

Abstract
Neuroendocrine (NE) cancers can cause significant patient morbidity. Besides surgery, there are no curative treatments for NE cancers and their metastases, emphasizing the need for the development of other forms of therapy. In this study, multifunctional unimolecular micelles were developed for targeted NE cancer therapy. The unimolecular micelles were formed by multi-arm star amphiphilic block copolymer poly(amidoamine)–poly(valerolactone)–poly(ethylene glycol) conjugated with KE108 peptide and Cy5 dye (abbreviated as PAMAM–PVL–PEG–KE108/Cy5). The unimolecular micelles with a spherical core–shell structure exhibited a uniform size distribution and excellent stability. The hydrophobic drug thailandepsin-A (TDP-A), a recently discovered HDAC inhibitor, was physically encapsulated into the hydrophobic core of the micelles. KE108 peptide, a somatostatin analog possessing high affinity for all five subtypes of somatostatin receptors (SSTR 1–5), commonly overexpressed in NE cancer cells, was used for the first time as an NE cancer targeting ligand. KE108 exhibited superior targeting abilities compared to other common somatostatin analogs, such as octreotide, in NE cancer cell lines. The in vitro assays demonstrated that the TDP-A-loaded, KE108-targeted micelles exhibited the best capabilities in suppressing NE cancer cell growth. Moreover, the in vivo near-infrared fluorescence imaging on NE-tumor-bearing nude mice showed that KE108-conjugated micelles exhibited the greatest tumor accumulation due to their passive targeting and active targeting capabilities. Finally, TDP-A-loaded and KE108-conjugated micelles possessed the best anticancer efficacy without detectable systemic toxicity. Thus, these novel TDP-A-loaded and KE108-conjugated unimolecular micelles offer a promising approach for targeted NE cancer therapy.
Keywords: neuroendocrine cancers, somatostatin receptor, KE108 peptide, unimolecular micelles, targeted drug delivery, HDAC inhibitor
1. INTRODUCTION
Neuroendocrine (NE) cancers are the second most prevalent gastrointestinal tract malignancy after colorectal cancer [1–4]. Surgical resection is the only curative option, but most patients are not candidates for operative intervention due to widespread metastases at the time of presentation [5, 6]. Moreover, other forms of therapy have had limited efficacy, emphasizing the need for the development of new therapy [7].
We have an ongoing interest in identifying effective approaches of reactivating tumor suppressor pathways and altering malignant NE phenotypes. In previous studies, we showed that the inhibition of histone deacetylases (HDACs) triggered activation of Notch signaling and induced cell cycle arrest and apoptosis, leading to NE cancer cell death [8–11]. HDAC inhibitors have hence emerged as a new type of anticancer agent. Up to now, three HDAC inhibitors—vorinostat (SAHA, Zolinza®) [12], belinostat (PXD-101, Beleodaq®) [13], and natural product FK228 (romidepsin, Istodax®) [14]—have been approved by the FDA. Thailandepsin A (TDP-A), a natural and potent HDAC inhibitor, was recently isolated by Cheng [15] from the bacterium Burkholderia thailandensis E264. TDP-A can selectively and strongly inhibit class I HDACs including HDAC1, HDAC2 and HDAC3 [15, 16]. It also showed potent cytotoxic activities against NE and endocrine cancer cell lines at low nanomolar concentrations [11, 17]. Its high potency and selective inhibition of class I HDACs makes TDP-A a promising candidate as an HDAC inhibitor. However, TDP-A is hydrophobic and lacks in vivo tumor-targeting capability when administered systemically, leading to dose-limiting toxicity and thereby limiting its application.
To overcome the poor water solubility, improve the biostability, and increase the tumor accumulation of TDP-A, we have developed a unique unimolecular micelle nanoparticle as a nanocarrier for TDP-A. Drug nanocarriers can not only improve the water solubility and stability of hydrophobic drugs, but also increase the therapeutic index of drugs by delivering them specifically to tumor sites via the enhanced permeability and retention (EPR) effect [18, 19]. Moreover, drug nanocarriers conjugated with specific tumor-targeting ligands (e.g., peptides, antibodies, aptamers, or small molecules) can further increase their accumulation at target sites and therefore significantly enhance a drug’s therapeutic effects [20–23].
Polymer micelles self-assembled from linear amphiphilic block copolymers have been widely studied as drug carriers [24–28]. Polymer micelles exhibit a core–shell architecture where the hydrophobic core serves as a natural carrier environment for hydrophobic drugs, while the hydrophilic shell imparts the micelle nanoparticle aqueous solubility. However, one major concern with the most widely studied drug delivery nanocarriers—including polymer micelles and vesicles, as well as liposomes, which are formed via the self-assembly of a large number of linear amphiphilic block copolymers or phospholipids—is their in vivo stability due to the dynamic nature of the self-assembly process. The in vivo stability of these self-assembled drug nanocarriers is affected by a variety of factors, including the concentration of the amphiphilic molecules, pH, temperature, ionic strength, or their interaction with proteins [29–31]. Premature dissociation of self-assembled drug nanocarriers during circulation in the bloodstream can cause a burst release of anticancer drugs into the bloodstream, which not only leads to potential systemic toxicity, but also undermines their tumor-targeting ability, thereby severely limiting their use for in vivo applications. Hence, there is a need to develop strategies to enhance the in vivo stability of self-assembled drug nanocarriers. Unimolecular micelles formed by individual/ single multi-arm star amphiphilic block copolymer molecules exhibit excellent stability in vitro and in vivo due to their unique chemical structure and covalent nature [32–40]. These unique unimolecular micelles have been successfully used to deliver various compounds to tumor tissues in a targeted manner [32–40].
The majority of NE cancers overexpress somatostatin receptors (SSTRs, a family of guanosine triphosphate-binding protein-coupled receptors, of which there are five subtypes, SSTR 1–5) [37, 41–43]. Our previous studies, as well as others, have shown that octreotide, a somatostatin analog with a strong binding affinity to SSTR, can be used as an effective tumor-targeting ligand for targeted NE cancer therapy [37, 41]. Octreotide displays a high binding affinity to SSTR2 and a moderate affinity to SSTR5 [44]. It was the first somatostatin analog used in clinic, but has very little affinity to the other three SSTR subtypes (i.e., SSTR1, SSTR3, and SSTR4) [44–46]. On the other hand, KE108, another somatostatin analog, shows a pan-somatostatin profile because it binds to all five SSTRs with high affinity (Fig. 1 (A)) [47], thus making it a superior targeting ligand for SSTRs. Despite its strong potential as an effective active NE tumor-targeting ligand, there have not been any reports on KE108-conjugated drug nanocarriers.
Fig. 1.

(A) The binding affinity of two somatostatin analogs, octreotide and KE108, to the five subtypes of somatostatin receptors (SSTR 1–5). KE108 displays a high affinity to all five SSTRs, while octreotide only has a high binding affinity to SSTR2 and a moderate affinity to SSTR5. (B) A schematic illustration of the multifunctional unimolecular micelle formed by multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108 for targeted NE cancer therapy. (C) A schematic illustration of the passive and active tumor targeting capabilities exhibited by the multifunctional unimolecular micelles after i.v. injection. Passive tumor targeting is achieved by extravasation of the unimolecular micelles as a result of leaky vessels of the tumor vasculature (i.e., the EPR effect). Active tumor targeting is achieved through the interaction between the KE108 targeting ligands present on the surface of the unimolecular micelles and the SSTRs overexpressed by the NE cancer cells triggering an enhanced cellular uptake of the micelles by the NE cancer cells via receptor-mediated endocytosis.
In this study, for the first time, a multifunctional unimolecular micelle nanoplatform using KE108 as the active tumor-targeting ligand was developed for targeted NE cancer drug delivery (e.g., TDP-A). The unimolecular micelles were formed by multi-arm star amphiphilic block copolymer poly(amidoamine)–poly(velarolactone)–poly(ethylene glycol) conjugated with KE108 peptide and Cy5 dye (abbreviated as PAMAM–PVL–PEG–KE108/Cy5) (Fig. 1 (B)). Cy5 dye was conjugated for visualization and detection of micelles both in vitro and in vivo. TDP-A was encapsulated inside of the hydrophobic core of the unimolecular micelles through hydrophobic interactions and hydrogen bonding. These drug-loaded unimolecular micelles are capable of both passive and active targeting for NE cancer therapy as illustrated in Fig. 1 (C).
2. METHODS
2.1 Materials
Poly(amidoamine) (1,4-diaminobutane; G4) dendrimer was purchased from NanoSynthons LLC (Mt. Pleasant, MI, USA). Dimethyl sulfoxide (DMSO), velarolactone (VL), and stannous (II) octoate (Sn(Oct)2) were purchased from Sigma–Aldrich (St. Louis, MO, USA). The heterobifunctional poly(ethylene glycol) (PEG) derivatives, methoxy–PEG–OH (mPEG–OH, Mn=5 kDa) and OH–PEG–N–hydroxylsuccinimide (HO–PEG–NHS, Mn=5 kDa) were purchased from JenKem Technology (Allen, TX, USA). 4-Dimethylamino pyridine (DMAP) and 1,3-dicyclohexylcarbodiimide (DCC) were purchased from ACROS and used without further purification. KE108 was purchased from Bachem Americas, Inc. (Torrance, CA, USA). Cy5 dye was obtained from Lumiprobe Corporation (Hallandale Beach, FL, USA). Other reagents were purchased from Thermo Fisher Scientific (Fitchburg, WI, USA) and used as received unless otherwise stated.
2.2 Synthesis of PAMAM–PVL–OH
PAMAM–PVL–OH was prepared by ring-opening polymerization of the VL monomer using PAMAM–OH as the macro-initiator (Scheme 1). A 50 ml two-neck flask equipped with an argon gas inlet was charged with PAMAM–OH (20 mg, 1.4 μmol) and placed in an oil bath. VL (277 mg, 2.7 mmol) and a catalytic amount of Sn(Oct)2 ([Sn(Oct)2]/[VL]=1/1000 mol/mol) was added subsequently. The reaction was carried out at 120 °C for 24 h. The resulting mixture was dissolved in THF and the solution was added dropwise into methanol to yield a pale yellow precipitate. The final product, PAMAMA–PVL–OH, was dried under vacuum.
Scheme 1.

Synthesis scheme of the multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108.
2.3 Synthesis of PAMAM–PVL–COOH
PAMAM–PVL–COOH was prepared by a reaction between PAMAM–PVL–OH (50 mg, 0.51 μmol) and succinic anhydride (8.2 mg, 82 μmol) in the presence of DMAP (12.5 mg, 98 μmol). The reaction was carried out in anhydrous DCM (10 mL) for 48 h at room temperature. Thereafter, the resulting solution was added into 10 fold of cold diethyl ether. The precipitate was redispersed in DI water and the impurities were removed by dialysis against DI water using cellulose tubing (molecular weight cut-off, 15 kDa) for 48 h. The final product was obtained after lyophilization.
2.4 Synthesis of PAMAM–PVL–PEG–OCH3/NHS
PAMAM–PVL–PEG–OCH3/NHS was synthesized by reacting PAMAM–PVL–COOH with mPEG–OH and NHS–PEG–OH in 10 mL of DMF in the presence of DCC and DMAP. The molar ratio of the reactants (PAMAM–PVL–COOH:mPEG–OH:NHS–PEG–OH:DCC:DMAP) was set at 1:48:16:70:7. The reaction was carried out at room temperature for 24 h and the byproduct, dicyclohexylcarbodiurea (DCU), was removed by filtration. The resulting solution was added dropwise into 10 fold of cold diethyl ether to yield the crude product. The impurities were then removed by dialysis against DMF for 48 h using cellulose tubing (molecular weight cut-off, 15 kDa). The resulting polymer, PAMAM–PVL–PEG–OCH3/NHS, was dried under vacuum. PAMAM–PVL–PEG–OCH3 was prepared following a similar procedure.
2.5 Synthesis of PAMAM–PVL–PEG–OCH3/KE108/Cy5
PAMAM–PVL–PEG–OCH3/NHS was first reacted with KE108 in DMF at a molar ratio of 1:6 under continuous stirring for 8 h. Thereafter, Cy5 dye (PAMAM–PVL–PEG–OCH3/NHS: Cy5 = 1:2) was added and the reaction was stirred for another 16 h. Then the impurities were removed by dialysis against DI water for 48 h using cellulose tubing (molecular weight cut-off, 15 kDa). The resulting polymer, PAMAM–PVL–PEG–OCH3/KE108/Cy5, was obtained after lyophilization. The multi-arm star amphiphilic block copolymers PAMAM–PVL–PEG–OCH3/ Cy5 and PAMAM–PVL–PEG–OCH3/KE108 were also prepared following a similar procedure.
2.6 Preparation of TDP-A-Loaded Unimolecular Micelles
To prepare the TDP-A-loaded targeted unimolecular micelles, PAMAM–PVL–PEG–OCH3/ KE108 (20 mg) and TDP-A (5 mg) were dissolved in 3 mL of acetonitrile under stirring. Thereafter, 9 mL of DI water was added dropwise into the solution under constant stirring. The resulting solution was stirred for 4 h and acetonitrile was then removed by rotary evaporation. The final TDP-A-loaded targeted unimolecular micelles were obtained after lyophilization. The TDP-A-loaded non-targeted unimolecular micelles were prepared following a similar method, using the PAMAM–PVL–PEG–OCH3 polymer instead.
2.7 Characterization
1H NMR spectra of all intermediate and final polymer products were recorded on a Varian Mercury Plus 300 spectrometer in DMSO–d6 or CDCl3 at 25 °C. Molecular weights (Mn and Mw) and polydispersity indices (PDI) of the polymers were determined by a gel permeation chromatographer (GPC) equipped with a refractive index detector, a viscometer detector, and a light scattering detector (Viscotek, USA). DMF with 0.1 mmol/L of LiBr was used as the mobile phase with a flow rate of 1 mL/min. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer. The sizes and morphologies of the unimolecular micelles were studied by dynamic light scattering (DLS, ZetaSizer Nano ZS90, Malvern Instruments, USA) and transmission electron microscopy (TEM, FEI Tecnai G2 F30 TWIN 300 KV, E.A. Fischione Instruments, Inc. USA). The TDP-A drug loading level, defined as the weight percentage of TDP-A in the TDP-A-loaded unimolecular micelles, was measured by high-performance liquid chromatography (HPLC) using ultraviolet (UV) detection at 200 nm. Water and acetonitrile (v/v 60/40) at a constant flow rate of 1 mL/min were used as the mobile phases.
2.8 Cellular Uptake Analyses
The cellular uptake behaviors of the micelles in NE cancer cell lines (human gastrointestinal carcinoid cell line BON and human medullary thyroid cancer cell line MZ–CRC–1) were analyzed using both flow cytometry and CLSM based on the Cy5 dye conjugated on the micelles. For flow cytometry analyses, cells were seeded in 6-well plates at a density of 3×105 cells/ml and incubated overnight in culture medium under standard conditions. Then the cells were treated with targeted (KE108-conjugated) or non-targeted (KE108-lacking) micelles at a concentration of 100 μg/ml and further incubated for 120 min. The culture medium was used as a blank control. Thereafter, the cells were washed with PBS and lifted using a non-enzymatic cell dissociation solution and stained with DAPI (10 ng/ml). The cellular uptake of the micelles was analyzed using a FACS Calibur four-color analysis cytometer (Becton Dickinson, San Jose, CA) and FlowJo analysis software (Tree Star, Inc., Ashland, OR). For each sample, at least 1×105 cells were analyzed and the Cy5 fluorescence was displayed on a linear scale. The cellular uptake behavior of the micelles in WI-38 (pulmonary fibroblasts), a cell line that does not overexpress SSTRs, was conducted following similar procedures and was used as the negative control. For the CLSM studies, cells were seeded (1×105 cells/ml) onto 96-well high-optical-quality plates and grown overnight. Cells were treated with either targeted or non-targeted micelles at a concentration of 100 μg/ml, followed by incubation for 120 min. The cells were then washed with PBS, fixed with 1.5% formaldehyde for 15 min, and washed again with double-distilled H2O. Then the cells on the optical plate were mounted with Prolong Gold anti-fade reagent with DAPI (Life Technologies, Carlsbad, CA) and the cellular uptake was observed with a Nikon A1R–Si high-speed spectral laser scanning confocal inverted microscope (Nikon, Melville, NY). Digital monochromatic images were acquired using NIS-Elements BR Software.
2.9 Quantitative Real-Time PCR of SSTR 1–5 Message Levels in NE Cancer Cells
Following the incubation with conditional media for two days, RNA was isolated from BON and MZ-CRC-1 cells using an RNeasy Mini-kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s directions and quantified using NanoDrop instrumentation (Thermo Scientific, Waltham, MA USA). Using 2 μg of RNA per sample, cDNA was synthesized with the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). The SSTR 1–5 primer sequences were as follows: SSTR1 forward, 5′-ATGGTGGCCCTCAAGGCCGG-3′ and reverse, 5′-CGCGGTGGCGTAATAGTCAA-3′; SSTR2 forward, 5′-CCAACACCTCAAACCAGAC-3′ and reverse, 5′-CATAGCCAAGAAAGGCAGAC-3′; SSTR3 forward, 5′-TCATCTGCCTCTGCTACCTG-3′ and reverse, 5′-GAGCCCAAAGAAGGCAGGCT-3′; SSTR4 forward, 5′-ATCTTCGCAGACACCAGACC-3′ and reverse, 5′-ATCAAGGCTGGTCACGACGA-3′; SSTR5 forward, 5′-CCGTCTTCATCATCTAACACGG-3′ and reverse, 5′-GGCCAGGTTGACGATGTTGA-3′. S27 was used as a housekeeping control to which SSTRs messages were normalized. The s27 primer sequences were as follows: forward, 5′-TCTTTAGCCATGCACAAACG-3′ and reverse, 5′-TTTCAGTGCTGCTTCCTCCT-3′. Quantitative real-time PCR reactions were performed using a CFX Thermal Cycler and SsoFast EvaGreen labeling system (Bio-Rad Laboratories). Each reaction was plated in triplicate. RNA expression levels were calculated using the comparative cycle threshold method (ΔCt), as described in the Real-Time PCR Applications Guide (Bio-Rad Laboratories). Once normalized to s27, relative SSTR1–5 expression for each cell line was plotted as a mean ± s.e.m.
2.10 Cell Proliferation Assays
The ability of TDP-A-loaded micelles to suppress NE cell proliferation was measured using 3-[4,5-dimethylthiazol-2-yl]–2,5-diphenyltetrazolium bromide (MTT) assays. BON cells were plated in quadruplicate on 96-well plates and incubated overnight. Cells were treated with either TDP-A-loaded targeted or non-targeted micelles, or one of the four controls; namely, empty targeted and empty non-targeted micelles (drug-free micelles), free TDP-A (5 nM), and pure medium. The equivalent TDP-A concentration for all TDP-A-loaded micelles was 5 nM. The cells were further incubated for 24 and 48 h, and the MTT assay was performed at each time point by aspirating the treatment medium and adding 25 μL of serum-free medium containing 0.5 mg/ml MTT reagent and incubating at 37 °C for 4 h. Thereafter, 75 μL of DMSO was added to each well and mixed thoroughly. The plates were then measured at 570 nm using a spectrophotometer (Quant, Bio-Tek Instruments, Winooski, VT), and the average absorbance and percent of cell viability relative to the control (pure medium) were calculated.
2.11 Tumor Marker Expression Assays
Western blot analysis was used to assess NE marker expression, including achaete–scute complex-like 1 (ASCL-1) and synaptophysin (SYP). BON cells were seeded into 100 mm culture plates and incubated overnight. The culture medium was removed and replaced with medium containing either TDP-A-loaded non-targeted or targeted micelles, or one of the four controls; namely, empty non-targeted or empty targeted micelles, free TDP-A (5 nM), or pure medium. The equivalent TDP-A concentration for TDP-A-loaded micelles was 5 nM. The plates were incubated for 48 h and protein was harvested according to a previously described protocol [41]. Protein concentrations were determined by a bicinchoninic acid assay (Thermo Scientific, Waltham, MA), and protein lysates (20 μg) were denatured and resolved on 10% SDS–PAGE gels. Protein bands in the gel were then transferred to a nitrocellulose membrane and incubated with the appropriate primary antibody with dilutions as follows: 1:2000 for ASCL-1, 1:1000 for SYP, and 1:1000 for β-actin. Membranes were incubated overnight at 4 °C and then washed with buffer (1×PBS, 0.05% Tween 20). Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies were used for secondary incubation. Proteins were then visualized with chemiluminescent substrates Immunstar (SYP and β-actin) or Femto (ASCL-1).
2.12 In Vivo Biodistribution Study
Four-week-old male athymic nude mice were obtained from Charles River (Wilmington, Maryland, USA). They were allowed to acclimate one week in the animal facility to reduce stress after arrival. Mice were maintained under specific pathogen-free conditions. A subcutaneous BON tumor xenograft mouse model was established by subcutaneously implanting 1 ×107 BON cells in 200 μL of Hanks balanced salt solution (Mediatech, Inc, Manassas, Virginia, USA) into the left flank of each mouse. Two weeks after inoculation, mice with palpable tumors were randomized into three groups (4 mice per group). The three groups of tumor-bearing mice were intravenously injected with 200 μL of either saline (control), Cy5-labeled non-targeted, or Cy5-labeled targeted micelles at a micelle concentration of 5.2 mg/ml. The mice were then scanned at different time points (5 h, 24 h, and 48 h) post-injection using an in vivo imaging system (IVIS) spectrum luminescence system with an excitation and emission wavelength of 675 nm and 710 nm, respectively.
2.13 In Vivo Anticancer Study
The same subcutaneous BON tumor xenograft mouse model as described above was used for the in vivo anticancer study. Two weeks after inoculation, mice with palpable tumors were divided into seven treatment groups (7 mice/per group). The seven groups of tumor-bearing mice were treated with (1) saline (control), (2) FK228 (an FDA approved HDAC inhibitor used as the second control, 3.125 mg/kg BW), (3) free TDP-A (3.125 mg/kg BW), (4) TDP-A-loaded non-targeted micelles, (5) TDP-A-loaded targeted micelles, (6) empty non-targeted micelles, and (7) empty targeted micelles. The equivalent TDP-A dose for the TPDA-A-loaded non-targeted and targeted micelles was 3.125 mg/kg BW, which was below the maximum tolerated dose (12.5 mg/kg BW, unpublished data; performed by NCI–DTP). The dosage of empty non-targeted or targeted micelles was 15.15 mg/kg BW, which corresponded to the dosage of the TDP-A-loaded micelles used in the in vivo studies. Each treatment group received five intravenous injections spaced 4 days apart. Tumor volumes were measured by an external caliper every four days and were then calculated by the modified ellipsoidal formula: Tumor volume = ½ (length × width2). Body weight change was monitored at the same time. At the end of the experiment, mice were sacrificed and postmortem examination of the lungs, liver, kidneys, and spleen were performed to confirm that there was no evidence of metastases or tumor growth outside of the inoculation site. Moreover, all major organs (liver, brain, heart, and leg muscles) of the mice treated with TDP-A-loaded targeted micelles were collected, fixed in 10% formalin, and embedded in paraffin blocks to prepare hematoxylin and eosin (H&E) stained sections for pathological assessment under the optical microscope. All experimental procedures were done in compliance with our animal care protocol, which was approved by the University of Wisconsin–Madison Animal Care and Use Committee.
2.13 Statistical Analysis
Weights and volumes were compared between groups using an analysis of variance (ANOVA), with pair-wise comparisons performed using Fisher’s protected least significant difference tests. Data were log-transformed prior to analyses in order to better meet the assumptions of ANOVA. P-values less than 0.05 were considered to be statistically significant.
3. RESULTS AND DISCUSSION
3.1 Synthesis and Characterization of the Multi-Arm Star Amphiphilic Block Copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108
The synthesis of multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108 was carried out following Scheme 1. First, PAMAM–PVL–OH polymer was synthesized by ring-opening polymerization of the valerolactone (VL) monomer using dendrimer PAMAM–OH (4th generation with 64 terminal hydroxyl groups) as the macro-initiator. The chemical structure was confirmed by 1H NMR (Fig. 2 (A)). The peaks at 1.65 ppm (a), 2.32 ppm (c), and 4.12 ppm (b) were ascribed to the protons of the PVL main chains. The signal at 3.65 ppm (b′), corresponding to the terminal methine protons of PVL, can also be identified. According to the 1H NMR spectrum, the number of repeat units in the PVL arms was determined to be 26 based on the relative intensity ratio of the peaks at 4.12 ppm (b) and 3.65 ppm (b′). The peaks at 2.55 ppm (f), 2.72 ppm (g), and 3.45 ppm (h) were attributed to the PAMAM core of the PAMAM–PVL polymer. The FT-IR spectrum shown in Fig. 2 (B) also confirmed the chemical structure of PAMAM–PVL–OH. The peaks located at 1648 cm–1 and 1542 cm–1 were ascribed to the C=O stretching and N–H bending of the amide groups present in PAMAM. The peak at 1720 cm–1 was assigned to the ester bond of the PVL arm. The average number of arms (# arm) per dendritic PAMAM–PVL polymer was calculated by comparing the molecular weights of PAMAM–OH and PAMAM–PVL–OH measured by GPC (Table 1). Based on Eq. 1, # arm per PAMAM–PVL–OH molecule was calculated as 33.
Fig. 2.

(A) 1H NMR and (B) FT-IR spectra of PAMAM–PVL–OH.
Table 1.
Molecular weights of all polymers.
| Polymers | Mn (g/mol) | Mw(g/mol) | Mw/Mn |
|---|---|---|---|
| PAMAM–OH | 14277 | ||
| PAMAM–PVL–OH | 99450 | 141616 | 1.424 |
| PAMAM–PVL–COOH | 102037 | 147035 | 1.441 |
| PAMAM–PVL–PEG–OCH3/NHS | 257017 | 354945 | 1.381 |
| Eq. 1 |
In the next step, the hydroxyl groups of PAMAM–PVL–OH were converted into carboxyl groups in the presence of succinic anhydride and DMAP. The chemical structure of PAMAM–PVL–COOH was also confirmed by 1H NMR. As shown in Fig. 3 (A), a new peak at 2.63 ppm (d) was observed, which was ascribed to the methylene groups of succinic anhydride. Afterwards, two types of hydrophilic PEG, mPEG–OH and NHS–PEG–OH, were conjugated to the PAMAM–PVL–COOH via esterification to form the multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/NHS. NHS groups were used to further conjugate the KE108-targeting ligand and Cy5 dye. The –OCH3 groups present in mPEG were used to control the molar ratio of the NHS group at the surface of the unimolecular micelle, which subsequently controlled the number of KE108 and Cy5 dye molecules per unimolecular micelle. The 1H NMR spectrum clearly demonstrated the formation of PAMAM–PVL–PEG–OCH3/NHS (Fig. 3 (B)). In addition to the peaks of PVL, the peak at 3.65 ppm (e) was observed due to the methylene protons of the oxyethylene units of PEG. The signal located at 3.35 ppm was ascribed to the NHS groups. GPC analyses further confirmed the successful synthesis of PAMAM–PVL–PEG–OCH3/NHS. As shown in Table 1, the molecular weight of PAMAM–PVL–PEG–OCH3/NHS was measured as 174,721 Da, which was significantly larger than that of PAMAM–PVL–COOH (54,721 Da), indicating the formation of the multi-arm star amphiphilic block polymer PAMAM–PVL–PEG–OCH3/NHS. Similarly, the # arm per PAMAM–PVL–PEG–OCH3/NHS was calculated as 32 using Eq. 2 based on the molecular weight measured by GPC. This result is in good agreement with previous reports on the number of arms in PAMAM-based dendritic amphiphilic block copolymers.
Fig. 3.

1H NMR spectra of (A) PAMAM–PVL–COOH, (B) PAMAM–PVL–PEG–OCH3/NHS, and (C) PAMAM–PVL–PEG–OCH3/Cy5/KE108.
| Eq. 2 |
Lastly, the KE108-targeting ligand and Cy5 dye were conjugated onto the distal ends of PEG arms of the multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/NHS. The molar ratio of PAMAM–PVL–PEG–OCH3/NHS, KE108, and Cy5 were set at 1:6:2 (corresponding to 6 KE108 peptide molecules and 2 Cy5 dye molecules per PAMAM–PVL–PEG–OCH3/KE108/Cy5). The 1H NMR confirmed the formation of PAMAM–PVL–PEG–OCH3/ KE108/Cy5 (Fig. 3 (C)). The new peaks ranging from 7.5 to 8.0 ppm were attributed to the protons of KE108 and Cy5.
3.2 Properties of Unimolecular Micelles
The multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG can form stable unimolecular micelles in an aqueous solution. The sizes and morphologies of the unimolecular micelles were characterized by both DLS and TEM. As shown in Fig. 4 (A), the hydrodynamic diameter of the unimolecular micelles ranged from 30 to 110 nm, with an average diameter of 56 nm. The TEM images of the unimolecular micelles (Fig. 4 (B)) showed a spherical morphology with a clear core–shell structure. The size measured by TEM ranged from 29 to 45 nm, with an average diameter of around 38 nm, which was smaller than that measured by DLS. This was because DLS measured the hydrodynamic diameter of the micelles with the hydrophilic PEG arms extending into the aqueous solution, while TEM measured the diameter of dried unimolecular micelles. The stability of the unimolecular micelles in aqueous solutions was also studied. Figure S1 shows the hydrodynamic size distribution of the freshly prepared unimolecular micelles (Day 1) and the unimolecular micelles after being stored for one day (Day 2), two days (Day 3), and 35 days (Day 36). It was found that the micelles were very stable in the aqueous solution since there were no obvious changes in either the appearance (e.g., no precipitations) or the size distribution of the micelles over a period of one month. The drug loading level and drug loading efficiency were 17.1% and 85.5%, respectively.
Fig. 4.

(A) The distribution of the hydrodynamic sizes of the unimolecular micelles measured by DLS, and (B) the morphology of the unimolecular micelles as measured by TEM.
3.3 In Vitro Cellular Uptake Studies
To evaluate the efficacy of KE108 as a SSTR-targeting ligand in vitro, the cellular uptake behaviors of KE108-conjugated (targeted) or KE108-lacking (non-targeted) unimolecular micelles conjugated with a fluorescent dye, Cy5, were examined in two SSTR-overexpressing NE cancer cell lines (i.e., human gastrointestinal carcinoid cell line BON and human medullary thyroid cancer cell line MZ–CRC–1). Based on the fluorescent intensity of the Cy5 dye, the effects of the KE108-targeting ligand on the cellular uptake of the micelles were measured by flow cytometry and confocal laser scanning microscopy (CLSM). Figure 5 shows the SSTR 1–5 expression levels in BON and MZ–CRC–1 cells. Our previous studies demonstrated that octreotide—which has a high affinity for SSTR2 and a moderate affinity for SSTR5—increased the cellular uptake of the micelles about 4-fold compared with non-targeted micelles in BON cells [48]. The KE108 peptide—which displays high affinity for all 5 subtypes of SSTRs (SSTR 1–5)—should exhibit much higher cellular uptake than octreotide. Indeed, the intracellular uptake of the KE108-conjugated micelles was 59 and 42 times higher than that of the non-targeted micelles in BON and MZ–CRC–1 cells, respectively (Fig. 6 (A) and (B)). Hence, the KE108 peptide exhibited a greatly superior SSTR-targeting ability than octreotide since it has high affinities for all five SSTR subtypes (SSTR 1–5). To further demonstrate the specific SSTR targeting ability of KE108, the cellular uptake behavior of both KE108-conjugated and KE108-lacking unimolecular micelles in a WI-38 cell line (pulmonary fibroblasts) that does not overexpress SSTRs was also studied. As shown in Fig. S2, no differences were observed in the levels of cellular uptake between the targeted and non-targeted micelles. As mentioned previously, this is the first report on using KE108 peptide as a targeting ligand for drug nanocarriers for targeted NE cancer therapy.
Fig. 5.
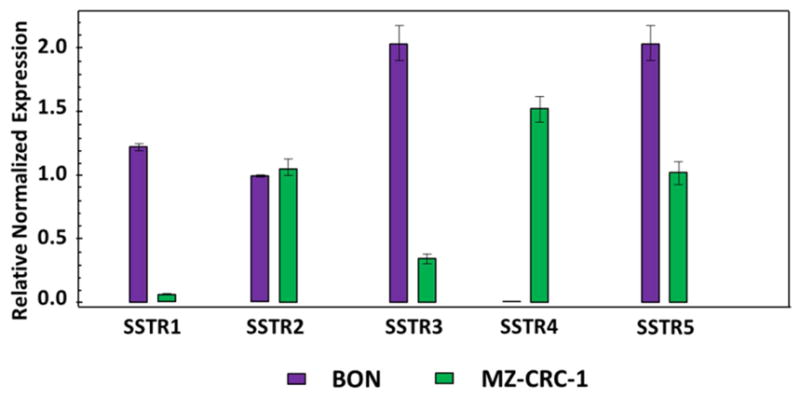
Expression of the SSTRs in NE cancer cells.
Fig. 6.

In vitro cellular uptake study of the micelles in NE cancer cells. Flow cytometry analysis of (A) BON cells, and (B) MZ–CRC–1 cells. CLSM images of (C) BON cells, and (D) MZ–CRC–1 cells. Cells were treated with pure medium (control) and non-targeted or KE108-conjugated (targeted) micelles (100 μg/ml) for 2 h at 37 °C. The KE108 peptide significantly enhanced the cellular uptake of micelles in both NE cancer cell lines.
The SSTR-targeting ability of KE108 was further analyzed and validated through CLSM analyses. As shown in Fig. 6 (C) and (D), as expected, KE108-conjugated micelles had significantly higher cellular uptake after 2 h of incubation, exhibiting much stronger red Cy5 fluorescent signals in both NE cancer cell lines compared to non-targeted micelles. Moreover, the micelles accumulated preferentially in the cytoplasm because they were taken up by the cells via endocytosis. Overall, the greater cellular uptake of the targeted micelles over the non-targeted ones indicated that the KE108-targeting ligand was effective at promoting SSTR-mediated endocytosis, while non-targeted micelles were taken up by the cells through non-specific endocytosis. Notably, besides NE cancers, these KE108-conjugated micelles can also be potentially effective drug delivery vehicles for several other types of cancers that overexpress SSTRs, including breast cancer [49] and melanoma [50].
3.4 In Vitro Evaluation of the Anti-Proliferative Efficacy of TDP-A-Loaded Micelles
The anti-proliferative efficacies of TDP-A-loaded micelles were evaluated in NE cancer cells using an MTT assay after 24 and 48 h of treatment. As shown in Fig. 7 (A), at each time point, TDP-A-loaded targeted micelles (4th bar) were much more effective at suppressing NE cancer cell proliferation than other groups. The enhanced cytotoxicity of TDP-A-loaded targeted micelles over non-targeted ones was attributed to their enhanced cellular uptake, via receptor-mediated endocytosis, in the NE cancer cells that overexpressed SSTRs. Interestingly, it was also observed that cells treated with TDP-A-loaded targeted micelles were more sensitive than free TDP-A. The difference observed in cytotoxicity may result from the different mechanism of cellular uptake for free drug versus the drug formulated in targeted micelles. As the cellular uptake of free TDP-A occurs through a passive diffusion mechanism, the drug may be pumped out through the P-glycoprotein (P-gp) pump on the cell membrane [51], while TDP-A-loaded targeted micelles were taken up by endocytosis, which may overcome the P-gp pumping action [52]. Notably, neither empty non-targeted micelles or empty targeted micelles exhibited any apparent cytotoxicity compared to the control group. To further investigate the effect of KE108 conjugation on the anticancer efficacy of TDP-A-loaded targeted micelles, Western blot analyses on NE marker expression in NE cancer cells were carried out. Achaete–scute complex-like 1 (ASCL-1) and synaptophysin (SYP) are well established NE cancer markers, and high levels of ASCL-1 and SYP are both linked to poor prognosis of NE cancers [53, 54]. Therefore, these markers were utilized to assess the hormone-producing activity of the NE cancer cells. As shown in Fig. 7 (B), the TDP-A-loaded targeted micelles (4th lane) were much more effective in reducing the expression of both NE markers, ASCL-1, and SYP than all other types of micelles containing fewer components as well as free TDP-A. Taken together, these results demonstrate that TDP-A-loaded targeted micelles optimally inhibited NE cancer cell proliferation and hormone secretion in vitro.
Fig. 7.

(A) MTT assays of BON cells treated with various types of micelles (TDP-A concentration 5 nM). TDP-A-loaded targeted micelles (4th bar) were much more effective in suppressing tumor cell proliferation than other types of micelles containing fewer components. All values are presented as a mean SD (n = 4); *: p < 0.05; **: p < 0.01; ***: p < 0.001; NS: not significant. (B) Western blot analyses for ASCL-1 and SYP of BON cells treated with various types of micelles (TDP-A concentration 5 nM). TDP-A loaded targeted micelles (4th lane) were much more effective in reducing NET markers ASCL-1 and SYP than other types of micelles containing fewer components. β-actin was used as a loading control.
3.5 In Vivo Biodistribution and Tumor Accumulation of Unimolecular Micelles
Encouraged by the promising in vitro results exhibited by the KE108-conjugated targeted micelles, in vivo biodistribution and tumor accumulation of the unimolecular micelles in BON-xenograft-bearing mice were studied via a near infrared fluorescence imaging (NIRF) approach using an IVIS microscopy. The mice were scanned at three time points, (i.e., 5, 24, and 48 h) after intravenous injection of the Cy5-conjugated targeted micelles or non-targeted micelles. The mice injected with saline served as a control. As shown in Fig. 8 (A), strong signals from the Cy5 dye were detected at the tumor sites (identified using red arrows) 5 h post-injection for mice injected with both non-targeted and targeted micelles. However, much stronger signals at the tumor sites were observed in mice treated with targeted micelles at all three time points. While the tumor accumulation of targeted micelles was clearly observed at 24 and 48 h post-injection, much weaker signals were detected in mice treated with non-targeted micelles. Tumor accumulation of non-targeted micelles was attributed to the EPR effect (i.e., the passive tumor-targeting ability), a well-known phenomenon in tumors (Fig. 1 (C)) [18, 19]. Other than the passive targeting ability, the targeted micelles were also capable of active targeting due to the KE108 peptide conjugated on their surface, which located and bound to SSTRs overexpressed by BON tumor cells, thereby significantly enhancing their tumor accumulation. Ex vivo imaging of the excised tumors (Fig. 8 (B)) further confirmed the much greater tumor uptake of the targeted micelles compared to the non-targeted ones.
Fig. 8.

(A) In vivo near-infrared fluorescence imaging of subcutaneous BON tumor-bearing mice treated with saline (control), non-targeted micelles conjugated with Cy5, and targeted micelles conjugated with KE108 and Cy5; arrows point to the tumor sites. (B) Ex vivo fluorescence imaging of the excised tumors 24 h post-injection.
3.6 In Vivo Evaluation of the Antitumor Efficacy of TDP-A-Loaded Micelles
After the superior tumor accumulation of the targeted micelles was validated in vivo, the anti-tumor efficacy of the TDP-A-loaded micelles in vivo was also studied. Mice bearing BON xenografts were intravenously injected with one of the following five agents: saline (control), pure FK228 (an FDA approved HDAC inhibitor), pure TDP-A, TDP-A-loaded non-targeted micelles, or TDP-A-loaded targeted micelles at an equivalent drug concentration (3.125 mg/kg BW) every five days for a total of five treatments. Based on the tumor volume changes shown in Fig. 9 (A), mice treated with empty non-targeted or empty targeted micelles did not show any anti-cancer effect when compared with the control group treated with saline suggesting that empty unimolecular micelles were not cytotoxic, which was consistent with the in vitro MTT assay findings. All of the other treatment groups exhibited inhibition of tumor growth as compared to the saline control group. However, TDP-A exhibited much better therapeutic efficacy than FK228 (p < 0.05) at the same dosage. Furthermore, free FK228 also demonstrated much higher systemic toxicity at the same dose in comparison with TDP-A since 3 out of 7 mice died during the treatment. Among other treatments, TDP-A-loaded non-targeted and targeted micelles clearly induced much better antitumor efficacy compared to free TDP-A. As expected, TDP-A-loaded targeted micelles induced much better antitumor efficacy than TDP-A-loaded non-targeted micelles. Overall, the TDP-A-loaded targeted micelles were the most effective at inhibiting tumor growth, resulting in a 92% of tumor reduction compared to the control, measured 20 days after the first injection. This observation can be largely contributed to the fact that the TDP-A-loaded, KE108-conjugated NE tumor-targeting unimolecular micelles had both passive (via the EPR effect) and active (via KE108 conjugation) tumor targeting abilities (Fig. 1 (C)) [18, 19]. In addition, we previously reported that TDP-A-loaded octreotide-conjugated unimolecular micelles induced 74% of tumor reduction compared to the control, also measured 20 days after the first injection using the same TDP-A dosage, same treatment schedule, and same tumor model [48]. This may be another indication that KE108 was more effective than octreotide as an NE tumor-targeting ligand for drug nanocarriers. During the whole treatment process, there were not any noticeable changes in body weight (Fig. 9 (B)). Pathological assessment of H&E-stained sections of different organs (i.e., brain, heart, liver, and leg muscles) of mice treated with TDP-A-loaded targeted micelles did not show any signs of acute or chronic inflammation, or apoptotic or necrotic regions (Fig. 9 (C)), indicating that the TDP-A-loaded targeted unimolecular micelle drug delivery system was safe for organs other than NE cancerous tissues, and that the potential off-target uptake of the micelles by normal tissues did not cause any detectable damage.
Fig. 9.

Therapeutic efficacy of the TDP-A-loaded micelles in the subcutaneous BON cell xenografts. Each mouse received five intravenous injections (3.125 mg/kg BW) spaced 4 days apart. (A) In vivo anticancer efficacy of different TDP-A formulations in BON tumor xenografts. (B) Change in the body weight of animals as a function of time. All values are presented as a mean SD (n = 7); **: P < 0.01; ***: P < 0.001; NS: not significant. Note: Three mice died in the FK228 group due to the severe toxicity of FK228. (C) Representative H&E-stained sections of brain, heart, liver, and leg muscles of a mouse treated with the control (baseline) and TDP-A-loaded targeted micelles. No signs of apoptotic or necrotic areas were observed for the TDP-A-loaded targeted micelles.
4. CONCLUSIONS
KE108-conjugated unimolecular micelles made of multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–KE108/Cy5 were synthesized and characterized for targeted NE cancer therapy. By employing KE108 peptide as a novel NE tumor targeting moiety, the cellular uptake of the targeted unimolecular micelles in NE cancer cells overexpressing SSTRs was dramatically enhanced compared to that of non-targeted micelles, owing to the strong binding affinity of KE108 to all five subtypes of SSTRs (SSTR 1–5). Moreover, TDP-A-loaded, KE108-conjugated unimolecular micelles were much more effective at suppressing NE cancer cell growth and hormone production. Finally, in vivo studies demonstrated that the KE108-conjugated micelles exhibited a significantly higher tumor accumulation than the non-targeted ones, and the TDP-A-loaded and KE108-conjugated micelles induced best antitumor efficacy. This is the first report utilizing KE108 as a targeting ligand for drug nanocarriers for targeted drug delivery. These findings may support the development of TDP-A-loaded NE tumor-targeting unimolecular micelles in the treatment and palliation of patients with NE cancers, which can significantly enhance the therapeutic outcome of cancer therapy while minimizing undesirable side-effects.
Supplementary Material
Acknowledgments
This project was financially supported by grants from the NIH (R01 CA121115 to H. Chen, 1K25CA166178 and R21CA196653 to S. Gong), the American Cancer Society (MEN2 Thyroid Cancer Professorship 120319–RPM–11–080–01–TBG to H. Chen and Research Scholar Award RSGM TBE–121413 to H. Chen), Layton F. Rikkers, MD, Chair in Surgical Leadership Professorship (H. Chen), the Caring for Carcinoids Foundation, and the AACR. The authors would like to thank Dr. Ricardo Lloyd for the pathological assessment of the mice tissues and Dr. Glen Leverson for the statistical analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen H. Therapeutic options for patients with metastatic gastrointestinal carcinoid. Journal of surgical oncology. 2008;97:203–4. doi: 10.1002/jso.20956. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: current approaches and future directions. The oncologist. 2008;13:779–93. doi: 10.1634/theoncologist.2008-0043. [DOI] [PubMed] [Google Scholar]
- 4.Frilling A, Akerstrom G, Falconi M, Pavel M, Ramos J, Kidd M, et al. Neuroendocrine tumor disease: an evolving landscape. Endocrine-related cancer. 2012;19:R163–85. doi: 10.1530/ERC-12-0024. [DOI] [PubMed] [Google Scholar]
- 5.Pitt SC, Moley JF. Medullary, anaplastic, and metastatic cancers of the thyroid. Seminars in oncology. 2010;37:567–79. doi: 10.1053/j.seminoncol.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Sippel RS, Kunnimalaiyaan M, Chen H. Current management of medullary thyroid cancer. The oncologist. 2008;13:539–47. doi: 10.1634/theoncologist.2007-0239. [DOI] [PubMed] [Google Scholar]
- 7.Lal A, Chen H. Treatment of advanced carcinoid tumors. Curr Opin Oncol. 2006;18:9–15. doi: 10.1097/01.cco.0000198018.53606.62. [DOI] [PubMed] [Google Scholar]
- 8.Jaskula-Sztul R, Eide J, Tesfazghi S, Dammalapati A, Harrison AD, Yu XM, et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Molecular cancer therapeutics. 2015;14:499–512. doi: 10.1158/1535-7163.MCT-14-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan M, et al. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. The oncologist. 2007;12:942–51. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- 10.Jaskula-Sztul R, Pisarnturakit P, Landowski M, Chen H, Kunnimalaiyaan M. Expression of the active Notch1 decreases MTC tumor growth in vivo. The Journal of surgical research. 2011;171:23–7. doi: 10.1016/j.jss.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinlander E, Somnay Y, Harrison AD, Wang C, Cheng YQ, Jaskula-Sztul R, et al. The novel histone deacetylase inhibitor thailandepsin A inhibits anaplastic thyroid cancer growth. The Journal of surgical research. 2014;190:191–7. doi: 10.1016/j.jss.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:2666–70. doi: 10.1158/1078-0432.CCR-14-3119. [DOI] [PubMed] [Google Scholar]
- 14.VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. The Journal of antibiotics. 2011;64:525–31. doi: 10.1038/ja.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Henkes LM, Doughty LB, He M, Wang D, Meyer-Almes FJ, et al. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. Journal of natural products. 2011;74:2031–8. doi: 10.1021/np200324x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao K, Li YP, Wang C, Ahmad S, Vu M, Kuma K, et al. Disulfide cross-linked micelles of novel HDAC inhibitor thailandepsin A for the treatment of breast cancer. Biomaterials. 2015;67:183–93. doi: 10.1016/j.biomaterials.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaskula-Sztul R, Dammalapati A, Korlesky C, Gong S, Cheng YQ, Chen H. Thailandepsin A, a new HDAC inhibitor, reduces cellular proliferation and activates the Notch pathway in human carcinoids cancer cells. Cancer research. 2013;73:1014. [Google Scholar]
- 18.Pan DJMK, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 19.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annual review of medicine. 2012;63:185–98. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 20.Pan L, He Q, Liu J, Chen Y, Ma M, Zhang L, et al. Nuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticles. J Am Chem Soc. 2012;134:5722–5. doi: 10.1021/ja211035w. [DOI] [PubMed] [Google Scholar]
- 21.Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev. 2013;42:1147–235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]
- 22.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17356–61. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, et al. Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug Delivery. Acs Nano. 2008;2:889–96. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nature reviews Drug discovery. 2005;4:145–60. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 25.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharmaceutical research. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 26.Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine : nanotechnology, biology, and medicine. 2010;6:714–29. doi: 10.1016/j.nano.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. Journal of pharmaceutical sciences. 2003;92:1343–55. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 28.Lavasanifar A, Samuel J, Kwon GS. Poly (ethylene oxide)-block-poly (L-amino acid) micelles for drug delivery. Advanced drug delivery reviews. 2002;54:169–90. doi: 10.1016/s0169-409x(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 29.Heise A, Hedrick JL, Frank CW, Miller RD. Starlike block copolymers with amphiphilic arms as models for unimolecular micelles. JACS. 1999;121:8647–8. [Google Scholar]
- 30.Lawrence MJ. Surfactant systems: their use in drug delivery. Chemical Society Reviews. 1994;23:417–24. [Google Scholar]
- 31.Kim S, Shi Y, Kim J, Park K, Cheng J. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 32.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Folate-conjugated amphiphilic hyperbranched block copolymers based on Boltorn H40, poly(L-lactide) and poly(ethylene glycol) for tumor-targeted drug delivery. Biomaterials. 2009;30:3009–19. doi: 10.1016/j.biomaterials.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials. 2009;30:5757–66. doi: 10.1016/j.biomaterials.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Guo J, Hong H, Chen G, Shi S, Zheng Q, Zhang Y, et al. Image-guided and tumor-targeted drug delivery with radiolabeled unimolecular micelles. Biomaterials. 2013;34:8323–32. doi: 10.1016/j.biomaterials.2013.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y, et al. Multifunctional unimolecular micelles for cancer-targeted drug delivery and positron emission tomography imaging. Biomaterials. 2012;33:3071–82. doi: 10.1016/j.biomaterials.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu W, Siddiqui IA, Nihal M, Pilla S, Rosenthal K, Mukhtar H, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34:5244–53. doi: 10.1016/j.biomaterials.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, Burke JF, Pilla S, Chen H, Jaskula-Sztul R, Gong S. Octreotide-functionalized and resveratrol-loaded unimolecular micelles for targeted neuroendocrine cancer therapy. Nanoscale. 2013;5:9924–33. doi: 10.1039/c3nr03102k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X, Grailer JJ, Pilla S, Steeber DA, Gong S. Tumor-Targeting pH-Responsive, and Stable Unimolecular Micelles as Drug Nanocarriers for Targeted Cancer Therapy. Bioconjugate Chemistry. 2010;21:496–504. doi: 10.1021/bc900422j. [DOI] [PubMed] [Google Scholar]
- 39.Guo J, Hong H, Chen G, Shi S, Nayak TR, Theuer CP, et al. Theranostic Unimolecular Micelles Based on Brush-Shaped Amphiphilic Block Copolymers for Tumor-Targeted Drug Delivery and Positron Emission Tomography Imaging. ACS applied materials & interfaces. 2014 doi: 10.1021/am5002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, Wang L, Cordie T, Vokoun C, Eliceiri KW, Gong S. Multi-functional self-fluorescent unimolecular micelles for tumor-targeted drug delivery and bioimaging. Biomaterials. 2015;47:41–50. doi: 10.1016/j.biomaterials.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao Y, Jaskula-Sztul R, Javadi A, Xu W, Eide J, Dammalapati A, et al. Co-delivery of doxorubicin and siRNA using octreotide-conjugated gold nanorods for targeted neuroendocrine cancer therapy. Nanoscale. 2012;4:7185–93. doi: 10.1039/c2nr31853a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun L-C, Coy HD. Somatostatin Receptor-Targeted Anti-Cancer Therapy. Current Drug Delivery. 2011;8:2–10. doi: 10.2174/156720111793663633. [DOI] [PubMed] [Google Scholar]
- 43.Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. The oncologist. 2008;13:1255–69. doi: 10.1634/theoncologist.2008-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimon L, Taylor JE, Dong JZ, Bitonte RA, SK, Morgan B, et al. Somatostatin Receptor Subtype Specificity in Human Fetal Pituitary Cultures. J Clin Invest. 99:789–98. doi: 10.1172/JCI119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Somatostatin analogues inhibit cancer cell proliferation in an SSTR2-dependent manner via both cytostatic and cytotoxic pathways. Oncology Reports. 1994 [PubMed] [Google Scholar]
- 46.Jiang H, Deng XF, Duan CM, Chen C, Xiang JL, Lu Y, et al. Somatostatin receptors SSTR2 and SSTR5 are experssed in the human thoracic duct. Lymphology. 2011;44:21–8. [PubMed] [Google Scholar]
- 47.Reubi JC, Eisenwiener K-P, Rink H, Waser B, Macke HR. A new peptidic somatostatin agonist with high affinity to all five somatostatin receptors. European Journal of Pharmacology. 2002;456:45–9. doi: 10.1016/s0014-2999(02)02651-1. [DOI] [PubMed] [Google Scholar]
- 48.Jaskula-Sztul R, Xu W, Chen G, Harrison A, Dammalapati A, Nair R, et al. Thailandepsin A-loaded and octreotide-functionalized unimolecular micelles for targeted neuroendocrine cancer therapy. Biomaterials. 2016 doi: 10.1016/j.biomaterials.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huo M, Zou A, Yao C, Zhang Y, Zhou J, Wang J, et al. Somatostatin receptor-mediated tumor-targeting drug delivery using octreotide-PEG-deoxycholic acid conjugate-modified N-deoxycholic acid-O, N-hydroxyethylation chitosan micelles. Biomaterials. 2012;33:6393–407. doi: 10.1016/j.biomaterials.2012.05.052. [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Alonso M, Llecha N, Mayorga M, Sorolla A, Dolcet X, Sanmartin V, et al. Expression of somatostatin receptors in human melanoma cell lines effect of two different somatostatin analogues, octreotide and SOM230, on cell proliferation. J Int Med Res. 2009;37:1813–22. doi: 10.1177/147323000903700617. [DOI] [PubMed] [Google Scholar]
- 51.Liang P, Zhao D, Wang CQ, Zong JY, Zhuo RX, Cheng SX. Facile preparation of heparin/CaCO3/CaP hybrid nano-carriers with controllable size for anticancer drug delivery. Colloids and surfaces B, Biointerfaces. 2013;102:783–8. doi: 10.1016/j.colsurfb.2012.08.056. [DOI] [PubMed] [Google Scholar]
- 52.Miller DW, Batrakova EV, Waltner TO, Alakhov VY, Kabanov AV. Interactions of Pluronic Block Copolymers with Brain Microvessel Enothelial Cells: Evidence of Two Potential Pathways for Drug Absorption. Bioconjugate Chemistry. 1997;8:649–57. doi: 10.1021/bc970118d. [DOI] [PubMed] [Google Scholar]
- 53.Oberg K, Janson ET, Eriksson B. Tumour markers in neuroendocrine tumours. Ital J Gastroenterol Hepatol. 1999;31:S160–S2. [PubMed] [Google Scholar]
- 54.Chen H, Udelsman R, Zeiger MA, Ball DW. Human achaete-scute homolog-1 is highly expressed in a subset of neuroendocrine tumors. Oncol Rep. 1997;4:775–8. doi: 10.3892/or.4.4.775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.