. At the end of the arrhythmia provocation experiments, the anaesthetized animals were killed by i.v. administration of pentobarbital sodium (RELEASE) in a dose of 300 mg·mL−1.
Transgene construct
The 4533 bp long rabbit β‐myosin heavy chain gene promoter (r‐MYH7) including three untranslated exons were amplified with the primers 5′‐ACA AAG CCC AGC TCC CTA AT‐3′(nt: 2509–2529) and 5′‐GGC TGT ACC TGT AGT GAG CG‐3′(nt: 7022–7042, Genbank Ac. No.: AF192306.1). PCR was performed with TaKaRa LA Taq polymerase. Conditions were 95°C for 1 min, 58°C for 45 s and 72°C for 5 min for 35 cycles, using New Zealand White genomic DNA as template. The PCR product was cloned into the TOPO site of the pCR®‐Blunt II‐TOPO vector with the Zero Blunt® TOPO® PCR Cloning Kit (Invitrogen™, Thermo Fischer Scientific, Waltham, MA, USA). The 410 bp human KCNE1 cDNA was isolated from a human cardiac myocyte cDNA library (Cat no: SC6204; 3H Biomedical AB, Uppsala, Sweden). The amplified KCNE1 cDNA was cloned into the PCR product at the insertion site of the pSC‐A‐amp/kan vector with the Strata Clone PCR Cloning Kit (Stratagene, an Agilent Technologies company, San Diego, CA, USA). At position 154, a guanine was changed to adenine with the Quick Change® (Agilent Technologies, San Diego, CA, USA) XL Site Directed Mutagenesis Kit, substituting an arginine for glycine at amino acid 52 (G52R‐KCNE1). The mutated KCNE1 cDNA was isolated with Eco RV and Sma I enzymes and blunt‐end cloned behind the r‐β‐MHC promoter at the EcoRV site of the pCR®‐Blunt II‐TOPO vector. The r‐β‐MHC promoter‐G52R‐KCNE1 fragment was isolated with BamHI and NotI enzymes and cloned into the pcDNA3.1 mammalian expression vector (Invitrogen) containing the bovine GH poly‐A signal. The ~5900 bp long microinjected fragment, which included the mutated human KCNE cDNA under the 4533 bp long rabbit β‐MHC promoter with three untranslated exons and the bovine GH polyA tail (MYH7‐G52R‐KCNE1‐BGHpolyA) was isolated with NheI‐SexAI digestion and purified with QIAquick gel extraction kit (Qiagen, Quiagen, Hilden, Germany, Cat no. 28704). The DNA concentration was set at 4 ng·μL−1.
Identification of transgenic animals and transgene copy number determination
Offspring derived from recipient does were screened for transgene integration by transgene‐specific PCR of genomic DNA purified from ear biopsies or blood. The forward primer complementary to r‐MYH7 (5′‐GCCTCAGCCAGAACCAGTTA‐3′; 5′‐GGC TGT ACC TGT AGT GAG CG‐3′) and the reverse oligonucleotides complementary to h‐KCNE1 (5′‐GCTTCTTGGAGCGGATGTAG‐3′; 5′‐TTA GCC AGT GGT GGG GTT CA‐3′) gave rise to a 450 bp diagnostic fragment. PCR conditions were 95°C for 30 s, 54°C for 30 s and 72°C for 45 s for 30 cycles.
Determination of the transgene integration site with nested PCR
Genomic DNA was digested with Sac I (Fermentas,Thermo Fischer Scientific, Waltham, MA, USA, Cat. No.: ER1131), and the fragments were purified and incubated with T4 ligase overnight to produce circular DNA fragments, which served as templates for nested PCR with primers designed to hybridize back to back in the 3′ sequence so that the entire circularized target fragment was amplified. Primers were: nested1‐F: CAACAGATGGCTGGCAACTA; nested1‐R: AGACAATAGCAGGCATGCTG; nested2‐F: AGGAAAGGACAGTGGGAGTG; nested2‐R: GACAGCAAGGGGGAGGAT.
Creation of transgenic LQT5 rabbits
The KCNE1 coding region of the transgene was amplified and analyzed by automated sequencing (Figure 1). The MYH7‐G52R‐KCNE1‐bGHpolyA insert was isolated and used to microinject rabbit embryos. Out of 497 injected embryos, 466 were transferred into 21 pseudopregnant recipients through laparoscopy. Thirty‐eight offspring (8%) were born alive, out of which four (10%) were transgenic by genomic PCR (Figure 2A). Three founders lived till sexual maturity. The female founder did not transmit the transgene to its offspring, suggesting that it was transgenic in somatic tissues but not the germ line. The remaining two transgenic founder males (# G52R‐JTJKK and #G52R‐JKK) transmitted the transgene to their offspring from wild‐type does at 52% (in the G52R‐JTJKK line) and 15% (in the G52R‐JKK line) ratio, respectively, which in case of the G52R‐JTJKK founder underlines a Mendelian type inheritance. For the #G52R‐JKK founder, the transgene transmission data indicate a high degree of germ‐line mosaicism. Therefore, except where it is otherwise indicated, for electrophysiological examinations, the F1 hemizygote offsprings of #G52R‐JTJKK line were used and are called transgenic animals in Table 2 and the Figures. The presence of G52R mutation was checked by sequencing the PCR product isolated from genomic DNA of the founder male #G52R‐JTJKK. In the #G52R‐JTJKK animal, the transgene was integrated at Σ 10 copies in two different loci on Chr11, namely at positions chr11:18,313,856 and chr11:41,935,523.
Figure 1.
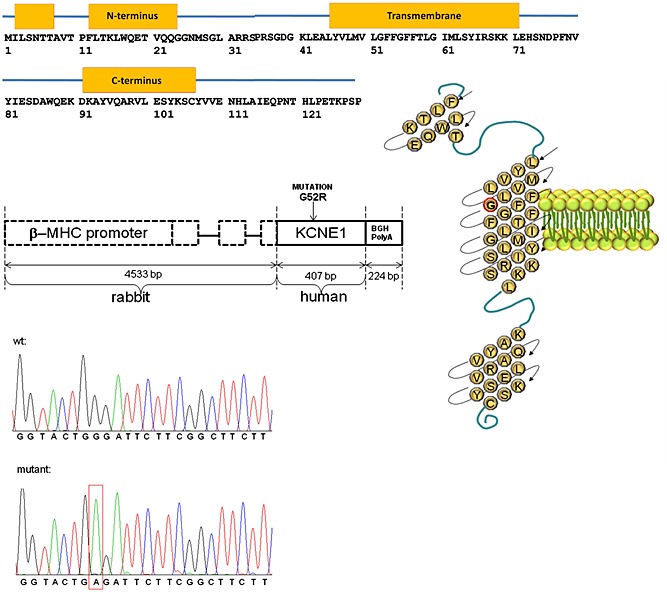
The G52R mutation and the transgene construct. Schematic drawing of the mutation in KCNE1 polypeptide (upper panel) and the transgene construct (middle panel). Mutation G to A at position 154 of human KCNE1 cDNA and wild‐type sequence. Sense‐strand sequences are shown (lower panel).
Figure 2.
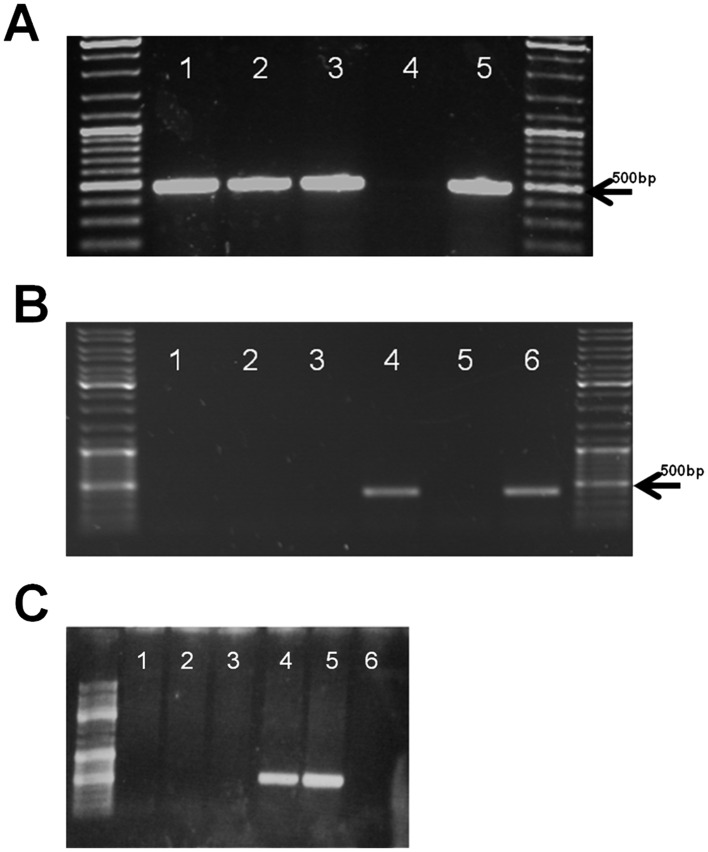
Identification of G52R founders and tissue specific transgene expression. (A) Transgene specific PCR of genomic DNA of three founders. Line 1: G52R‐JTJKK; line 2: female founder without transgenic offspring; line 3: G52R‐14‐JKK; line 4: negative control; and line 5: positive control (injected construct). (B) Expression profile of hKCNE1 specific mRNAs detected by RT‐PCR in ventricular myocardium and diverse tissues from transgenic (tr) rabbit #G52R‐JTJKK. β‐actin specific PCR reaction was carried out to control the quality of RNA and the RT step in all sets of samples (data not shown). Molecular weight marker: Fermentas Gene Ruler DNA Ladder Mix (Cat.No.: SM0331); Line 1: transgenic rabbit (tr) smooth muscle; 2: tr striated muscle; 3 tr atrial myocardium; 4: tr ventricular myocardium; 5: tr brain; 6: human ventricle. (C) Expression profile of hKCNE1 specific mRNAs detected by RT‐PCR in ventricular myocardium and diverse tissues of transgenic rabbit #G52R‐JKK. β‐actin specific PCR reaction was carried out to control the quality of RNA and the RT step in all sets of samples (data not shown). Molecular weight marker: Fermentas Gene Ruler DNA Ladder Mix (Cat.No.: SM0331). Line 1: tr smooth muscle; line 2: tr striated muscle; line 3: tr brain; line 4: tr right ventricle; line 5: tr left ventricle; line 6: tr atrium.
Table 2.
Different ECG parameters in WT and TG rabbits at baseline and following the administration of the IKr blocker dofetilide
| WT baseline | WT prior to arrhythmia | WT DOF | TG baseline | TG prior to arrhythmia | TG DOF | |
|---|---|---|---|---|---|---|
| PQ (ms) | 69.6 ± 2.83 | 70.8 ± 2.49 | 71.9 ± 3.04 | 67.9 ± 1.67 | 69.1 ± 1.48 | 71.6 ± 3.86 |
| QRS (ms) | 36.2 ± 1.28 | 39.2 ± 2.13 | 40.6 ± 3.26 | 34.6 ± 1.10 | 35.4 ± 1.71 | 40.1 ± 2.79 |
| RR (ms) | 226.6 ± 7.93 | 222.5 ± 9.61 | 228.2 ± 8.96 | 230.1 ± 5.44 | 235.8 ± 6.45 | 255.1 ± 9.78# |
| HR (1 min‐1) | 267.9 ± 9.11 | 268.9 ± 8.88 | 267.3 ± 10.1 | 262.9 ± 5.47 | 255.8 ± 6.12 | 244 ± 9.04# |
| QT (ms) | 139.3 ± 4.54 | 145.5 ± 4.77 | 159.9 ± 5.97# | 140.2 ± 2.49 | 146.7 ± 5.22 | 172.9 ± 5.24# |
| QTc (ms) | 155.8 ± 4.09 | 162.1 ± 2.81 | 175.4 ± 3.70# | 154.5 ± 2.86 | 156.3 ± 2.07 | 175.5 ± 3.63# |
Dof, measured at 10 min after the end of dofetilide infusion; HR: heart rate; TG, transgenic; WT, wild type.
P < 0.05 versus baseline in the same group
Organ‐specific expression of the transgene
One adult female and a male of #G52R‐JTJKK line and the G52R‐JKK founder and its F1 offspring were killed and samples collected to evaluate the tissue specificity of transgene expression. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen, Cat. No: 74134). A tissue sample isolated from a healthy human heart ventricle served as positive control. A total of 100 ng RNA per sample was used for RT‐PCR with High Capacity cDNA Reverse Transcription Kit (Applied Biosystem, Thermo Fischer Scientific, Waltham, MA, USA, Cat. No.: 4368814). The program for first strand synthesis was 25°C for 10 min, 37°C for 120 min and 85°C for 5 min. The human G52R‐KCNE1 specific 407 bp amplicon was produced with the primers (5′‐ATG ATC CTG TGT AAC ACC ACA GAG‐3′) (5′‐TTA GCC AGT GGT GGG GTT CA‐3′) and a 385 bp endogenous control amplicon from the rabbit α‐actin mRNA with the primer pair: (5′‐TGTGACATCGACATCAGGAAGG‐3′; nt 917–938 and 5′‐TAGGTAATGAGTCAGAGCTTTGG‐3′; nt 1277–1299; MYH6; Genbank: AF192305). Amplified products were resolved on 1% agarose gel.
The human G52R‐KCNE1 specific 407 bp amplicon was detected only in the mRNA from the ventricular part of hemizygote rabbit's heart from the two independent lines (#G52R‐JTJK and #G52R‐JKK) but was not detected either in the atrium or other examined organs of the transgenic rabbits. It was not detected in non‐transgenic, littermate rabbits (Figure 2B and C).
Quantitative RT‐PCR
Heart samples from four F1 hemizygotes (#G52R‐JTJKK line) and four littermate controls were dissected manually to 2 mm tissue sections, and the samples immediately homogenized for 1 min with Bullet Blender Storm 24 homogenizer. Total RNA was isolated from the tissue using an RNAzol RT kit (Molecular Research Center Inc., Cincinnati, OH, USA). Total RNA was quantified with a Nanodrop ND1000 spectrophotometer (Thermo Fischer Scientific, Waltham, MA, USA). First‐strand cDNA was synthesized from 100 ng RNA using Tetro cDNA Synthesis Kit (Bioline, London, UK) reverse transcriptase in a 20 μL reaction, using oligo(dT)18 priming. Aliquots of 1 μL of the resulting cDNA were used in the qPCR reactions.
The relative abundance of cDNA fragments was determined with qPCR using a Maxima SYBR green/ROX qPCR Master Mix (Thermo Scientific) and custom‐designed primers (0.3 μM final concentration) [Integrated DNA Technologies (IDT), Coralville, IA, USA] (Table 1) in 20 μL reactions. A two‐step cycling protocol was performed: initial denaturation 95°C, 10 min; denaturation 95°C,15 s; annealing/extension 60°C, 60 s. Each run consisted of 40 cycles, and each sample was run in triplicate. Gene expression levels were determined with qRT‐PCR and the Maxima SYBR green/ROX qPCR Master Mix (Thermo Scientific) with custom‐designed primers (0.3 μM final concentration, IDT for primer sequences see Table 1) in 20 μL reactions. Initial denaturation was at 95°C, 10 min; denaturation 95°C, 15 s; annealing/extension 60°C, 60 s. Each run consisted of 40 cycles, and each sample was run in triplicate. Primer specificity was verified with dissociation curve analyses. Gene expression levels were normalized to the geometric average of the expression of two housekeeping genes (RPS5 and 28S ribosomal RNA) according to (Vandesompele et al., 2002).
Table 1.
List of primers used in quantitative mRNA determination
| Gene | Origin | Primers | Annealing temperature | Reference |
|---|---|---|---|---|
| KCNE1 | rabbit | 5′‐CTACATCCGCTCCCAGAAACT‐3′ 5′‐GCTGGTTTTCAAGGACGTAGC‐3′ | 60°C | (Atkinson et al., 2011) |
| RPS5 | rabbit | 5′‐TAC ATT GCG GTG AAG GAG AA‐3′ 5′‐TCA TCA TGG AGT TGG TGA GG‐3′ | 60°C | (Atkinson et al., 2011) |
| KCNQ1 | rabbit | 5′‐AGGAGCTGATCACCACCCTGTA‐3′ 5′‐ATCTGCGTAGCTGCCGAACTC‐3′ | 60°C | (Atkinson et al., 2011) |
| MYH7 | rabbit | 5′‐GCCTCAGCCAGAACCAGTTA‐3′; 5′‐GGC TGT ACC TGT AGT GAG CG‐3′ | 60°C | This publication |
| 28S | rabbit | 5′‐GTTGTTGCCATGGTAATCCTGCTCAGTACG‐3′; 5′‐TCTGACTTAGAGGCGTTCAGTCATAATCCC‐3′ | 60°C | (Atkinson et al., 2011) |
Relative expression of transgene and endogenous mRNAs
The high expression of the human transgene led to up‐regulation of the endogenous rabbit KCNQ1 and KCNE1 mRNAs (Figure 3). The expression level of the rabbit β‐myosin heavy chain mRNA (MYH7), whose promoter was directing the G52R‐KCNE1 transgene expression, also slightly increased the endogenous mRNA levels in the transgenic rabbits compared with wild type (Figure 3).
Figure 3.
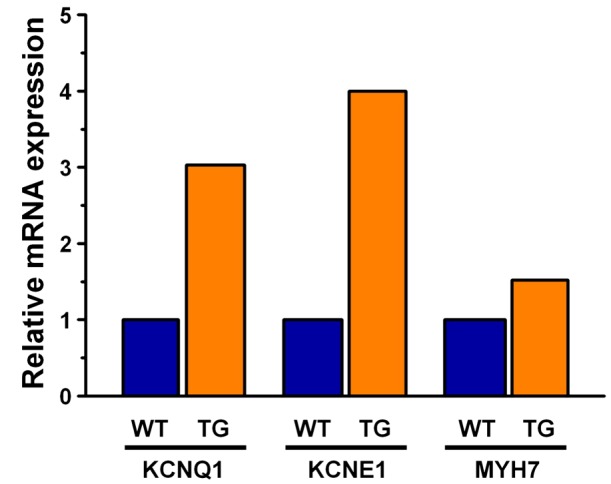
Quantitative determination of KCNE1 specific and related mRNAs in ventricular tissue of transgenic rabbits. G52R‐JTJKK and littermate controls. Relative quantities of mRNAs in the left ventricular apex of transgenic rabbit hearts. The data shown derive from the pooled cDNA of two females and two bucks from both the transgenic and the littermate control groups. The rabbits were sexually mature adults, weighed 3–3.5 kg and were 5 months old. Gene expression levels were normalized to the geometric average of the expression of two housekeeping genes (RPS5 and 28S ribosomal RNA).
Western blotting
Left ventricular tissue samples were snap‐frozen in liquid N2 and stored at −80°C. To obtain sufficient tissue for protein extraction, samples from six to seven hearts were pooled in three groups: a wild‐type (n = 7 hearts) and two transgenic groups (transgenic 1 and 2, n = 6 hearts each). Tissue samples were homogenized in liquid N2, and 200 mg was homogenized in ice‐cold lysis‐buffer (20 mM Tris‐HCl pH 7.4, 250 mM sucrose, 0.1% protease inhibitor cocktail (Sigma, St. Louis, MO, USA). After homogenization and centrifugation (2000 g, 15 min), the supernatant was centrifuged at 100 000 g (45 min), and the pellet re‐suspended in lysis buffer. Protein content was determined with the Lowry method.
Membrane protein samples were separated on a 12% polyacrylamide denaturing Tris‐glycine gel and transferred to PVDF membranes (Merck Millipore, Billerica, MA, USA). After blocking in 5% non‐fat milk, membranes were labelled overnight at 4°C with the anti‐KCNE1 antibody (raised against amino acids 67–130 of human KCNE1 (Abcam) (1:2000). The bound anti‐KCNE1 antibody was detected with HRP‐conjugated goat anti‐mouse secondary antibody (1:8000) at room temperature for 1 h. Blots were developed with ECL‐plus (GE Healthcare, Little Chalfont, UK) and developed on X‐ray films (Amersham Biosciences, GE Healthcare, Little Chalfont, UK). For loading verification, Ponceau staining was performed. Band‐intensities were measured using imagej software (NIH), and signals were normalized to corresponding Ponceau stains.
NanoPro analysis
Cell and tissue samples were lysed with RIPA lysis buffer, DMSO Inhibitor Mix and Aqueous Inhibitor Mix (Protein Simple, Wallingford, CT, USA). Protein concentration was determined by NanoDrop (NanoDrop Technologies, Thermo Fischer Scientific, Waltham, MA, USA) and diluted to 0.1 mg·mL−1. Lysates were mixed with ampholyte premix (3–10 pI) and fluorescently labelled isoelectric‐point standards for analysis (pI Standard Ladder 1) on the NanoPro100 system (Cell Biosciences, Protein Simple, Wallingford, CT, USA). The Nanopro protein data were obtained from 40 weeks old, male rabbit heart tissue samples from the same litter. The pI for rabbit KCNE1 was determined with wild‐type rabbit heart‐tissue samples. Capillary isoelectric focusing electrophoresis was carried out at 15 000 microwatts for 30 min. The separated proteins were immobilized to the capillary wall by exposing to UV light for 100 s. Immunoprobing was with the same anti‐human KCNE1 primary antibody (50‐times dilution) as for Western blotting and then probed with the HRP‐conjugated goat anti‐mouse secondary antibody (ProteinSimple). Luminol and peroxidase mixture (ProteinSimple) was added to generate chemiluminescent light, captured by a charge‐coupled device camera. The digital image was analysed with compass software (v1.8.0; Cell Biosciences).
Immunohistochemistry and confocal microscopy
Rabbit ventricular cardiomyocytes were enzymically isolated as described previously (Varro et al., 1996) and fixed with acetone. Samples were rehydrated and blocked for 1 h in PBST (PBS with 0.01% Tween) with 5% BSA (Sigma) at room temperature.
Cells were incubated for 1.5 h at room temperature with primary antibody (mouse monoclonal anti‐human KCNE1, Abcam) at 1:2000 followed by three 1 min washes with PBS and 1 h incubation with secondary antibody (fluorescein isothiocyanate (FITC)‐labelled anti‐mouse IgG) (Sigma‐Aldrich) at 1:300. Cover slips containing the samples were mounted in Aqua Poly/Mount (Polysciences Inc., Warrington, PA, USA). Fluorescent images were captured by Olympus FV1000 (Tokyo, Japan) confocal laser‐scanning microscope with identical parameter settings.
Voltage clamp measurements
Single ventricular myocytes were obtained by enzymic dissociation of isolated wild‐type and transgenic rabbit hearts. Following heparin administration (400 NE·kg−1, i.v.), sedation with xylazine (1 mg·kg−1, i.v.) and thiopental (30 mg·kg−1, i.v.)‐induced anaesthesia, each heart was rapidly removed through a right lateral thoracotomy and placed into cold (4–8°C) solution with the following composition (in mM): NaCl 135, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, HEPES 10, NaHCO3 4.4, glucose 10, taurine 20, CaCl2 1, (pH 7.2). The heart was then mounted on a modified, 60 cm high Langendorff column and perfused with oxygenated perfusate of the same composition warmed to 37°C. After 3–5 min of perfusion to flush blood from the coronary vasculature, the perfusate was switched to one having no exogenously added calcium (i.e. to one that was nominally Ca2 +‐free) until the heart ceased to contract (~8–10 min). Enzymic digestion was accomplished by perfusion with the same, nominally Ca2 +‐free solution with 290 U·mL−1 collagenase (Worthington Type 2) and 30 μM CaCl2. After 10‐15 min perfusion, the heart was removed from the aortic cannula and placed into enzyme‐free solution containing 1 mM CaCl2 warmed to 37°C for 10 min. Then, the tissue was minced into small chunks, and following gentle agitation, myocytes were separated by filtering the resulting slurry through a nylon mesh. Myocytes were finally harvested by gravity sedimentation. Once the majority of individual myocytes settled to the bottom of the container, the supernatant was decanted and replaced with Tyrode's solution, and the myocytes were resuspended by gentle agitation. This procedure was repeated twice more, and the resulting myocyte suspension was stored in HEPES‐buffered Tyrode's solution at room temperature.
One drop of cell suspension was placed within a transparent recording chamber mounted on the stage of an inverted microscope (Olympus IX51, Olympus), and individual myocytes were allowed to settle and adhere to the bottom of the chamber for at least 5 min before superfusion was initiated. HEPES‐buffered Tyrode's solution was used as the normal superfusate. This solution contained (in mM): NaCl 144, NaH2PO4 0.4, KCl 4.0, CaCl2 1.8, MgSO4 0.53, glucose 5.5, and HEPES 5.0 at pH of 7.4. Patch clamp micropipettes were fabricated from borosilicate glass capillaries using a P‐97 Flaming/Brown micropipette puller (Sutter Co., Novato, CA, USA). These electrodes had resistances between 1.5 and 2.5 MΩ after filling with pipette solution containing (in mM): KOH 110, KCl 40, K2ATP 5, MgCl2 5, EGTA 5, GTP 0.1 and HEPES 10, when measuring K+ current. The pH of the pipette solution was adjusted to 7.2 by aspartic acid.
Membrane currents were recorded with Axopatch 200B patch‐clamp amplifiers (Molecular Devices, Sunnyvale, CA, USA) using the whole cell configuration of the patch clamp technique. Membrane currents were digitized and recorded under software control (digidata 1440A, pClamp 10, Molecular Devices, Sunnyvale, CA, USA) after low‐pass filtering at 1 kHz.
The inward rectifier (IK1), transient outward (Ito), rapid (IKr) and slow (IKs) delayed‐rectifier potassium currents were recorded in rabbit ventricular myocytes. Nisoldipine, gift from Bayer AG, Leverkusen, Germany (1μM) was added to the bath solution to block the L‐type Ca2 + current (ICa,L). When IKr was recorded, IKs was inhibited by using the selective IKs blocker HMR 1556 (0.5 μM). During IKs measurements, IKr was blocked by 0.5 μM dofetilide (Sequoia Research Products Ltd., Pangbourne, UK), and the bath solution contained 0.1 μM forskolin (Sigma, St. Louis, MO, USA). All experiments were performed at 37°C.
ECG recording and evaluation
Since different types of anaesthetics can influence cardiac ion channels, rabbits were anaesthetized either with ketamine‐S/xylazine i.m. (12.5 and 3.5 mg·kg−1) or thiopental (50 mg·kg−1) in the marginal ear‐vein. A catheter was inserted into the carotid artery for blood‐pressure measurement. The right jugular vein was cannulated for i.v. drug administration.
Blood pressures and electrocardiograms were continuously recorded, digitized and stored for off‐line analysis. RR and QT intervals were measured as the average of 30 beats. For QT‐interval measurements, published guidelines were followed (Farkas et al., 2004). The frequency‐corrected QT interval (QTc) was calculated by a specific formula for thiopental‐anaesthetized rabbits (Batey and Coker, 2002): QTc = QT–[0.704 * (RR‐250)]. In ketamine/xylazine anaesthetized animals, heart rate‐corrected QT‐indices were calculated as: QTi (%) = QTmeasured * 100/QTexpected; QTexpected = 86 + 0.22 * RR) as described previously (Brunner et al., 2008).
I.v. infusions were administered with a programmable infusion pump (Terufusion TE‐3, Terumo Europe, Leuven, Belgium). Both wild‐type and transgenic rabbits received dofetilide (Gedeon Richter Ltd., Budapest, Hungary) 20 μg·kg−1 over 10 min.
Short‐term variability of RR and QT intervals
The short‐term beat‐to‐beat variability of the QT interval was calculated as a marker that reliably predicts arrhythmia‐propensity in animal‐models (Thomsen et al., 2004; Lengyel et al., 2007) and patients (Hinterseer et al., 2009, 2010).
Poincaré plots were constructed by plotting all RR and QT values against their preceding values (Figure 9A) to characterize the temporal instability of beat‐to‐beat heart rate and repolarization. In case of TdP arrhythmia, the measurements were taken prior to TdP. Beat‐to‐beat short‐term variability (STV) of RR or QT intervals was calculated as: STV = ∑ |D n+1 − D n| (30x√2)−1, with D the duration of QT or RR interval. STV represents the mean orthogonal distance to the line of identity on the Poincaré plot; the estimation of beat‐to‐beat instability of RR and QT intervals is as described previously (Brennan et al., 2001).
Figure 9.
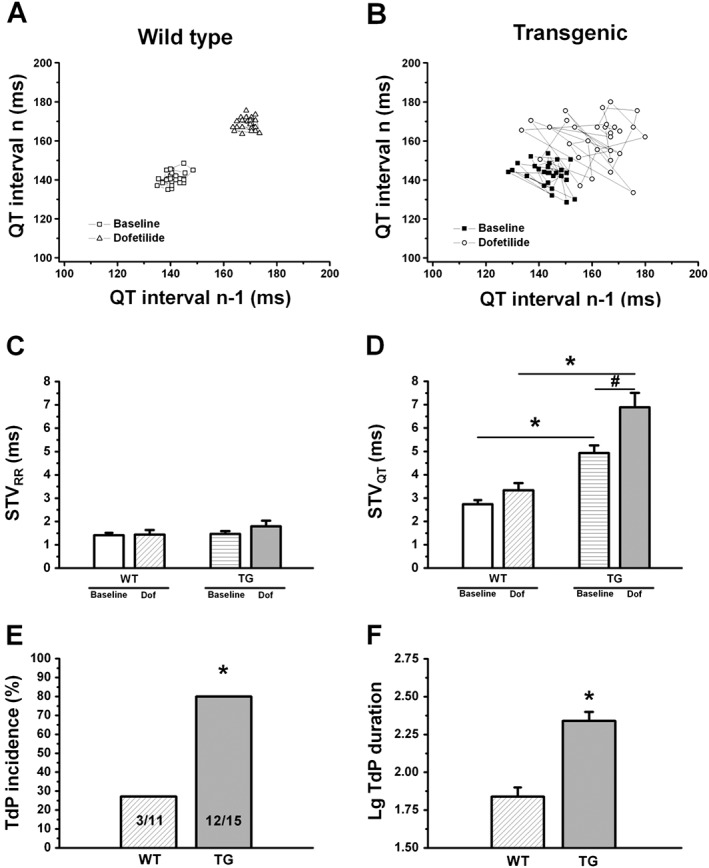
Short‐term variability of the RR and QT intervals and Torsade‐de‐Pointes in thiopental anaesthetized rabbits. (A–B) Representative Poincaré plots demonstrate higher short‐term beat‐to‐beat variability of the QT interval (STVQT) in anaesthetized transgenic (TG) rabbits at baseline conditions compared with wild‐type (WT) animals. Following the administration of the IKr blocker dofetilide, STVQT further and markedly increased only in transgenic animals. (C) There were no differences in short‐term variability of the RR interval (STVRR) between WT and TG rabbits either at baseline conditions or following the administration of the IKr blocker dofetilide. (D) Short‐term variability of the QT interval (STVQT) was higher in TG animals at baseline conditions and following dofetilide infusion, indicating increased temporal instability of repolarization in TG rabbits. (E) Accordingly, TG animals exhibited Torsade‐de‐Pointes (TdP) with significantly higher incidence. (F) The duration of TdP episodes was significantly longer in TG rabbits, expressed as the log10 of duration in seconds (to allow statistical comparison of data with normal distribution). Dof: dofetilide (20 μg·kg−1, i.v.); n = 11 and 15 animals in WT and TG groups, respectively on panels (C) to (E); n = 3 and 12 animals on panel (F); * P < 0.05 versus wild type; # P < 0.05 versus baseline in the same group.
Data and statistical analysis
All studies in this manuscript followed the editorial on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are shown as means ± SEM. Graphical and statistical data analysis was carried out using origin 8.1 (Microcal Software, Northampton, MA, USA). Results were compared using Student's t‐tests for paired and unpaired data as appropriate. The incidence of arrhythmias was compared using χ2‐test with Yates' correction. Differences were considered significant when P < 0.05.
Results
G52R‐KCNE1 localization
Histological examination indicated an absence of structural abnormalities in transgenic hearts. Ventricular cardiomyocytes from a transgenic rabbit showed intense KCNE1/minK fluorescence (Figure 4D). The membrane localization of G52R‐KCNE1 is consistent with observations that G52R‐KCNE1 assembles and traffics correctly (Harmer et al., 2010).
Figure 4.
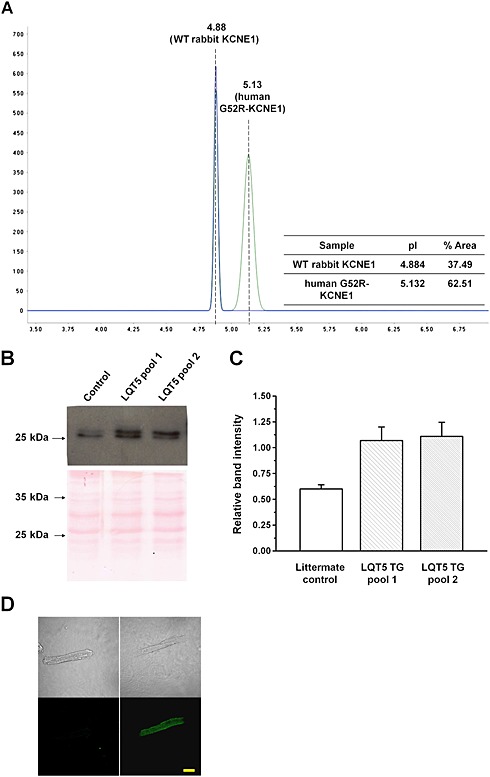
Detection and quantitation of the mutant human minK in transgenic rabbit heart. (A) Representative profile of NanoPro analysis and quantitation of the G52R mutant and wild‐type rabbit minK proteins in the F1 hemizygote rabbit heart ventricles (#G52R‐JTJKK). (B) Changes in the membrane protein content of KCNE1. Representative Western blots (upper panel) and quantitative evaluation (lower panel) of the membrane protein extracts isolated from the pooled non‐transgenic and two pools of G52R transgenic ventricles. (C) Due to the highly homologous regions of the human and rabbit KCNE1 amino acid sequences, the anti‐KCNE1 antibody detected an approximately 28 kDa protein both in the non‐transgenic littermate control and G52R transgenic rabbit heart samples. (D) Representative immunofluorescent images of rabbit ventricular cardiomyocytes, labelled with anti‐human KCNE1 antibody. Left panel shows an image of a cardiomyocyte isolated from the non‐transgenic rabbit and right panel from a #G52R‐JTJKK transgenic animal labelled with both the primary and secondary antibodies. Bar represents 25 μm.
Cellular electrophysiology
Myocytes isolated from transgenic rabbits expressed moderately but significantly larger transient outward current (Ito) than wild‐type rabbits (Figure 5A). There was no difference in inward‐rectifier potassium current (IK1) density between wild‐type and transgenic myocytes (Figure 5B). There was no difference in the amplitude (Figure 6A) of the IKr tail current at −40 mV between wild‐type and transgenic myocytes. Similarly, the activation time constant of IKr was not different between the wild‐type and transgenic myocytes. The fast deactivation time constant of IKr (at −40 mV, test potential: 30 mV) was slightly faster in transgenic rabbit cells compared with wild‐type (Figure 6B).
Figure 5.
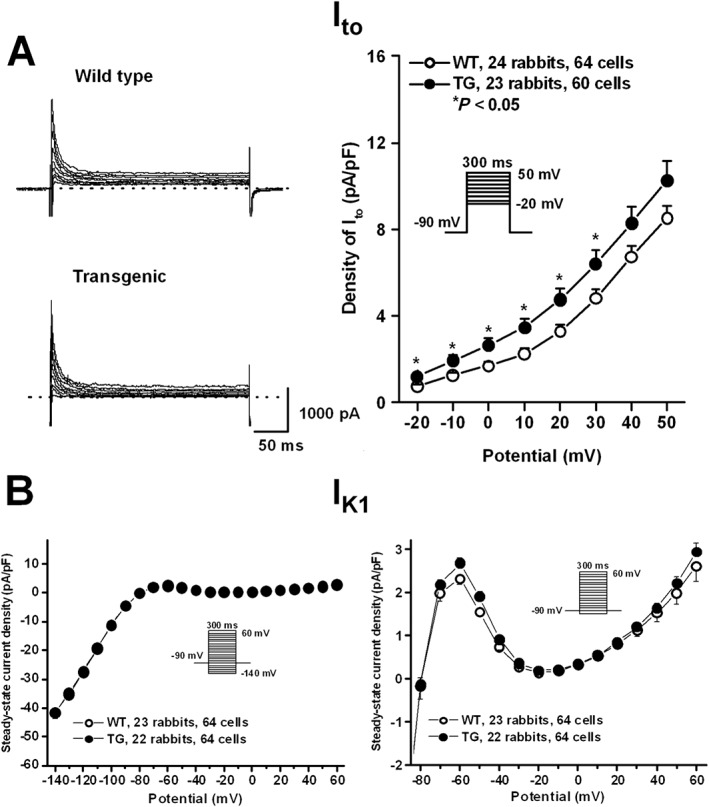
The density of Ito (A) and IK1 (B) in rabbit ventricular myocytes. Representative Ito current traces recorded from wild‐type (WT) and transgenic (TG) ventricular myocytes are shown in panel A (left). Current–voltage relations (right) demonstrate higher Ito density in TG rabbits compared with WT rabbits. Panel (B) illustrates steady‐state current–voltage curves (left) demonstrating no differences in density of IK1 between WT and TG rabbits. The outward current sections of the curves are enlarged on the right. Insets show the applied voltage protocols. Values are means ± SEM. * P < 0.05 versus WT littermates.
Figure 6.
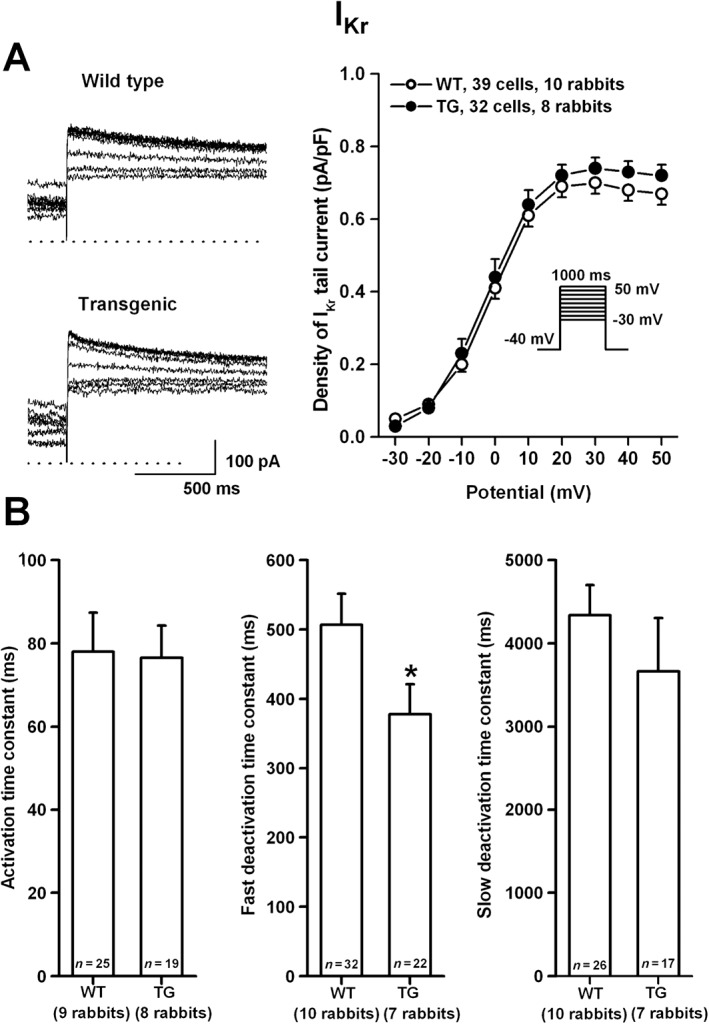
The density of IKr (A) in rabbit ventricular myocytes. Representative IKr tail current traces recorded from wild‐type (WT) and transgenic (TG) ventricular myocytes are shown on the left. Current–voltage relations (right) demonstrate no differences in density of IKr between WT and TG rabbits. Inset shows the applied voltage protocol. Bar diagrams in panel (B) illustrate the activation and deactivation (fast and slow) time constants in WT and TG ventricular myocytes. Values are means ± SEM. * P < 0.05 versus WT littermates.
No difference was observed in IKs tail‐current amplitude between transgenic and wild‐type rabbits (Figure 7). There were no differences between the stimulating effect of forskolin in wild‐type versus transgenic rabbits (Figure 7A). The activation kinetics of IKs were not different, but the deactivation kinetics (at −40 mV, test potential: 50 mV) was markedly and significantly faster, in transgenic‐rabbit myocytes compared with wild‐type rabbits (Figure 7B). These changes would be expected to reduce phase‐3 repolarizing current, particularly in the presence of a repolarization stress. The subtle repolarization abnormalities associated with our model were reflected in cellular action‐potential recordings (Supplemental Figure 1), which showed a significant increase in APD‐variability but not in APD‐increase resulting from dofetilide exposure in transgenic versus wild‐type rabbits.
Figure 7.
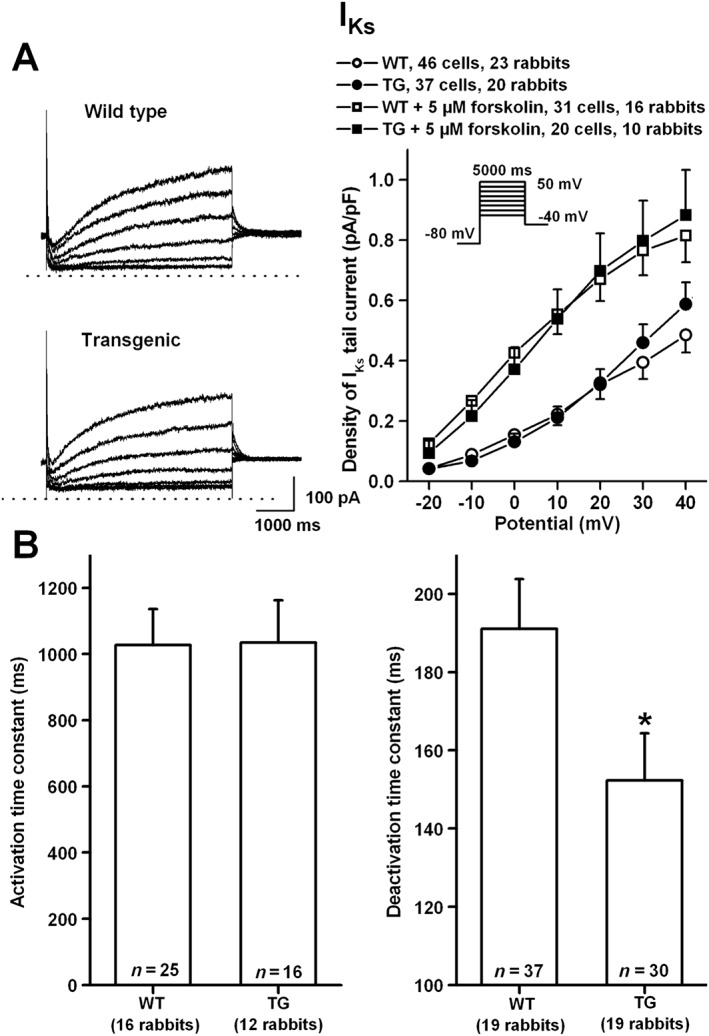
The density of IKs (A) in absence and presence of 5 μM forskolin in rabbit ventricular myocytes. Representative IKs current traces recorded from wild‐type (WT) and transgenic (TG) ventricular myocytes are shown in panel A (left). Current–voltage relations (right) demonstrate no differences in density of IKs between WT and TG rabbits. Inset shows the applied voltage protocol. Bar diagrams in panel (B) illustrate the activation and deactivation time constants in WT and TG ventricular myocytes. Values are means ± SEM. * P < 0.05 versus WT littermates.
ECG parameters and arrhythmias
During anaesthesia with ketamine/xylazine, which does not affect cardiac repolarization (Odening et al., 2008), there was a slightly but significantly longer QTi in LQT5‐transgenic versus wild‐type littermates (104 ± 0.9% vs. 98.9 ± 1.0%, P < 0.001, Figure 8). No sex‐related differences were observed.
Figure 8.
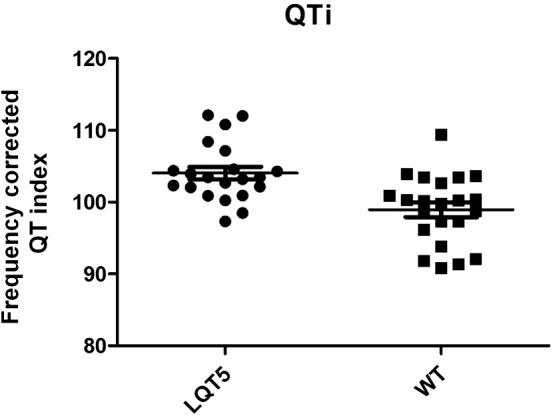
Heart rate corrected QT index in anaesthetized rabbits. Plots indicating the heart‐rate corrected QT‐indices (QTi) in LQT5 transgenic (n = 21) and wild‐type littermate (WT; n = 22) rabbits under anaesthesia with ketamine/xylazine. Mean ± SEM are indicated as horizontal lines. P < 0.001.
Examples of ECG recordings are shown in Supplemental Figure 2. In arrhythmia provocation experiments under thiopental anaesthesia, which blocks IKs (Sakai et al., 1997), there were no significant differences in any ECG intervals between wild‐type and transgenic animals at baseline (Table 2). The short‐term variability of the RR interval (STVRR) was also similar in both groups, both at baseline and following dofetilide (Figure 9C). Figure 9A and B shows Poincaré plots of QT‐intervals of representative individual wild‐type and transgenic rabbits. As representative plots (Figure 9A and B) and grouped data show (Figure 9D), the short‐term QT‐interval variability (STVQT) was already greater in transgenic rabbits at baseline. Administration of dofetilide (20 μg·kg−1, i.v.) prolonged the QT and QTc intervals significantly in both groups (Table 2). However, dofetilide significantly decreased heart rate only in transgenic animals (Table 2). Dofetilide caused a further and significant increase in STVQT in transgenic animals (Figure 9D). While dofetilide led to similar QTc prolongation in both groups, its STVQT‐increasing effect was more pronounced and accompanied by more arrhythmias in transgenic animals. Dofetilide provoked TdP arrhythmias in 3/11 (27%) wild‐type animals versus 12/15 (80%) of transgenic rabbits (P < 0.001, Figure 9E). In transgenic animals, the durations of TdP episodes were also significantly longer than those observed in wild‐type rabbits (Figure 9F).
Discussion
This is the first study to report the creation and primary characterization of a transgenic LQT5 pro‐arrhythmia model in rabbits. The reliable prediction of pro‐arrhythmic risk associated with pharmacological therapy is essential to reduce the incidence of serious drug‐induced arrhythmias in the clinical setting. Cardiac electrophysiological safety assessment remains unsatisfactory (Farkas and Nattel, 2010). Current in vitro and in vivo proarrhythmia models mostly rely on investigating effects in healthy tissues, animals and volunteers (ICHE14, 2005; ICH‐S7B, 2005). New models with better predictive value for proarrhythmic risk are needed, to better reflect clinical situations at risk of drug‐induced arrhythmias, particularly in patients with remodelled myocardium (Nattel et al. 2007) exhibiting impairment of repolarization reserve (Varro and Baczko, 2011).
Already existing in vivo proarrhythmia models with impaired repolarization reserve include a dog model with chronic atrioventricular block (Vos et al., 1998). In this model, ventricular hypertrophy and bradycardia develop, and the consequent down‐regulation of IKs leads to reduced repolarization reserve and increased arrhythmia susceptibility (Vos et al., 1998; Volders et al., 1999). Another approach is the acute pharmacological inhibition of IKs by HMR‐1556 (Lengyel et al., 2007) mimicking reduced repolarization reserve. Loss‐of‐function mutations in repolarizing and gain‐of‐function mutations in depolarizing currents cause congenital long QT syndromes with impaired repolarization and increased sudden death risk (Moss and Kass, 2005). The two most common forms of congenital LQT (LQT1 and LQT2) were modelled by expression of human mutant KCNQ1 and KCNH2 genes in transgenic rabbits in a landmark study by Brunner et al. (2008). Both LQT1 and LQT2 rabbits exhibited markedly prolonged QT intervals, and LQT2 transgenic rabbits showed a high incidence of sudden death (Brunner et al., 2008; Odening et al., 2010). These two transgenic rabbit models mimic the human LQT1 and LQT2 phenotypes and are extremely useful tools for studying mechanisms of sudden death, as shown by at least 13 additional publications since the original paper (see (Duranthon et al., 2012). On the other hand, the importance of ‘silent’ or ‘subclinical’ LQT cases should be emphasized, where impairment of repolarization reserve does not necessarily result in clinically obvious QT‐prolongation, but the heart is more susceptible to arrhythmia development (Roden, 2006; Varro and Baczko, 2011).
The present study aimed at the creation and primary characterization of a novel transgenic rabbit model with latent impairment of repolarization reserve and a weaker phenotype compared with the LQT1 and LQT2 models.
Pronuclear microinjection results in concatemers with multicopy transgene integrations. Our data shows that the transgenic line examined in detail carries the transgene integrated at two different loci on Chr 11 at high copy numbers. The quantity in this line of the mutant human minK protein was twice that of the endogenous minK (Figure 4A, B, C). The data confirmed our earlier observations, where a minigene type transgene construct integrated at high copy numbers and resulted in a relatively high recombinant protein expression in transgenic rabbit milk (Bodrogi et al., 2006), despite the known phenomenon that the presence of multiple transgene homologous copies within a concatemer array can have a repressive effect upon gene‐expression (Garrick et al., 1998).
In the present study, functional effects of the G52R‐KCNE1 mutation were investigated in left ventricular myocytes obtained from wild‐type and transgenic rabbit hearts. There were no differences in the amplitude of the IK1 current and tail currents of IKr and IKs. Current amplitudes and densities can be influenced by the cell isolation procedure, which may attenuate differences between study groups, in spite of relatively large numbers of rabbits (8–24) and cells (20–64). On the other hand, IKr/IKs deactivation kinetics were significantly affected by the transgene, potentially accounting for the in vivo phenotype.
The functional importance of the moderate augmentation of Ito in transgenic rabbits is difficult to interpret in the rabbit heart, in which the Ito is carried mainly, but not exclusively, by Kv1.4 channels (Zicha et al., 2003), which have slow (>1 s time‐constant) recovery kinetics from inactivation. The exact role of Ito in ventricular repolarization at normal rabbit heart rates ‐ in the range of 200–250 beats min‐1 at rest ‐ is uncertain because rabbit Ito may be largely inactivated at physiological heart rates.
The most important cellular finding in this study is the accelerated deactivation kinetics of IKs and IKr in the transgenic rabbits, compared with their wild‐type littermates. Accelerated deactivation causes more rapid channel closure and consequently lower densities of IKs and IKr during the action potential. These functional changes impair repolarization reserve and might explain the increased susceptibility of transgenic rabbits to arrhythmias, in our in vivo studies. The accelerated deactivation of IKs in native ventricular cardiomyocytes expressing the mutant G52R‐KCNE1 in our study contrasts with the findings obtained in Xenopus oocytes co‐expressing wild‐type KCNE1 and G52R‐KCNE1 with KCNQ1, where the IKs current amplitude was decreased by 50% (Ma et al., 2003). In CHO cells, co‐expression of KCNQ1 with G52R‐KCNE1 produced a current devoid of the characteristic slow activation of IKs, and the current density was similar to those in cells expressing only the KCNQ1 without any type of KCNE1 (Harmer et al., 2010). The reasons for these conflicting results in native rabbit cardiomyocytes and various cellular expression systems are not known. However, accumulating evidence suggests that KCNE1 is not the only regulatory subunit that associates with KCNQ1, and KCNE2, KCNE3, possibly KCNE4 and KCNE5 also form channel complexes with KCNQ1, and these complexes play potential physiological roles (Bendahhou et al., 2005; Lundquist et al., 2005). In addition, although KCNE1 is established primarily as a KCNQ1 subunit, there is evidence that it may also play a role in IKr (Yang et al., 1995; McDonald et al., 1997; Ohno et al., 2007). The varying contributions of different accessory subunits may account for the different effects of mutations in different expression systems.
During in vivo characterization, no spontaneous sudden deaths were observed in LQT5‐transgenic rabbits. ECG studies in xylazine/ketamine‐anaesthetized rabbits revealed a slight but significant prolongation of the heart rate‐corrected QT index in LQT5‐rabbits, indicating mild prolongation of repolarization (Figure 8). Contrasting with observations during anaesthesia with ketamine/xylazine, which does not affect repolarizing ion‐currents (Odening et al., 2008), during thiopental‐anaesthesia, there were no differences in the QT or frequency‐corrected QTc intervals between LQT5 rabbits and their wild‐type littermates (Table 2). Because thiopental has IKs‐blocking effects (Sakai et al., 1997.), unlike xylazine/ketamine (Baum, 1993), it is likely that the small baseline repolarization differences disappeared following thiopental anaesthesia. Even under thiopental anaesthesia; however, the short‐term variability of the QT interval that is an ECG biomarker for proarrhythmic risk (Atiga et al., 1998; Varkevisser et al., 2012) was significantly greater in LQT5 rabbits (Figure 9 A‐B, D), suggesting increased repolarization instability already at baseline. To compare the arrhythmia‐susceptibility of LQT5 rabbits with reduced repolarization reserve to wild‐type littermates, the IKr blocker dofetilide was added. As expected, dofetilide prolonged the QT interval in both groups (Table 2). Dofetilide further increased STVQT in LQT5‐rabbits and provoked typical drug‐induced polymorphic ventricular tachycardia, Torsade de Pointes, in a much larger number of LQT5‐animals compared with wild type (Figure 9E), and these episodes lasted significantly longer (Figure 9F). These observations agree with other proarrhythmia studies in models with reduced repolarization reserve, such as dogs with chronic AV‐block (Thomsen et al., 2004) and anaesthetized rabbits subjected to pharmacological block of IKs (Lengyel et al., 2007). Although we did not observe any evidence of cardiac structural or functional disturbances in transgenic rabbits, formal studies with echocardiographic assessment of cardiac and structure should be performed in the future to exclude any subtle alterations occurring in the model.
Further studies are needed to detail the cellular electrophysiological mechanisms responsible for the increased arrhythmia susceptibility observed in LQT5‐rabbits, especially in the light of data indicating that (i) chronic loss of function of one repolarizing current can lead to compensatory up‐regulation of other K+‐channels (Xiao et al., 2008) and (ii) different KCNE‐encoded proteins do not exclusively associate with KvLQT1 to form IKs channels, but may be involved in the assembly of other ion channels (Abbott and Goldstein 2002; Um and McDonald, 2007). Future studies should also evaluate all important ionic currents in a large number of native ventricular cardiomyocytes isolated from LQT5 transgenic and wild‐type littermate animals at different voltage levels focusing on channel kinetics, but such work is beyond the scope of the present study due to the limited availability of such animals. In addition, it would be interesting to create a study involving wild‐type KCNE1 overexpression, to compare the resulting phenotype with that of mutant KCNE1 overexpressing rabbits. Furthermore, studies of the effects of additional repolarization‐stresses, such as adrenergic stimulation, would be appropriate. Finally, it would be useful to examine whether changes occur in other ion‐currents like INa and ICa,L, because they might be altered as part of the cellular adaptation to changes in electrical function induced by the mutation.
In conclusion, we report the successful creation of a novel transgenic LQT5 rabbit model based on the cardiac‐specific overexpression of human KCNE1 carrying a G52R missense. These rabbits exhibit reduced repolarization reserve but no striking repolarization disturbances or serious ventricular arrhythmias at baseline. However, LQT5 rabbits are more susceptible to arrhythmias than wild‐type littermates upon repolarization stress, indicating that they may be suitable to model the challenging clinical situation of ‘silent’ LQT. This model can also provide further insights into the mechanisms underlying arrhythmias and sudden cardiac death based on repolarization disturbances and may represent a novel model for testing the pro‐arrhythmic potential of new drugs under development.
Author contributions
P.M. and L. H. created the transgene construct and transgenic rabbits. I.B. conceived and designed the experiments, wrote the paper and analysed data. K.E.O. worked on in vivo and in vitro rabbit characterization and manuscript editing. V.J., Z.K., A.H., G.S., M.K. and Z.D. focussed on rabbit phenotyping. Z.K., L.V., N.J., J.P. and B.Ö. performed patch‐clamp experiments. L.V. and N.J. also worked on data analysis. B.Ö. carried out data evaluation. S.N. provided advice on experiment design and interpretation and edited the manuscript. A.V. also conceived and designed the experiments. Z.B. also conceived and designed the experiments and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Effect of 100 nM dofetilide on the action potential duration and on the short‐term variability of the action potential duration in rabbit ventricular myocytes. The upper panels show representative action potential recordings in the absence and presence of 100 nM dofetilide from wild type (left) and transgenic (right) ventricular myocytes. Bar graphs in the lower panels illustrate APD90 (left) and the short‐term variability of APD90 (right) in control conditions and in the presence of 100 nM dofetilide in wild type (WT) and transgenic (TG) ventricular myocytes. Values are means ± errors of the means. The measurements were taken from 6 WT and 6 TG rabbit hearts, “n” values on the graph represent the number of cells in each group.
Figure S2 Representative ECG recordings from thiopental anaesthetized wild type littermate (upper panel) and transgenic (lower panel) rabbits at baseline (control) and following the i.v. administration of dofetilide (20 μg kg‐1).
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
The authors gratefully acknowledge Dr Ilona Bodi for performing initial patch clamping experiments of CHO cells transfected with the transgenic constructs and for transfection experiments to set up the NanoPro analysis of human and rabbit KCNE. This work was supported by the Hungarian Scientific Research Fund (OTKA CNK 77855 and OTKA NK 104331 to A.V., OTKA NN 110896 to I.B., OTKA NN 109904 to N.J. and OTKA NK 104397 to Z.B.), by the National Office for Research and Technology – Baross Programme (REG‐DA‐09‐2‐2009‐0115‐NCXINHIB), by the Hungarian National Development Agency (TÁMOP‐4.2.6‐15/1‐2015‐0002 and TÁMOP‐4.2.1.C‐14/1/KONV‐2015‐0013 projects) in the framework of Szechenyi 2020 programme supported by the European Union and co‐financed by the European Social Fund and State of Hungary and by the Hungarian Academy of Sciences, the Canadian Heart Foundation and the Canadian Institutes of Health Research. M.K. was supported by the MTA Postdoctoral Research Programme of the Hungarian Academy of Sciences.
Major, P. , Baczkó, I. , Hiripi, L. , Odening, K. E. , Juhász, V. , Kohajda, Z. , Horváth, A. , Seprényi, G. , Kovács, M. , Virág, L. , Jost, N. , Prorok, J. , Ördög, B. , Doleschall, Z. , Nattel, S. , Varró, A. , and Bősze, Z. (2016) A novel transgenic rabbit model with reduced repolarization reserve: long QT syndrome caused by a dominant‐negative mutation of the KCNE1 gene. British Journal of Pharmacology, 173: 2046–2061. doi: 10.1111/bph.13500.
References
- Abbott GW, Goldstein SA (2002). Disease‐associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J 16: 390–400. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atiga WL, Calkins H, Lawrence JH, Tomaselli GF, Smith JM, Berger RD (1998). Beat‐to‐beat repolarization lability identifies patients at risk for sudden cardiac death. J Cardiovasc Electrophysiol 9: 899–908. [DOI] [PubMed] [Google Scholar]
- Atkinson A, Inada S, Li J, Tellez JO, Yanni J, Sleiman R et al. (2011). Anatomical and molecular mapping of the left and right ventricular His‐Purkinje conduction networks. J Mol Cell Cardiol 51: 689–701. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384: 78–80. [DOI] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ (2002). Proarrhythmic potential of halofantrine, terfenadine and clofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol 135: 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum VC (1993). Distinctive effects of three intravenous anesthetics on the inward rectifier (IK1) and the delayed rectifier (IK) potassium currents in myocardium: implications for the mechanism of action. Anesth Analg 76: 18–23. [DOI] [PubMed] [Google Scholar]
- Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V et al. (2005). In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res 67: 529–538. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Begenisich TB (1987). Catecholamines modulate the delayed rectifying potassium current (IK) in guinea pig ventricular myocytes. Pflugers Arch 410: 217–219. [DOI] [PubMed] [Google Scholar]
- Besenfelder U, Brem G (1993). Laparoscopic embryo transfer in rabbits. J Reprod Fertil 99: 53–56. [DOI] [PubMed] [Google Scholar]
- Bodrogi L, Brands R, Raaben W, Seinen W, Baranyi M, Fiechter D et al. (2006). High level expression of tissue‐nonspecific alkaline phosphatase in the milk of transgenic rabbits. Transgenic Res 15: 627–636. [DOI] [PubMed] [Google Scholar]
- Brennan M, Palaniswami M, Kamen P (2001). Do existing measures of Poincare plot geometry reflect nonlinear features of heart rate variability? IEEE Trans Biomed Eng 48: 1342–1347. [DOI] [PubMed] [Google Scholar]
- Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR et al. (2008). Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest 118: 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demolombe S, Lande G, Charpentier F, van Roon MA, van den Hoff MJ, Toumaniantz G et al. (2001). Transgenic mice overexpressing human KvLQT1 dominant‐negative isoform. Part I: Phenotypic characterisation. Cardiovasc Res 50: 314–327. [DOI] [PubMed] [Google Scholar]
- Drici MD, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G et al. (1998). Involvement of IsK‐associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange‐Nielsen syndrome. Circ Res 83: 95–102. [DOI] [PubMed] [Google Scholar]
- Duranthon V, Beaujean N, Brunner M, Odening KE, Santos AN, Kacskovics I et al. (2012). On the emerging role of rabbit as human disease model and the instrumental role of novel transgenic tools. Transgenic Res 21: 699–713. [DOI] [PubMed] [Google Scholar]
- Farkas A, Batey AJ, Coker SJ (2004). How to measure electrocardiographic QT interval in the anaesthetized rabbit. J Pharmacol Toxicol Methods 50: 175–185. [DOI] [PubMed] [Google Scholar]
- Farkas AS, Nattel S (2010). Minimizing repolarization‐related proarrhythmic risk in drug development and clinical practice. Drugs 70: 573–603. [DOI] [PubMed] [Google Scholar]
- Garrick D, Fiering S, Martin DI, Whitelaw E (1998). Repeat‐induced gene silencing in mammals. Nat Genet 18: 56–59. [DOI] [PubMed] [Google Scholar]
- Han W, Wang Z, Nattel S (2001). Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol Heart Circ Physiol 280: H1075–H1080. [DOI] [PubMed] [Google Scholar]
- Harmer SC, Wilson AJ, Aldridge R, Tinker A (2010). Mechanisms of disease pathogenesis in long QT syndrome type 5. Am J Physiol Cell Physiol 298: C263–C273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterseer M, Beckmann BM, Thomsen MB, Pfeufer A, Dalla Pozza R, Loeff M et al. (2009). Relation of increased short‐term variability of QT interval to congenital long‐QT syndrome. Am J Cardiol 103: 1244–1248. [DOI] [PubMed] [Google Scholar]
- Hinterseer M, Beckmann BM, Thomsen MB, Pfeufer A, Ulbrich M, Sinner MF et al. (2010). Usefulness of short‐term variability of QT intervals as a predictor for electrical remodeling and proarrhythmia in patients with nonischemic heart failure. Am J Cardiol 106: 216–220. [DOI] [PubMed] [Google Scholar]
- ICH‐S7B (2005). International Conference on Harmonisation; guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals; availability. Notice Fed Regist 70: 61133–61134. [PubMed] [Google Scholar]
- ICHE14 (2005). International Conference on Harmonisation: Guidance on E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs; availability. Notice Fed Regist 70: 61134–61135. [PubMed] [Google Scholar]
- Jost N, Papp JG, Varro A (2007). Slow delayed rectifier potassium current (IKs) and the repolarization reserve. Ann Noninvasive Electrocardiol 12: 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P et al. (2005). Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 112: 1392–1399. [DOI] [PubMed] [Google Scholar]
- Kupershmidt S, Yang T, Roden DM (1998). Modulation of cardiac Na + current phenotype by beta1‐subunit expression. Circ Res 83: 441–447. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Ackerman MJ, Benson DW Jr, Brugada R, Clancy CE, Donahue JK et al. (2007). Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation 116: 2325–2345. [DOI] [PubMed] [Google Scholar]
- Lengyel C, Varro A, Tabori K, Papp JG, Baczko I (2007). Combined pharmacological block of I(Kr) and I(Ks) increases short‐term QT interval variability and provokes torsades de pointes. Br J Pharmacol 151: 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist AL, Manderfield LJ, Vanoye CG, Rogers CS, Donahue BS, Chang PA et al. (2005). Expression of multiple KCNE genes in human heart may enable variable modulation of I(Ks). J Mol Cell Cardiol 38: 277–287. [DOI] [PubMed] [Google Scholar]
- Ma L, Lin C, Teng S, Chai Y, Bahring R, Vardanyan V et al. (2003). Characterization of a novel Long QT syndrome mutation G52R‐KCNE1 in a Chinese family. Cardiovasc Res 59: 612–619. [DOI] [PubMed] [Google Scholar]
- McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW et al. (1997). A minK‐HERG complex regulates the cardiac potassium current I(Kr). Nature 388: 289–292. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ, Kass RS (2005). Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest 115: 2018–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai T, Kakizuka A, Takumi T, Ohkubo H, Nakanishi S (1989). Molecular cloning and sequence analysis of human genomic DNA encoding a novel membrane protein which exhibits a slowly activating potassium channel activity. Biochem Biophys Res Commun 161: 176–181. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, Yeh YH (2007). Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87: 425–456. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM (2000). Molecular basis of functional voltage‐gated K+ channel diversity in the mammalian myocardium. J Physiol 525 (Pt 2): 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253. [DOI] [PubMed] [Google Scholar]
- Nishio H, Kuwahara M, Tsubone H, Koda Y, Sato T, Fukunishi S et al. (2009). Identification of an ethnic‐specific variant (V207 M) of the KCNQ1 cardiac potassium channel gene in sudden unexplained death and implications from a knock‐in mouse model. Int J Leg Med 123: 253–257. [DOI] [PubMed] [Google Scholar]
- Odening KE, Hyder O, Chaves L, Schofield L, Brunner M, Kirk M et al. (2008). Pharmacogenomics of anesthetic drugs in transgenic LQT1 and LQT2 rabbits reveal genotype‐specific differential effects on cardiac repolarization. Am J Physiol Heart Circ Physiol 295: H2264–H2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odening KE, Kirk M, Brunner M, Ziv O, Lorvidhaya P, Liu GX et al. (2010). Electrophysiological studies of transgenic long QT type 1 and type 2 rabbits reveal genotype‐specific differences in ventricular refractoriness and His conduction. Am J Physiol Heart Circ Physiol 299: H643–H655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Zankov DP, Yoshida H, Tsuji K, Makiyama T, Itoh H et al. (2007). N‐ and C‐terminal KCNE1 mutations cause distinct phenotypes of long QT syndrome. Heart Rhythm 4: 332–340. [DOI] [PubMed] [Google Scholar]
- Panaghie G, Tai KK, Abbott GW (2006). Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J Physiol 570 (Pt 3): 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B et al. (2008). Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res 103: 298–306. [DOI] [PubMed] [Google Scholar]
- Roden DM (2006). Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med 259: 59–69. [DOI] [PubMed] [Google Scholar]
- Roden DM (1998). Taking the "idio" out of "idiosyncratic": predicting torsades de pointes. Pacing Clin Electrophysiol 21: 1029–1034. [DOI] [PubMed] [Google Scholar]
- Sakai F, Hiraoka M, Amaha K (1997). Comparative actions of propofol and thiopentone on cell membranes of isolated guinea pig ventricular myocytes. Br J Anaesth 77: 508–516. [DOI] [PubMed] [Google Scholar]
- Salama G, London B (2007). Mouse models of long QT syndrome. J Physiol 578 (Pt 1): 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384: 80–83. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL et al. (2000). Spectrum of mutations in long‐QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102: 1178–1185. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J et al. (2004). Increased short‐term variability of repolarization predicts d‐sotalol‐induced torsades de pointes in dogs. Circulation 110: 2453–2459. [DOI] [PubMed] [Google Scholar]
- Um SY, McDonald TV (2007). Differential association between HERG and KCNE1 or KCNE2. PLoS One 2: e933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al (2002). Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: research0034.1–0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkevisser R, Wijers SC, van der Heyden MA, Beekman JD, Meine M, Vos AM (2012). Beat‐to‐beat variability of repolarization as a new biomarker for proarrhythmia in vivo. Heart Rhythm 9: 1718–1726. [DOI] [PubMed] [Google Scholar]
- Varro A, Baczko I (2011). Cardiac ventricular repolarization reserve: a principle for understanding drug‐related proarrhythmic risk. Br J Pharmacol 164: 14–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varro A, Balati B, Iost N, Takacs J, Virag L, Lathrop DA et al. (2000). The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol 523 (Pt 1): 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varro A, Virag L, Papp JG (1996). Comparison of the chronic and acute effects of amiodarone on the calcium and potassium currents in rabbit isolated cardiac myocytes. Br J Pharmacol 117: 1181–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders PG, Sipido KR, Vos MA, Spatjens RL, Leunissen JD, Carmeliet E et al. (1999). Downregulation of delayed rectifier K(+) currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation 100: 2455–2461. [DOI] [PubMed] [Google Scholar]
- Vos MA, de Groot SH, Verduyn SC, van der Zande J, Leunissen HD, Cleutjens JP et al. (1998). Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation 98: 1125–1135. [DOI] [PubMed] [Google Scholar]
- Xiao L, Xiao J, Luo X, Lin H, Wang Z, Nattel S (2008). Feedback remodeling of cardiac potassium current expression: a novel potential mechanism for control of repolarization reserve. Circulation 118: 983–992. [DOI] [PubMed] [Google Scholar]
- Yang T, Kupershmidt S, Roden DM (1995). Anti‐minK antisense decreases the amplitude of the rapidly activating cardiac delayed rectifier K+ current. Circ Res 77: 1246–1253. [DOI] [PubMed] [Google Scholar]
- Zicha S, Moss I, Allen B, Varro A, Papp J, Dumaine R et al. (2003). Molecular basis of species‐specific expression of repolarizing K+ currents in the heart. Am J Physiol Heart Circ Physiol 285: H1641–H1649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of 100 nM dofetilide on the action potential duration and on the short‐term variability of the action potential duration in rabbit ventricular myocytes. The upper panels show representative action potential recordings in the absence and presence of 100 nM dofetilide from wild type (left) and transgenic (right) ventricular myocytes. Bar graphs in the lower panels illustrate APD90 (left) and the short‐term variability of APD90 (right) in control conditions and in the presence of 100 nM dofetilide in wild type (WT) and transgenic (TG) ventricular myocytes. Values are means ± errors of the means. The measurements were taken from 6 WT and 6 TG rabbit hearts, “n” values on the graph represent the number of cells in each group.
Figure S2 Representative ECG recordings from thiopental anaesthetized wild type littermate (upper panel) and transgenic (lower panel) rabbits at baseline (control) and following the i.v. administration of dofetilide (20 μg kg‐1).
Supporting info item
Supporting info item
Supporting info item
Supporting info item