

Abstract
Hepatitis E virus (HEV) is a significant health problem in developing countries causing sporadic and epidemic forms of acute viral hepatitis. Hepatitis E is a self-limiting disease; however, chronic HEV infections are being reported in immunocompromised individuals. The disease severity is more during pregnancy with high mortality (20–25%), especially in third trimester. Early cellular responses after HEV infection are not completely understood. We analyzed innate immune responses associated with genotype-I HEV replication in human hepatoma cell lines (Huh7, Huh7.5 and HepG2/C3A) using HEV replicon system. These cells supported HEV replication with different efficiencies due to the cell type specific innate immune responses. HepG2/C3A cells were less supportive to HEV replication as compared to Huh7.5 and S10-3 cells. Reconstitution of the defective RIG-I and TLR3 signaling in Huh7.5 cells enabled them to induce higher level antiviral responses and restrict HEV replication, suggesting the involvement of both RIG-I and TLR3 in sensing HEV RNA and downstream activation of interferon regulatory factor 3 (IRF3) to generate antiviral responses. Inhibition of IRF3 mediated downstream responses in HepG2/C3A cells by pharmacological inhibitor BX795 significantly improved HEV replication efficiency implying the importance of this study in establishing a better cell culture system for future HEV studies.
Hepatitis E virus (HEV) is a single-stranded positive-sense RNA virus classified in the genus Orthohepevirus of the family Hepeviridae1. The genome is ~7.2 kb long with short 5′-and 3′-noncoding regions (NCRs), 5′-methylguanine cap, 3′-poly (A) stretch and three open reading frames (ORF1, ORF2 and ORF3)2,3. ORF1 is translated from genomic RNA and encodes non-structural polyprotein with major domains such as methyltransferase, protease, helicase and RNA-dependent RNA polymerase that are required for virus replication4,5. ORF2 encodes capsid protein, while ORF3 encodes a small accessory protein. Both ORF2 and ORF3 are translated from a single bicistronic (subgenomic) mRNA6. HEV is the causative agent of hepatitis E, an acute and self-limiting disease caused by enteric transmission of the virus. Severe manifestation of hepatitis E is more common in pregnant women with high mortality rates (20–25%)7. Persistent HEV infections have been documented in immunosuppressed individuals8.
Cellular antiviral response is mediated by interferon (IFN) system and interferon induced antiviral effector genes known as interferon stimulated genes (ISGs). Two major pathways involved in sensing viral infections are Toll Like Receptor (TLR) dependent pathways and the cytosolic Retinoic acid-Inducible Gene I (RIG-I) like receptor (RLR) dependent pathways. TLRs sense pathogen components on cells surface and endosomal compartments, while, RLRs survey cytoplasm for the presence of viral double-stranded RNA (a replication intermediate) and 5′-triphosphate group containing single stranded RNA molecules9,10,11,12. Type I IFNs initiate expression of numerous ISGs, in autocrine or paracrine manner to induce antiviral state in infected cells and neighbouring cells10.
Molecular mechanisms associated with HEV replication and cellular antiviral responses against HEV are still not completely understood. We have previously shown that HEV can elicit inflammatory responses in human lung adenocarcinoma (A549) cells via TLR adaptors, TRIF and MyD88 and activate interferon regulatory factor 3 (IRF3) and NF-κB13. Dong et al.14 have demonstrated that HEV has ability to inhibit IFN-α signaling and replicate in presence of IFN-α, in A549 cell culture system. HEV mediated interferon antagonism has been shown to be mediated by macro- and papain like cysteine protease- domains of ORF1 polyprotein15. However, it is not yet known how liver cells detect HEV infection to initiate antiviral responses. The major obstacle has been lack of efficient liver cell culture system. Several groups have used HEV replicons and human hepatoma cell lines such as HepG2, Huh7 and PLC/PRF5 to shed some light on these aspects of HEV biology16,17,18.
In the present study, we used HEV replicon based cell culture system to analyse HEV induced antiviral responses. We used three hepatoma cell lines, HepG2/C3A, Huh7.5 and S10-3, which were supporting HEV replication, albeit with differential efficiencies. There was a direct correlation between induction levels of different interferon stimulated genes and HEV replication efficiencies in these cells. RIG-I and TLR3 were found to be major sensing molecules for HEV.
Materials and Methods
Cells
HepG2/C3A cell line was purchased from American Type Culture Collection (ATCC), S10-3 cells (a subclone of human hepatoma Huh7 cells) were a kind gift from Dr. S. Emerson (NIH, USA) and Huh7.5 cell line was from Apath LLC (Brooklyn, New York). HepG2/C3A cells were grown in Dulbecco’s modified Eagle’s medium and minimal essential medium (MEM) (mixed in 1:1 proportion) with 10% heat inactivated fetal bovine serum (FBS), 100 U penicillin/ml, and 0.1 mg streptomycin/ml while S10-3 and Huh7.5 cells were grown in Dulbecco’s modified Eagle’s medium containing 10% heat inactivated FBS, 100 U penicillin/ml, and 0.1 mg streptomycin/ml. Stocks were maintained at 37 °C while transfected cultures were incubated at 34.5 °C.
Plasmids
HEV Rluc replicon encoding Renilla luciferase (Rluc) gene was a kind gift from Dr. X. J. Meng (Virginia Tech, Blacksburg, USA). This subgenomic clone has been developed from pSKHEV-2 (genotype 1 HEV infectious cDNA clone, GenBank accession No. AF444002) (19). Using HEV-Rluc replicon as template, the mutant HEV Rluc GAA was constructed (by changing conserved RdRp GDD motif to GAA) with QuickChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). This change is known to completely stop HEV replication18,19,20. Plasmids bearing human RIG-I and TLR3 gene, pUNO1-hRIG-I, pUNO-hTLR3, pZERO-TLR3 (TLR3-ΔTIR; a TIR-less form of TLR3 gene) and Poly (I:C) (HMW)/Lyovec were from InvivoGen, USA.
Generation of capped RNA transcripts and cell transfection
HEV Rluc replicon plasmid was linearized by utilizing unique Bgl II site located immediately downstream of the poly (A) tract of the HEV sequence and capped RNA transcripts were synthesized by in vitro transcription using mMessage mMachine T7 ultra kit (Ambion). Following transcription, DNA template was removed by DNase I treatment, transcribed RNA was purified by lithium chloride precipitation method as per the manufacturer’s instructions and quantified on Nanodrop spectrophotometer (ND-1000, Nanodrop technologies). Integrity of the transcripts was checked by doing denaturing agarose gel electrophoresis. For each experiment, cells were grown up to 60–70% confluence in 24-well cell culture plates and washed with serum free medium, OptiMEM (Invitrogen, Life technologies) prior to transfection. Cells were transfected with capped RNA transcripts, diluted appropriately in OptiMEM (2 μg/well of the 24 well plate) using 1,2-dimyristyl Rosenthal inhibitor ether (DMRIE-C) reagent (Invitrogen) as per the manufacturer’s instructions. Cells were co-transfected with Firefly luciferase plasmid DNA (pGL-3 promoter vector, 100 ng/well) along with HEV-Rluc RNA to normalize cell transfection efficiency and Renilla luciferase signals. For gene expression analysis, transfections were carried out similarly without including firefly luciferase plasmid DNA. After 4 h of incubation at 34.5 °C, transfection mixture was replaced with DMEM containing 10%FBS. All cell transfections were carried out in triplicates and each set of experiments was repeated twice/thrice. For plasmids, cell transfections were carried out with Lipofectamine 2000 transfection reagent (invitrogen) as per the manufacturer’s instructions.
Reporter gene assay
Monolayer of the cells transfected with RNA was washed two times with phosphate buffered saline, cells were lysed in 100 μl of 1X Passive Lysis Buffer (Promega) and lysates were immediately frozen at −80 °C until use. For the assay, samples were thawed, centrifuged at 10,000 RPM for 2 min and 20 μl cell lysates were used for measuring dual luciferase activities (Renilla luciferase: Rluc and firefly luciferase: FLuc) using Dual luciferase assay system (Promega) and readings were taken on the Perkin Elmer 2030 Reader (Victor X3). Rluc values were normalized with FLuc values at respective time points.
Treatment of the cells with IFN-α and BX795 inhibitor
Before transfection with RNA, cells were pre-treated for 2 h with 1 μM BX795 (InvivoGen) while IFN-α (500–1,000 U/ml) (Sigma) was added to the culture medium after 4 h of cell transfection with RNA. Cell treatment with BX795 or IFN-α was continued after transfection till the end point of the respective experiment. Cells remained untreated during the 4 h transfection period.
Gene Expression profiling by TaqMan Low Density Array (TLDA)
Antiviral pathway genes (n = 95) and 18 s rRNA as endogenous control were chosen and the array cards were procured from Applied Biosystems (USA). Gene expression profiling was carried out as described previously13.
Quantitative real-time PCR (qRT-PCR)
Individual SYBR green-based quantitative reverse transcription PCR assays were performed for selective genes. The cDNAs prepared as described previously13 were analyzed on 7300 Real-Time PCR system (Applied Biosystems, USA). GAPDH was used as a housekeeping gene to normalize the RNA input. RNA from mock transfected cells was used as the calibrator and relative gene expression analysis was carried out using SDS2.2 software (Applied Biosystems, USA).
Immunoblotting
Immunoblotting was carried out as described previously13. The primary antibodies used were anti-RIG-I (IMGENEX), mAb anti-phospho IRF3 (Ser396), anti-TLR3 (Cell Signaling Technology, Beverly, MA), anti-IRF3 and anti-actin (Sigma).
Results
Differential replication efficiencies of HEV in different hepatoma cell lines
Human hepatoma cell lines HepG2/C3A and Huh7 derived clonal cell lines S10-3 and Huh7.5 were transfected with capped RNA transcripts generated from HEV Rluc (original wild type) and GDD mutant (HEV Rluc GAA) replicons and processed to measure luciferase activities which would reflect viral RNA replication. HEV Rluc is subgenomic replicon which expresses Rluc in place of ORF2 (capsid) protein. Detection of Rluc activity means successful negative strand RNA intermediate synthesis, followed by positive strand genomic and subgenomic RNA synthesis. As seen from the Rluc activities, all three hepatoma cell lines were supporting HEV RNA replication, however, replication efficiencies differed. HepG2/C3A cells were found to be less supportive as compared to S10-3 (p = 0.0057, for the 6th day) and Huh7.5 (p = 0.0026, for the 6th day) cells. Renilla luciferase activity of HEV Rluc GAA remained negligible till 6 days without any increase from the base level values seen after 24 h in Huh7 derived cell lines S10-3, Huh7.5 (data not shown) and HepG2/C3A cells (Fig. 1a–c).
Figure 1. Differential replication efficiencies of HEV in different human hepatocyte derived cell lines.

S10-3 (a), Huh7.5 (b) and HepG2/C3A (c) cells were transfected with either wild-type HEV Rluc or HEV Rluc GAA (GDD mutant) RNA (2 μg/well). Cells were co-transfected with firefly luciferase plasmid DNA (pGL-3 promoter vector, 100 ng/well) to normalize cell transfection efficiency and the Renilla luciferase signal. Cell associated Renilla luciferase activity was checked to monitor HEV replication from 1-6 days. The data represents mean ± SD of three independent triplicate set of experiments, [** and ***represent p < 0.01 and p < 0.001 respectively, statistical comparisons were done by one-way analysis of variance (ANOVA) between mock transfected cells, and cells transfected with HEV-Rluc RNA].
Cell-specific antiviral responses
Since there was a significantly low level HEV replication in HepG2/C3A cells as compared to other two cell lines, we decided to analyze antiviral status of these cells. For that, TaqMan Low Density Array (TLDA)13 cards were used to analyze gene expression levels of different innate immune pathway genes in the transfected cells. Total RNA was isolated at 24 h and 96 h post-transfection and processed for the gene expression analysis as described previously13 (Table 1). Results obtained with low density arrays showed differential expression of genes which were functionally categorized as follows-.
Table 1. Gene expression profiling in Hepatoma cell lines transfected with HEV Rluc and HEV Rluc GAA RNA.
| Pathway/ Function | Gene Name | RQ Values (Fold change) determined by TLDA |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HepG2/C3A | S10-3 | Huh7.5 | |||||||||||
| 24 h | 96 h | 24 h | 96 h | 24 h | 96 h | ||||||||
| HEV Rluc | GAA | HEV Rluc | GAA | HEV Rluc | GAA | HEV Rluc | GAA | HEV Rluc | GAA | HEV Rluc | GAA | ||
| PRRs | DDx58 | 32 | 15.2 | 11.8 | 4.2 | 4.4 | 1.1 | 0.7 | 0.9 | 1.5 | 1.3 | 0.9 | 0.8 |
| IFIH1 | 142 | 80 | 7.2 | 2.6 | 5.8 | 1.5 | 0.8 | 1.2 | 2.1 | 1.5 | 1.2 | 1.2 | |
| TLR3 | 157 | 124 | 19.7 | 0.6 | 3.5 | 0.7 | 2.2 | 1.3 | 3.9 | 1.8 | 2.2 | 1.5 | |
| Interferons | IFNA1 | 3 | 1.5 | 7.5 | 0.5 | 3.2 | 0.7 | 1.2 | 0.7 | 3.8 | 1.4 | 3.7 | 0.3 |
| IFNB1 | 3.3 | 2 | 5.7 | 0.6 | 2.6 | 0.7 | 1 | 0.8 | 2.2 | 1.7 | 2.1 | 0.4 | |
| IFNW1 | 3.5 | 2 | 8.1 | 0.5 | 3 | 0.7 | 1.2 | 0.8 | 3.4 | 2.6 | 2.7 | 0.4 | |
| Interferon regulatory factors (IRFs) | IRF1 | 4.6 | 2.7 | 1.4 | 0.4 | 2.1 | 0.6 | 0.9 | 0.4 | 1.7 | 1.8 | 1 | 1 |
| IRF3 | 2 | 1.2 | 0.99 | 0.2 | 1.3 | 0.7 | 0.4 | 0.3 | 1.2 | 1.4 | 0.7 | 0.6 | |
| IRF7 | 3.9 | 2.6 | 1.1 | 0.1 | 3.2 | 1.1 | 1.9 | 0.6 | ND | ND | ND | ND | |
| IRF9 | 18.3 | 9.8 | 6.9 | 1.2 | 9.2 | 2.7 | 1.3 | 1.6 | 1.3 | 1.1 | 0.6 | 1.3 | |
| Interferon stimulated genes (ISGs) | ISG15 | 108 | 80.2 | 6.8 | 1.7 | 6 | 4 | 1.6 | 1.37 | 1.3 | 1.2 | 0.7 | 1 |
| ISG20 | 24.3 | 16.5 | 2 | 0.5 | 2.8 | 1.4 | 2.9 | 1 | 1.4 | 1.4 | 0.9 | 1 | |
| EIF2AK2/PKR | 10.2 | 6.3 | 3.7 | 1.3 | 4.3 | 1.1 | 0.7 | 0.6 | 1.5 | 1.1 | 1 | 0.7 | |
| GBP1 | 78.3 | 52 | 1.2 | 1.3 | 7.4 | 1.7 | 3.2 | 1.6 | 2.3 | 2.1 | 5.1 | 1 | |
| IFI27 | 901 | 600 | 140.7 | 63.4 | 6.7 | 2.5 | 3.5 | 1.3 | 1 | 0.7 | 1.9 | 0.7 | |
| IFIT1/ISG56 | 582 | 344.7 | 5.6 | 1.4 | 28.6 | 9.5 | 7.1 | 2 | 1.3 | 0.9 | 5.9 | 0.73 | |
| IFIT2/ISG54 | 13990 | 7424 | 120 | 65.2 | 15.9 | 3.6 | 1.4 | 1.5 | 2.9 | 3.1 | 2.9 | 1 | |
| MX1 | 865.5 | 372 | 98 | 39.6 | 50.4 | 14.7 | 9.3 | 2.7 | 1.1 | 0.9 | 7.3 | 0.6 | |
| MX2 | 16.6 | 10.2 | 1.8 | 0.9 | 1.3 | 0.4 | 0.2 | 0.3 | 1 | 1 | 0.3 | 0.6 | |
| OAS1 | 446 | 315 | 27.5 | 9.6 | 15.5 | 5.7 | 4.5 | 1.4 | 1.2 | 1 | 1.4 | 1.3 | |
| RSAD2/Viperin | 6873 | 4987 | 23.5 | 0.9 | 4.6 | 1.3 | 0.9 | 0.7 | ND | ND | ND | 0.9 | |
| SOCS1 | 4.5 | 2.4 | 3.8 | 1.2 | 1.2 | 0.6 | 0.5 | 0.3 | 1.3 | 1.1 | 1 | 0.4 | |
| STAT1 | 25 | 13.3 | 1.3 | 2.7 | 2 | 0.8 | 0.6 | 0.6 | 1.1 | 1.4 | 0.8 | 0.7 | |
| STAT2 | 4.5 | 2.2 | 1.2 | 0.7 | 1 | 0.4 | 0.2 | 0.4 | 1.3 | 1.3 | 0.6 | 0.5 | |
| STAT3 | 1.2 | 0.7 | 2.8 | 1.5 | 2.7 | 0.7 | 0.3 | 0.8 | 2 | 1.5 | 1.1 | 0.9 | |
| B2M | 7.3 | 5.2 | 4.9 | 0.8 | 1.9 | 0.7 | 0.6 | 0.7 | 0.9 | 1 | 0.9 | 0.6 | |
| TNFSF10/TRAIL | 34 | 20 | 2.5 | 0.8 | 2 | 0.4 | 0.9 | 0.3 | 1.2 | 0.8 | 1 | 0.9 | |
Gene expression results are given as averages from duplicate cultures and expressed as fold change (RQ value) compared to the mock transfected cells (Fold change ≥ 2 considered to be up-regulated genes) (ND, Not determined).
Pattern Recognition Receptors (PRRs)
Transcription levels of viral RNA sensing receptors RIG-I (DDx58), Mda5 (IFIH1) and TLR3 were higher in HepG2/C3A cells transfected with wild-type HEV Rluc RNA (~2 folds higher) as compared to HEV Rluc GAA RNA at 24 h. This difference in gene expression was more noticeableat 96 h post-RNA transfection suggesting the replication specific effect. However, expression levels of these genes in S10-3 and Huh7.5 cells, transfected with both HEV Rluc and HEV Rluc GAA RNA were significantly low. This suggested that S10-3 and Huh7.5 cells were either unable to detect replicating HEV RNA or had impaired downstream antiviral pathways.
Interferon regulatory factors (IRFs)
Activation of IRFs is the downstream signaling component after sensing of virus associated PAMPs. The transcription factors such as IRF1, IRF3, IRF7 and IRF9 are known to play important role in initiating cellular antiviral responses21. HepG2/C3A cells showed increased expression levels of IRF1, IRF3, IRF7 and IRF9 genes (4.6, 2.0, 3.9 and 18.3 folds respectively) as compared to mock transfected cells at 24 h. Expression levels of IRFs reverted back to basal level at 96 h, except for IRF9 which remained induced ~6 fold compared to GAA mutant RNA. IRF9 is the component of ISGF3 complex and aids in amplification of ISG response. This clearly corroborated with ongoing viral RNA replication and amplification of innate response in HepG2/C3A cells. There was no change in the expression levels of these genes in S10-3 and Huh7.5 cells.
Type I Interferons (IFNs) and Interferon stimulated genes (ISGs)
There was no significant increase in the levels of transcripts for the type I IFN genes, IFN-α, IFN-β and IFN-ω in any of the hepatoma cells transfected with HEV Rluc or HEV Rluc GAA.
Interferon stimulated genes such as ISG15, ISG20, PKR, IFI27, IFIT1 (ISG56), IFIT2 (ISG54), MX1, MX2, OAS1, RSAD2/ viperin, STAT1, STAT2, STAT3 and TNFSF10/TRAIL showed significantly higher induction in HepG2/C3A cells transfected with HEV Rluc RNA as compared to HEV Rluc GAA (Table 1). Point noteworthy here was that HepG2/C3A cells showed up regulation of above genes in response to HEV GAA Rluc, as in vitro transcribed capped RNA transcripts are known to have some amount of uncapped RNA (with 5′ phosphate ends), which is a potential inducer of innate immune response. However, expression levels of all ISGs were significantly higher in cells transfected with HEV Rluc at 24 h and 96 h. These observations indicated that HepG2/C3A cells were sensitive in detecting both single stranded RNA and double stranded RNA intermediates. On the other hand, S10-3 and Huh7.5 cells were less sensitive.
IFN-α inhibits HEV RNA replication in Huh7.5 cells
To confirm direct correlation of HEV replication with ISGs, we exposed Huh7.5 cells to IFN-α (1,000 U ml−1) 4 h after transfection and measured luciferase activity. Cells exposed to IFN showed significant suppression of HEV replication as compared to unexposed cells (Fig. 2a). These results were in agreement with previous reports that have shown functional impairment of pathogen recognition receptor/s in these cells22,23. To confirm antiviral status of IFN exposed Huh7.5 cells, we analysed expression levels of representative ISGs by SYBR green based quantitative real time PCR assay and detected considerably higher levels of ISG56, IFIH1 (Mda5) and Mx1 in cells transfected with HEV Rluc and then treated with IFN (Fig. 2b). These results showed that downstream antiviral response pathways are intact in Huh7.5 cells since externally added IFN-α could induce ISGs and restrict HEV replication.
Figure 2. Interferon-α treatment inhibits HEV replication in Huh7.5 cells.

(a) Huh7.5 cells transfected with HEV Rluc replicon RNA were treated with IFN-α (1,000 U/ml) 4 h post transfection. Cells were assayed for luciferase activity to monitor HEV RNA replication as described in Fig. 1, [* and ***represent p < 0.05 and p < 0.001 respectively, statistical comparisons were done by one-way analysis of variance (ANOVA) between cells transfected with HEV-Rluc RNA, and cells transfected with HEV-Rluc RNA and treated with IFN-α]. (b) Expression of ISGs; ISG56, Mda5, Mx1 and PKR genes in Huh7.5 cells was checked by SYBR-green based qRT-PCR, 24 h post RNA transfection (mRNA levels (fold) were calculated in relation to the levels of respective genes in mock transfected cells). The data represents mean ± SD of two independent triplicate sets of experiments [*represents p < 0.05, comparison between IFN-α treated cells and cells transfected with HEV-Rluc RNA and treated with IFN-α].
Reconstitution of RIG-I and TLR3 signaling pathways in Huh7.5 cells can restrict HEV replication
Huh7.5 cell line is known to have defective RIG-I signaling due to a point mutation within its CARD-like homology domain23. To assess whether RIG-I has any role in detecting double stranded RNA during HEV replication, we decided to reconstitute RIG-I pathway by transfecting these cells with plasmid containing RIG-I expression cassette, confirmed the expression (Fig. 3a) and assessed HEV replication in these cells. As shown in Fig. 3b, HEV replication remained low at 2 and 4 days post HEV RNA transfection in cells expressing functional RIG-I, whereas, cells transfected with empty vector showed significantly higher HEV replication at 4 days. Thus, RIG-I complementation in Huh7.5 cells resulted in phenotypic switch from hyper permissive to a relatively non permissive phenotype characterized by suppression of viral RNA replication. These results suggested important role of RIG-I as pathogen recognition receptor in detecting HEV RNA.
Figure 3. RIG-I and TLR3 are the pattern recognition receptors in sensing HEV RNA replication.

(a) RIG-I expression in Huh7.5 cells: Cells either mock transfected or transfected with RIG-I expression plasmid (pUNO-hRIG-I) were analyzed by immunoblotting for RIG-I protein levels at 24, 48, 72 and 96 h respectively (lanes 1–4). Actin was used as a loading control. (b) RIG-I complementation in Huh7.5 cells inhibits HEV RNA replication: Huh7.5 cells were either mock transfected or transfected with empty vector or with RIG-I expression plasmid. After 24 hours, cells were transfected with HEV Rluc RNA and monitored by luciferase assay at 2 and 4 days post RNA transfection respectively. (c) TLR-3 and TLR3-ΔTIR expression in Huh7.5 cells: Cells either mock transfected or transfected with TLR3 expression plasmids (pUNO-hTLR3 and pZERO-TLR3) were analyzed by immunoblotting for TLR3 protein levels at 24 and 48 h respectively. TLR3-ΔTIR is a TIR domain-less form of the TLR3 gene showing lower molecular weight compared to wild-type TLR3 (TLR3-ΔTIR recognizes its ligands but is unable to induce the signaling). Actin was used as a loading control. (d) TLR3 complementation in Huh7.5 cells inhibits HEV RNA replication: Huh7.5 cells were transfected with TLR3 or TLR3-ΔTIR expression plasmids 24 h prior to transfection with HEV Rluc RNA and monitored by luciferase assay at 2 and 4 days post transfection respectively [**represents statistical comparison by one-way analysis of variance (ANOVA) with cells transfected with empty vector or TLR3-ΔTIR expression plasmid and showed p < 0.01 in figures (b,d)]. (e) RIG-I and TLR3 complementation restore antiviral response in Huh7.5 cells: Cells expressing RIG-I and TLR3 were transfected with HEV Rluc RNA and expression of ISGs ISG56, MDA5, Mx1 and PKR were assessed by SYBR-green based qRT-PCR at 24 h post RNA transfection. (f) Antiviral response in HepG2/C3A cells: HepG2/C3A cells transfected with HEV Rluc RNA were analyzed for ISG expression similarly as in (e) with Huh7.5 cells as a control. The data represents mean ± SD of three independent triplicate sets of experiments. Cropped blots are used in the main figure and full length blots are included in Supplementary Figure 1.
Similar to RIG-I, Huh7 cells are reported to be defective in TLR3 signaling due to lower expression of TLR322. By reconstituting TLR3 pathway in Huh7.5 cells (Fig. 3c) we assessed the role of TLR3 in sensing HEV RNA replication. It was observed that elevated expression of functional TLR3 in Huh7.5 cells significantly inhibited HEV replication as compared to defective TLR3 (TLR3-ΔTIR), lacking the TIR signaling domain (Fig. 3d), however this inhibition was not as pronounced as that seen with RIG-I. These results indicated that both RIG-I (in the cytoplasm) and TLR3 (in the endosomal compartments) can function as sensors in hepatocytes for HEV RNA.
To confirm antiviral status of the cells we assessed levels of representative ISGs in the RIG-I expressing cells. As expected, the levels of ISG56 (105 fold), Mda5 (47 fold), Mx1 (227 fold) and PKR (10 fold) were significantly increased only in cells transfected with plasmid containing functional RIG-I gene and not with the empty vector. Similar analyses with TLR3 complementation showed comparatively lower levels of ISGs (~2.5 to 4 folds) as compared to RIG-I complemented cells (Fig. 3e) indicating TLR3 being less effective in detecting HEV replication as compared to RIG-I. HepG2/C3A cells transfected with HEV Rluc RNA showed increased expression of ISGs in agreement with the TLDA results (Fig. 3f). Overall, these results showed involvement of RIG-I and TLR3 pathways in restricting HEV replication in hepatoma cells.
Inhibition of pattern recognition receptor signaling enhances HEV replication
After confirming the role of RIG-I and TLR3 in sensing HEV RNA, we checked the effect of inhibition of IKK related kinase, TBK1 (TANK-binding kinase 1) and IKKε complex, which acts as the common mediator in downstream signaling for both RLRs (RIG-I/Mda5) and TLR3. HepG2/C3A cells were exposed to BX795, a cell permeable inhibitor which is relatively specific for TBK1/IKKε, known to block phosphorylation, nuclear translocation and transcription activity of IRF324. To analyse possible changes in the IRF3 phosphorylation status of BX795 treated cells, cells were transfected with either double stranded RNA analogue, poly (I:C) or HEV Rluc. There was a significant reduction in the levels of IRF3 phosphorylation in BX795 treated cells as compared to untreated cells (Fig. 4a,b). Importantly, there were comparable levels of IRF3 phosphorylation at the early time points after transfection with HEV Rluc or HEV Rluc GAA (at 10 and 24 h) in BX795 untreated cells, which was also evident from the elevated expression levels of antiviral genes. However, there was a selective increase in IRF3 phosphorylation at the later stages (48 h) only in cells transfected with HEV Rluc, indicating requirement of ongoing viral RNA replication (double stranded RNA formation). BX795 treatment also inhibited poly (I:C) triggered IRF3 phosphorylation at 10 and 24 h (Fig. 4c). Inhibition of IRF3 phosphorylation was also evident from the down regulation of transcripts for type I IFNs and ISGs such as PKR and Mx1 in BX795 treated cells as compared to untreated cells (Fig. 4d–f).
Figure 4. BX795 blocks the phosphorylation and activation of IRF3 leading to suppression of interferon stimulated genes (ISGs).

HepG2/C3A cells, untreated or treated with BX795, were transfected with (a) HEV Rluc RNA, (b) HEV Rluc GAA and (c) double stranded RNA analogue (poly I:C) and processed for immunoblotting for the detection of phosphorylated IRF3 (P-IRF3, Ser396). The blot was reprobed for the detection of total IRF3 (T-IRF3) and actin at indicated time points.The relative protein band density for p-IRF3 was normalized with actin and compared with mock treated and BX795 treated cells at different time points and mentioned as relative fold (RF) values. Cropped blots are used in the main figure and full length blots are included in Supplementary Figure 1. (d–f) HepG2/C3A cells left untreated or treated with BX795 were transfected with HEV Rluc RNA and processed for qRT-PCR of type I IFN genes (IFNA and IFNB) and ISGs (PKR and Mx1) at 24 h post transfection. The data represents mean ± SD of three independent experiments [*, ** and ***represent p < 0.05, p < 0.01 and p < 0.001 respectively, statistical comparisons were done by one-way analysis of variance (ANOVA) between HEV Rluc and BX795 + HEV Rluc].
Treatment of HepG2/C3A cells with 1 μM BX795 showed significant improvement in HEV replication. On monitoring of the cells that were transfected with HEV Rluc further, a steady increase in the replication was observed from the days 1–5 in BX795 treated cells as compared to untreated cells (Fig. 5). Overall, these results confirmed the role of IRF3 mediated antiviral pathways in restricting HEV RNA replication in HepG2/C3A cells.
Figure 5. Inhibition of IFN signaling enhances HEV replication efficiency.
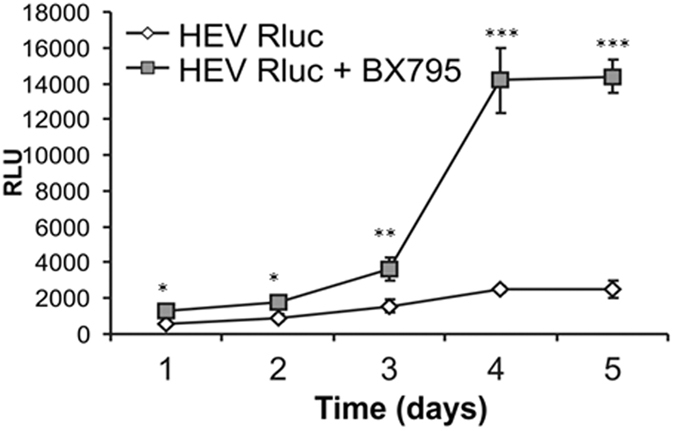
HepG2/C3A cells untreated or treated with BX795 were transfected with HEV Rluc RNA and viral RNA replication was monitored by luciferase assay from 1–5 days post transfection. The data represents mean ± SD of three independent triplicate sets of experiments [*, ** and ***represent p < 0.05, p < 0.01 and p < 0.001 respectively, statistical comparisons were done by one-way analysis of variance (ANOVA) between untreated and BX795 treated cells at indicated time points].
Discussion
Though there are reports on efficient cell culture models for genotype 3 and 4 viruses25, better cell culture systems are still lacking for genotype 1 HEV studies. We used HEV replicons and human hepatoma cells such as HepG2/C3A, Huh7.5 and S10-3 for HEV replication analysis and observed that these cells do not support HEV replication at comparable levels. In the present study, we addressed this issue by utilizing luciferase reporter based subgenomic HEV replicon (HEV Rluc) that allowed quantitation of HEV replication in terms of Renilla luciferase activity. Real-time PCR based assay could not be used for the quantitation due to two reasons, high background of replicon RNA used for the transfections and very low replication efficiency of the replicon. The quantitative luciferase assay showed that HEV replicates more efficiently in Huh-7 derived cell lines, S10-3 and Huh7.5, as compared to HepG2/C3A cells (Fig. 1). Similar observations have been reported by Emerson et al.17. Cellular antiviral responses were the most obvious cause for the restricted virus replication. It is also known that Huh7 cells have impaired TLR3 pathway while, Huh7.5 are defective in both RIG-I and TLR3 pathways. Hence, it was understood that these cells were able to restrict HEV replication less efficiently as compared to HepG2/C3A cells.
Our gene expression analysis experiments confirmed that there were comparatively higher levels of the pattern recognition receptor (PRRs) and type I IFN pathway gene transcripts in HepG2/C3A cells as compared to S10-3 and Huh7.5 cells (Table 1). Though expression levels were higher in HepG2/C3A cells transfected with HEV Rluc as well as HEV Rluc GAA (mutant) in comparison with mock transfected cells, levels were further elevated in cells transfected with HEV Rluc. This indicated that cells could sense secondary structures in RNA immediately upon transfection and triggered antiviral responses and upon sensing double stranded RNA replication intermediates enhanced this induction further. To confirm involvement of type I IFNs in eliciting antiviral environment, we exposed Huh7.5 to IFN-α, and detected significant inhibition of HEV replication, due to induction of ISGs (Fig. 2). This confirmed presence of intact downstream pathway/s in these cells. Huh7 cells have proven to be nearly unique in their ability to support autonomous replication of hepatitis C virus26 and hepatitis A virus27 RNA replicons.
Pattern recognition receptors (PRR) recognizing HEV associated molecular patterns are still unknown. Considering presence of defective RIG-I/TLR3 signaling and HEV permissiveness of Huh7.5 cells, we used these cells further to identify HEV PRRs. We reconstituted RIG-I and TLR3 signaling by expressing functional proteins exogenously in Huh7.5 cells and observed that both viral RNA sensing pathways are involved in recognizing HEV RNA. It was seen that RIG-I acted as an immediate sensor of HEV RNA (at 2 days) while, TLR3 sensed replicative intermediates comparatively later (at 4 days) (Fig. 3b,d). This was expected since, ssRNA with 5′-phosphate groups and dsRNA are both potential RIG-I ligands12, while TLR3 senses only dsRNA. Reconstitution of the RIG-I signaling in Huh7.5 cells resulted in comparatively higher levels of ISGs that resulted in efficient virus inhibition as compared to TLR3 reconstitution (Fig. 3e). The difference could be also due to specific cellular locations of these RNA sensors, as TLR3 mainly resides in endosomal compartments while RLRs remain in cytoplasm. Some RNA viruses release their viral RNA genomes into cytoplasm allowing cytosolic PRRs to be activated12. However, activation via endosomal TLRs remains limited, since most of the viral RNA remains membrane-associated in viral replication complexes, and probably safeguarded from the TLR3 surveillance. Proposed site of HEV replication is assumed to be endoplasmic reticulum28 and hence less accessible to TLR3.
Following infection, latent IRF3 in cytoplasm is known to get translocated into nucleus upon TBK1/IKKε mediated phosphorylation and result into transcription of IFN and IFN stimulated genes29. Both TLR3 and RIG-I pathways are known to converge into downstream IRF3 activation. Since both RIG-I and TLR3 were detected as HEV RNA sensors, we decided to inhibit downstream IRF3 activation. When HepG2/C3A cells were exposed to BX795, an inhibitor of TBK1/IKKε24, there was a significant decrease in IRF3 phosphorylation, low level induction of ISGs and these resulted finally in improving HEV replication. Similar enhancement in lentiviral transduction efficiency has been shown in BX795 treated human and mouse cell lines30.
Various human hepatoma cell lines HepG2, PLC/PRF5 and Huh7 and non-hepatoma cell lines Caco2 (human colon carcinoma) and A549 are known to support HEV replication (13, 17, 25, 31–33). However, none of these cell culture systems can provide high titers of infectious virus in culture supernatants. Amongst these cells, Huh7 derived cell lines are comparatively better, however, HEV infection needs to be initiated by transfecting these cells with replicon RNA instead of direct infection with native virus particles. Although PLC/PRF5 cell line was reported to propagate genotype 3 HEV32,33, this cell line contains hepatitis virus B surface antigen (HBsAg) and thus cannot be used to study HEV specific innate immune response. On the other hand, HepG2/C3A cells can be directly infected with virus particles but they restrict virus replication. HepG2/C3A cells are thought to have higher density of HEV receptors on their surface as compared to Huh7 derived cell lines31 and hence are a better system to assess virus infectivity. With present observations, we feel BX795 treated HepG2/C3A cells could be used as a cell culture model for genotype 1 HEV studies.
In conclusion, we observed direct correlation between HEV replication and levels of antiviral responses in different hepatoma cell lines. RIG-I and TLR3 pattern recognition receptors were mainly responsible for HEV RNA detection in these cells. Inhibition of IRF3 activation by BX795 in HepG2/C3A cells prevented synthesis of interferon stimulated genes and improved HEV replication in these cells. BX795 could be used to improve HEV replication in HepG2/C3A cells for its use as a model system to study HEV biology.
Additional Information
How to cite this article: Devhare, P. B. et al. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 6, 26827; doi: 10.1038/srep26827 (2016).
Supplementary Material
Acknowledgments
The work described here was supported by Indian Council of Medical Research (ICMR) and National Institute of Virology (NIV), India. We thank Dr. S Emerson (NIH, USA), Dr. X. J. Meng (Virginia Tech, Blacksburg, USA) for providing us the S10-3 cell line and HEV luciferase subgenomic replicon respectively.
Footnotes
Author Contributions P.B.D. and K.S.L. conceived the idea, planned the experiments and wrote the manuscript. P.B.D. and S.D. conducted the experiments and analyzed the results. All authors reviewed the manuscript.
References
- Smith D. B. et al. Consensus proposals for classification of the family Hepeviridae. J Gen Virol 95, 2223–2232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad I., Holla R. P. & Jameel S. Molecular virology of hepatitis E virus. Virus Res 161, 47–58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holla R. P., Ahmad I., Ahmad Z. & Jameel S. Molecular virology of hepatitis E virus. Semin Liver Dis 33, 3–14 (2013). [DOI] [PubMed] [Google Scholar]
- Koonin E. V. et al. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. ProcNatlAcadSci USA 89, 8259–8263 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perttila J., Spuul P. & Ahola T. Early secretory pathway localization and lack of processing for hepatitis E virus replication protein pORF1. J Gen Virol 94, 807–816 (2013). [DOI] [PubMed] [Google Scholar]
- Graff J., Torian U., Nguyen H. & Emerson S. U. A bicistronicsubgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J Virol 80, 5919–5926 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalimar, & Acharya S. K. Hepatitis E and Acute Liver Failure in Pregnancy. J ClinExpHepatol 3(3), 213–224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamar N., Dalton H. R., Abravanel F. & Izopet J. Hepatitis E virus infection. Clinical microbiology reviews 27, 116–138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S., Uematsu S. & Takeuchi O. Pathogen recognition and Innate Immunity. Cell 124, 783–801 (2006). [DOI] [PubMed] [Google Scholar]
- Randall R. E. & Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89, 1–47 (2008). [DOI] [PubMed] [Google Scholar]
- Thompson M. R., Kaminski J. J., Kurt-Jones E. A. & Fitzgerald K. A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S. & Thomsen A. R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86, 2900–2910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devhare P. B., Chatterjee S. N., Arankalle V. A. & Lole K. S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 8(5), e63793 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. et al. Suppression of interferon-α signaling by hepatitis E virus. HEPATOLOGY 55, 1324–1332 (2012). [DOI] [PubMed] [Google Scholar]
- Nan Y. et al. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J Virol 88, 11924–11932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. K., Ansari I. H., Durgapal H., Agrawal S. & Jameel S. The in vitro synthesized RNA from a cDNA clone of hepatitis E virus is infectious. J Virol 74, 2430–2437 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson S. U. et al. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J Virol 78, 4838–4846 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J. et al. In vitro and in vivo mutational analysis of the 3′-terminal regions of hepatitis E virus genomes and replicons. J virol 79, 1017–1026 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D., Huang Y. W. & Meng X. J. The nucleotides on the stem-loop RNA structure in the junction region of the hepatitis E virus genome are critical for virus replication. J Virol 84, 13040–13044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devhare P., Sharma K., Mhaindarkar V., Arankalle V. & Lole K. Analysis of helicase domain mutations in the hepatitis E virus derived from patients with fulminant hepatic failure: Effects on enzymatic activities and virus replication. Virus Res 184, 103–110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K. & Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 6(9), 644–658 (2006). [DOI] [PubMed] [Google Scholar]
- Li K., Chen Z., Kato N., Gale M. Jr. & Lemon S. M. Distinct Poly(I-C) and virus-activated signaling pathways leading to interferon production in Hepatocytes. J BiolChem 280, 16739–16747 (2005). [DOI] [PubMed] [Google Scholar]
- Sumpter R. Jr. et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79, 2689–2699 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Plater L., Peggie M. & Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J BiolChem 284, 14136–14146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H. Culture systems for hepatitis E virus. Journal of Gastroenterol, 48(2), 147–158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999). [DOI] [PubMed] [Google Scholar]
- Yi M. & Lemon S. M. Replication of subgenomic hepatitis A virus RNAs expressing firefly luciferase is enhanced by mutations associated with adaptation of virus to growth in cultured cells. J Virol 76, 1171–1180 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S., Kapur N., Durgapal H. & Panda S. K. Subcellular localization of hepatitis E virus (HEV) replicase. Virology 370, 77–92 (2008). [DOI] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J BiolChem 282, 15325–15329 (2007). [DOI] [PubMed] [Google Scholar]
- Sutlu T. et al. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Human gene therapy. 23, 1090–1100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson S. U. et al. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J Virol 84, 9059–9069 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Takahashi M., Kusano E. & Okamoto H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J Gen Virol 88, 903–911 (2007). [DOI] [PubMed] [Google Scholar]
- Takahashi M. et al. Hepatitis E virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: characterization of HEV virions in blood circulation. J ClinMicrobiol 48, 1112–1125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.