

Abstract
Recent progress in high-speed sequencing technology has revealed that tumors harbor novel mutations in a variety of genes including those for molecules involved in epigenetics and splicing, some of which were not categorized to previously thought malignancy-related genes. However, despite thorough identification of mutations in solid tumors and hematological malignancies, how these mutations induce cell transformation still remains elusive. In addition, each tumor usually contains multiple mutations or sometimes consists of multiple clones, which makes functional analysis difficult. Fifteen years ago, it was proposed that combination of two types of mutations induce acute leukemia; Class I mutations induce cell growth or inhibit apoptosis while class II mutations block differentiation, co-operating in inducing acute leukemia. This notion has been proven using a variety of mouse models, however most of recently found mutations are not typical class I/II mutations. Although some novel mutations have been found to functionally work as class I or II mutation in leukemogenesis, the classical class I/II theory seems to be too simple to explain the whole story. We here overview the molecular basis of hematological malignancies based on clinical and experimental results, and propose a new working hypothesis for leukemogenesis.
Keywords: Epigenetics, Hematological malignancy, MDS, MPN, AML
The first gene aberration identified in hematological malignancies is translocation between chromosome 9 and 22, t(9;22) which was found in most patients with chronic myelogeneous leukemia CML. A decade later, the first fusion kinase BCR-ABL was identified from the translocation site (1). In 1990s, BCR-ABL expression was shown to induce CML-like diseases both in the transgenic mouse (2) and in the bone marrow transplant (BMT) model (3, 4), where bone marrow cells transduced with BCR-ABL are transplanted to irradiated mice. These results indicated that BCR-ABL is a sole cause of CML pathogenesis. However, most of other chromosomal translocations identified in acute leukemia did not efficiently induce acute leukemia by themselves despite the fact that some of the translocations such as AML1-ETO or PML-RARa are identified in specific types of acute leukemia AML-M2 or M3, respectively. These results suggested that multiple mutations are required for leukemogenesis. In this article, we first introduce the ‘classical’ two-step leukemogenesis model (5) and the experimental results that support two-step leukemogenesis model, and then propose a new working hypothesis for leukemogenesis based on the recent experimental results and clinical information.
Two-Step Leukemogenesis Model
About 15 years ago, Dr. Gilliland and his colleagues proposed a 2-step leukemogenesis model (5). They categorized gene mutations into two groups, class I and class II mutations. The following works indicated that combination of class I and class II mutations would induced acute leukemia. Activating mutations of kinases, receptor kinases and oncogenes and inactivating mutations of tumor suppressors are categorized as class I mutations, and induce cell growth or inhibit apoptosis. On the other hand, class II mutations affect transcription factors, and disrupt normal differentiation process. In addition to inactivating or loss-of-function mutations of transcription factors, MLL-fusions could be also categorized to class II mutations (Fig. 1). Combination of class I and class II mutations are supposed to induce acute leukemia (Table I and Fig. 1). In principle, the hematopoietic stem or progenitor cells, whose differentiation was blocked by class II mutations, proliferate with class I mutations. Thus, class I and II mutations collaborate in inducing acute leukemia. This hypothesis was proposed based on their experimental result that the bone marrow cells of PML-RARα (class II mutation) transgenic mice transduced with FLT3-ITD (class I mutation) efficiently induced acute myeloid leukemia (AML) resembling APL (7) while the PML-RARα transgene alone induced AML only in 30% of the mice with much longer latencies (8). Experimental results from multiple groups later supported this two-step leukemogenesis model (9–12). For example, we demonstrated that combination of MLL-Sept6 (class II) and either FLT3-ITD or an activating Ras mutant G12V (class I) efficiently induced acute myeloid leukemia in the transplanted mice (9, 11). Not every combination of class I and class II mutations may induce leukemia and there are some frequent combinations such as MLL-fusions and Ras mutations, or Runx1-ETO and c-KIT mutations.
Fig. 1.
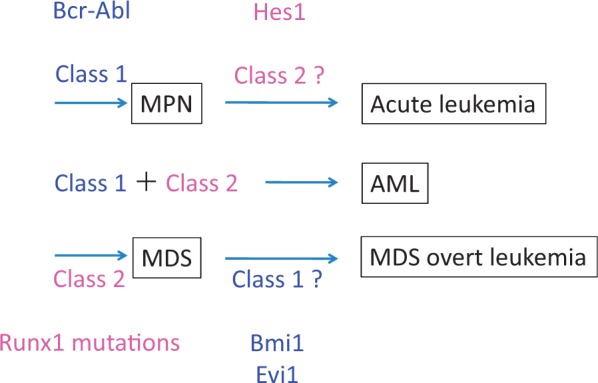
Molecular basis of hematological malignancies; modified version of classical class I and class II theory. Combination of class I and II mutations induces leukemogenesis. Class I mutations including Bcr-Abl or JAK2 induce MPN including CML. With additional class II mutations, MPN undergoes leukemic change including blast crisis of CML (CML-BC). On the other hand, a class II mutation Runx 1 mutation induces MDS, which progresses to MDS/AML with additional class I mutations. Modified from Kitamura et al. (6).
Table I.
Frequent combination of class I and class II mutations in acute myeloid leukemia: Not all combinations of class I and class II mutations would induce leukemia
| Class I | Class II | Disease |
|---|---|---|
| FLT3-ITD | Runx1 mutations | AML |
| PML-RARa | AML:M3 | |
| FLT3-TKD | MLL-fusions | pediatric ALL |
| Ras mutations | MLL-fusions | pediatric ALL |
| c-Kit mutations | Runx1-ETO | AML:M2(M1,4) |
| N-Ras mutations | Runx1 mutations | MDS/AML |
| P53 mutations | MLL amplification | tMDS/AML |
| FLT3 mutants | NPM1 mutations | AML |
| C/EBPα(N-term?) | C/EBPα(C-term) | AML (mostly M1 or M2) |
Some frequent combinations are shown.
Class I Mutations Induce Myeloproliferative Neoplasm
As stated earlier, a fusion kinase BCR-ABL generated by aberrant translocation of chromosomes could induce a CML-like disease in transgenic mice as well as in a BMT model (Fig. 1). In myeloproliferative neoplasm (MPN), a tyrosine kinase JAK2 is frequently activated by the mutation in the pseudokinase domain (95% for PV, 40–60% for both MF and ET) (13–16). Activating mutations of the thrombopoietin receptor (MPL) (17) and frame-shift mutations of CALR (18, 19) have also been identified. These mutations are mutually exclusive and all activate the JAK-STAT pathway, in particular STAT5, indicating that CALR mutation is also a class I mutation. Both JAK2 and MPL mutations have been reported to induce MPN in mouse models. Other class I mutations including RAS mutations, FLT3-ITD and FIP1L1-PDGFRα also induce MPN-like disease in mouse models (7, 9, 20, 21) although these mutations have not been so far identified in patients with MPN. These results suggest the heterogeneity of class I mutations as we discuss later.
Class II Mutations Induce Myelodysplastic Syndromes (MDS)
The first mutation identified in MDS was the activating mutation of RAS which seems to be associated with leukemic transformation of MDS to AML. It is known that MDS is frequently associated with a variety of chromosomal abnormalities, including 5q-, 7q-, 20q- and inv(3)(q21;26). Until recently only few mutations had been identified in MDS patients including RUNX1 (22, 23) and C/EBPα (24) which belong to the class II mutation. Recent progress in high-speed sequencing technologies revealed many novel mutations in patients with MDS including TET2, ASXL1 and EZH2 (25, 26), most of which do not belong to the classical class II mutations.
There are several mouse MDS models. NUP98-HOXD13, which is caused by t(2;11) and found in MDS and AML patients, induced a typical MDS in transgenic mice (27), some of which undergo leukemic transformation. It was also reported that combination of NUP98-HOXD13 and FLT3-ITD induced AML in the transplanted mice (28), suggesting that NUP98-HOXD13 plays class II mutation-like roles in inducing MDS and AML. We previously reported that mouse bone marrow cells transduced with RUNX1/AML1 mutants induced an MDS-like disease with occasional leukemic transformation (29). In this model, it was found that overexpression of EVI1 collaborates with a RUNX1 mutant D171N in inducing leukemic transformation of MDS. In fact, some MDS patient who underwent leukemic transformation with high expression of EVI1 harbored D171G mutation of RUNX1 (30). Expression profiles of patients with MDS and MDS/AML with and without Runx1 mutations identified that Bmi1 is highly expressed in a significant fraction of MDS/AML patients harboring Runx1 mutations but not in those without Runx1 mutations. In fact, in the BMT model, Bmi1 collaborated with Runx1-D171N in inducing AML with relatively short latencies (30). Over expression of EVI1 or BMI1 inhibited expression of PTEN or p16/p19, respectively, thus inducing cell cycle progression. These results suggest that overexpression of Evi1 and Bmi1 plays class I-like roles in collaborating with Runx1 mutants to induce leukemic transformation of MDS.
The mutations of another transcription factor C/EBPα, which is required for granulocytic differentiation, are unique. There are two types of C/EBPα mutations; one in the N-terminal domain which disrupt the full-length form of C/EBPα leaving a naturally occurring dominant-negative form lacking the N-terminal transcriptional activating domain, and the other in the C-terminal DNA binding domain. It is known that the two types of C/EBPα mutations are frequently found in the same patients (31), and that they collaborate with each other in inducing AML in mouse models (32, 33). This may be related to dual functions of C/EBPα as a transcription factor for differentiation and a tumor suppressor. We have shown that the C-terminal mutant strongly inhibited the granulocytic differentiation of 32D cells and collaborate with a typical class I mutation FLT3-ITD in inducing AML in the mouse BMT model, suggesting that the C-terminal mutant of C/EBPα plays class II roles. However, there is no evidence showing that the C-terminal C/EBPα mutant induces an MDS-like disease. Instead, this mutation induced AML by itself with a long latency in a mouse BMT model (34).
Novel Mutations; Class I-Like or Class II-Like?
Recent progress of high-speed sequence technologies has identified many novel mutations in a variety of genes encoding for epigenetic factors, signaling molecules, molecules of splicing machinery, and molecules of the cohesion complex in patients with malignant diseases including hematological malignancies (26, 35–37). It is difficult to categorize all these mutations to class I/II mutations, however some of the mutations are equivalent to either class I or class II mutations. We will not encompass such mutations here, but will present some examples.
Mutations of ASXL1 are frequently found in a variety of hematological malignancies, and are always associated with poor prognosis. In particular, the frequencies of the ASXL1 mutation are high in CMML (45%) and MDS (15–25%) (25, 26, 38, 39). Gene disruption of asxl1 induced an MDS-like disease in mice, suggesting loss-of-function feature of the ASXL1 mutation (40, 41). However, most of the mutations localize to the 5’ end of the last exon, causing frameshift and early termination of the transcript (38, 39), which will probably escape nonsense-mediate decay (NMD) of the transcript and result in the production of a C-terminal truncated protein. In addition, the mutations are always heterogyzous. These results strongly indicate that a truncated ASXL1 protein with a dominant-negative function or a gain-of-function is indeed expressed from the mutated ASXL1 allele. In relation to this, expression of the truncated form induced an MDS-like disease in the transplanted mice (42). Since gene disruption induce a similar disease, truncation mutants probably play dominant-negative roles. We have recently identified the C-terminal truncated ASXL1 protein in several leukemic cell lines (manuscript in preparation).
Concerning the functions of ASXL1 mutants, in 32D cells or HL60 cells, either expression of truncated form of ASXL1 (ASXL1-MT) or knockdown of ASXL1 (ASXL1-KD) inhibits the granulocytic differentiation of these cells induced by G-CSF. The molecular mechanisms underlying this phenomenon are as follows; ASXL1-MT suppressed the EZH2/PRC2 function, leading to derepression of oncogenes HOXA9/10 and miR125a via decreased levels of H3K27me3. Then, miR125a repressed the expression of CLEC5A, required for granulocytic differentiation of 32D cells, and inhibited G-CSF-induced differentiation of 32D cells. Importantly, forced expression of CLEC5A at least partially rescued the reduced differentiation of 32D cells caused by ASXL1-MT. Thus, ASXL1-MT plays class II mutation-like roles in the development of MDS by inhibiting cell differentiation. This notion is further enhanced by the clinical data and experimental results. In clinicals, typical class I mutations, RAS and JAK2 active forms frequently coexist with ASXL1 mutations in the advanced MDS. In addition, the presence of SETBP1 mutations is closely associated with ASXL1 mutations. Mutations of SETBP1 stabilize the SETBP1 protein, leading to stabilization of an oncoprotein SET followed by inactivation of PP2A and activation of AKT, suggesting the class I-like feature of this mutation. In fact, the combination of either RAS or SETBP1 mutation with ASXL1-MT quickly induced AML in the mouse BMT model (43).
On the other hand, tet2 knockout expanded the hematopoietic stem cells and progenitors, resulting in the development of MPN or CMML (44, 45). Although the underlying mechanisms remain elusive, the phenotype induced by tet2 knockout suggests that loss-of-function mutation of TET2 is a class I-like mutation. However, whether TET2 mutations identified in hematological malignancies are loss-of-function type mutations is not clear. Thus, whether tet2 knockout can recapitulate the human disease is also unclear. Mutations of IDH1/2 have similar effects with TET2 mutations because the mutant IDH1 and 2 convert a-KG to 2-HG while wild type IDH1 and 2 convert isocitrate to a-KG, and 2-HG inhibit TET2 (Fig. 2) (46, 47). In fact, the TET2 mutation and IDH1/2 mutation are mutually exclusive. Despite these facts, curiously, IDH1/2 mutations behave differently in mouse models when compared with the tet2 knockout. IDH1-R132H, the most frequent mutant, collaborated with HOXA9 in a mouse BMT model in accelerating development of an MPN-like disease although it did not induce any disease by itself (48). Similarly, another mutant IDH2-R140Q did not induce disease by itself, but collaborated with HOXA9/MEIS1 or FLT3-ITD in inducing leukemia (49). These results suggest that IDH1/2 mutations work class II-like roles unlike TET2 mutations. This discrepancy could be explained by the fact that 2-HG inhibition of TET2 activity is not enough, thus making IDH1/2 mutation weaker than tet2 knockout. Alternatively, 2-HG inhibition of other enzymes such as dioxygenases and histone methyltransferases (47) may affect the disease phenotypes.
Fig. 2.
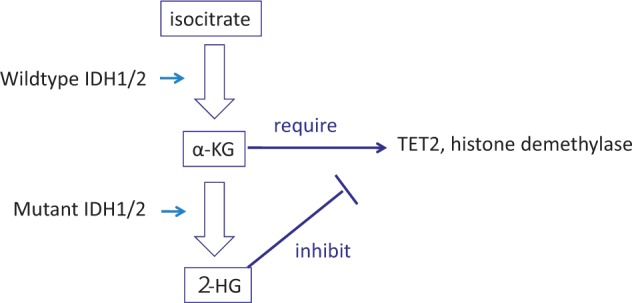
An example of gain-of-function mutation. Most of the gene mutations identified in patients with hematological malignancies. IDH1/2 gains a new function to convert α-KG to 2-HG, leading to inhibition of TET2 as well as some histone demethylase. Quoted from Kitamura et al. (6).
A DNA methyltransferase mutant DNMT3A-R882H, lacking its enzymatic activity, binds and inhibits the intact DNMT3A in a dominant-negative fashion. In mouse models, this mutant induced expression of several genes including mpl via aberrant methylation of genes, and induced CMML with thrombocytosis in a mouse BMT model (50). In addition, DNMT3A binds cyclin-dependent kinase 1 (CDK1), resulting in its increased expression levels and enhanced cell cycle progression (51). These results suggest a class I-like feature of this mutation. However, like other epigenetic mutations including MLL-fusions, the DNMT3A mutation would affect a large set of genes, and may play class II-like roles as well depending on the cellular context.
In summary, except some mutations such as the SETBP1, it would not be appropriate to categorize newly identified mutations to the classical class I and II mutations.
How Many Mutations Are Required for the Development of Myeloid Malignancies?
Deep sequencing technologies have revealed a variety of mutations in cancers and leukemias. It has been reported that leukemic cells harbor fewer mutations (13 mutations on average per genome) than cancer cells (52). Patients with MDS harbor even fewer mutations; the number of the mutations increases with the advancement of MDS, from 1.5 for patients with refractory anemia (RA) to 4 for those with refractory anemia with excess blast (RAEB) (26). As we reported, mouse bone marrow cells transduced with RUNX1 mutants or ASXL1 mutants induced similar MDS-like disease in the transplanted mouse. However, patients with MDS frequently harbored both RUNX1 and ASXL1 mutations, suggesting the co-operability of these two mutations in MDS pathogenesis. It is possible that overexpression models could be more potent to induce diseases. In fact, although we did not observe significant collaborative effects between Runx1 and ASXL1 mutations in the BMT model (53), the ASXL1-mutant knock-in and a Runx1 transgene seem to collaborate in inducing MDS in a preliminary experiment (unpublished results). This indicates that forced overexpression would result in exaggeration of the phenotypes. In relation to this, it has been recently reported that knock-in of bcr-abl in the proper site of the genome enhanced bone marrow engraftment but never induced the CML-like disease by itself, indicating that even BCR-ABL requires some other event(s) for leukemogenesis (54).
It is obvious that multiple events are necessary for development of hematological malignancies. In fact, many mutations have been identified from each patient with hematological malignancy. However, it is not clear how many of those are required for disease development. In mouse leukemia models, mouse leukemia virus could induce leukemia in a couple of weeks probably by itself. On the other hand, combination of class I and class II mutations usually induces leukemia with longer latencies, several months, suggesting that additional mutations or events are required for leukemogenesis. Some combination quickly induces AML. For example, combination of a C-terminal mutation of C/EBPα and FLT3-ITD are able to induce leukemia just in two weeks in the BMT model (33), suggesting that this combination does not require additional mutation in the induction of leukemia.
Modified Working Hypothesis for Leukemogenesis
As stated above, although the ‘class I/II hypothesis’ is with the truth in a significant portion, it is too simple to explain the whole picture. To complement this, two amended hypotheses have been proposed.
Rocquain et al. (55) proposed 4 class mutations; the first class includes RUNX1 and TET2 mutations which may cause clonal dominance of hematopoietic stem cells. The second class includes ASXL1 and NPM1 mutations which, they insisted, promote a pathway to primary or secondary AML. The third class is equivalent with the former class I mutations including mutations of genes involved in signaling pathways and proliferation. The last class they proposed includes TET2, IDH1, IDH2 and WT1 mutations that are exclusive with each other. It has recently demonstrated that WT1 binds TET2 and plays important roles in hydroxymethylation of genome DNA (56), thus supporting to form this group of gene mutations. However, WT1 is a sequence-specific transcription factor, and it is not clear whether WT1 mutations have equal effects on DNA hydroxymethylation with the effects of IDH1/2 and TET2 mutations that would work for the whole genome. In addition, many mutations including C/EBPα and EZH2 are not included in either group.
Thiede (57) proposed the third class of mutations of epigenetic factors in addition to the class I and II mutations and he also proposed class IV mutations for adhesion molecules and class V mutations for DNA repair/RNA splicing molecules in the comment for the paper dealing with the DNMT3A mutations (58). However, this classification is based on the categorization of the molecules harboring mutations and therefore will not be useful in investigating functional aspects of leukemogenesis.
Cancer Genome Altas Research Network categorized mutations derived from 200 adult cases with acute myeloid leukemia into 9 groups based on the functions of the mutated genes, including transcription factor-fusions, NPM1, tumor suppressors, DNA methylation-related genes, chromatin modifiers, myeloid transcription factors, cohesion-related genes and genes involved in splicing (52). Correlation between the combinations of these mutations and disease phenotypes may lead to new leukemogenesis models.
A Novel Working Hypothesis for Leukemogenesis
The disease in itself is caused by abnormal gene expression, and the disease phenotypes are determined by changes in gene expression patterns. The changes in gene expression patterns are brought by a variety of gene mutations. One particular mutation may induce changes in the expression levels of a set of genes, leading to some phenotypic changes of the cells. In the comprehensive understanding of multi-step leukemogenesis, we probably need to focus on the cellular phenotypes induced by each gene mutation rather than the categories of the molecules that are mutated. We categorize the cellular phenotypes to (i) induction of proliferation, (ii) survival or block of differentiation, (iii) block of differentiation and (iv) immortalization (Fig. 3). Among them, induction of proliferation is caused by ‘strong’ class I mutations while survival or block of apoptosis may be caused by ‘weak’ class I mutations. On the other hand, ‘weak’ class II mutations may disturb normal differentiation and ‘strong’ class II mutations such as MLL fusions are supposed to immortalize the hematopoietic cells. For example, MLL-AF9 is highly oncogenic and induces AML by itself in BMT models within several months. It has been reported that MLL-AF9 is able to induce many oncogenic genes including HOXA9, MEIS 1 and PLZF. Thus, in the mouse model, MLL-AF9 is able to induce not only the immortalization and block of differentiation but also proliferation. Nonetheless, in human AML, MLL-fusions are frequently associated with activating mutations of Ras that inhibits apoptosis, suggesting that its anti-apoptotic effect is required for full transformation of AML.
Fig. 3.

A new working hypothesis on the molecular bases of hematological malignancies: Combination of mutations-induced cellular phenotypes determines the disease (cMIP-DD). In addition to the classical class I and II mutations, mutations have been found in a variety of genes involved in epigenetics, splicing, signal transduction and cohesin complex. Each gene mutation leads to disregulated gene expression. The disregulated gene expression induces cellular phenotypes including proliferation, survival, block of differentiation, and immortalization. The combination of the phenotypes determines the disease. Modified from Kitamura et al. (6).
We here propose that combination of mutation-induced cellular phenotypes determines the disease phenotypes (cMIP-DD). Cell immortalization and block of differentiation would induce MDS while combination of immortalization and proliferation would induce MPN. Combination of the four cellular phenotypes, including proliferation, block of apoptosis, block of differentiation and immortalization, should induce acute leukemia. Each mutation eventually induces changes in gene expression patterns depending on the cell context, leading to the induction of cellular phenotypes. For example, FLT3-ITD or N-RAS-G12V induces proliferation or blocks apoptosis, respectively, through changes in gene expression patterns. Thus, we think that molecular bases of leukemogenesis could be explained by cellular phenotypes induced by gene mutations rather that the categories of the mutations.
Concluding Remarks
In the classical 2-step leukemogenesis model, the class I or class II mutation is supposed to correspond to growth promotion/apoptosis block or differentiation block/immortalization, respectively, we here propose that the disease phenotypes are determined by the combinations of cellular phenotypes including proliferation, block of apoptosis, block of differentiation and immortalization. These cellular phenotypes are induced by the aberrant gene expression caused by gene mutations or deletions. We call this theory as cMIP-DD standing for combination of the mutation-induced cellular phenotypes determines the disease. It would be possible to categorize this aberrant gene expression profile by gene set enrichment analysis (GSEA) or pathway analyses.
There are two important aspects that we did not discuss in this review. First, it is important which stage of the cell gains mutation, hemopoietic stem cells or some progenitor cells. BCR-ABL, a causative mutation of CML, can transform KSL including hematopoietic stem cells but not committed progenitors CMP and GMP. It is possible that KSL needs less mutation to develop hematologic malignancy because it has a self-renewal ability in itself and may not need an immortalization process in leukemogenesis. On the other hand, it is believed that most AML arise from committed progenitors CMP or GMP. Second, it is now recognized that multiple clones harboring different sets of gene mutations co-exist in one patient. This complexity also makes it difficult to interpret functional aspects of combination of mutations.
In conclusion, clarification of molecular mechanisms responsible for each cellular phenotype induced by each gene mutation is required for comprehensive understanding of etiologies of hematological malignancies, and will be useful in developing therapies for hematological malignancies, which will be informative for the research of other cancers.
Funding
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas and Grants-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Conflict of Interest
None declared.
References
- 1.Bartram C.R. de Klein A. Hagemeijer A. van Agthoven T. Geurts van Kessel A. Bootsma D. Grosveld G. Ferguson-Smith M.A. Davies T. Stone M. Heisterkamp N. Stephenson J.R., and Groffen J (1983) Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature 306, 277–280 [DOI] [PubMed] [Google Scholar]
- 2.Honda H., Oda H., Suzuki T., Takahashi T., Witte O.N., Ozawa K., Ishikawa T., Yazaki Y., Hirai H. (1998) Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcr/abl: a novel transgenic model for human Ph1-positive leukemias. Blood 91, 2067–2075 [PubMed] [Google Scholar]
- 3.Wong S., Witte O.N. (2001) Modeling Philadelphia chromosome positive leukemias. Oncogene 20, 5644–5659 [DOI] [PubMed] [Google Scholar]
- 4.Van Etten R.A. (2001) Retroviral transduction models of Ph+ leukemia: advantages and limitations for modeling human hematological malignancies in mice. Blood Cells Mol. Dis. 27, 201–205 [DOI] [PubMed] [Google Scholar]
- 5.Gilliland D.G., Griffin J.D. (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100, 1532–1542 [DOI] [PubMed] [Google Scholar]
- 6.Kitamura T., Inoue D., Okochi-Watanabe N., Kato N., Komeno Y., Lu Y., Enomoto Y., Doki N., Uchida T., Kagiyama Y., Togami K., Kawabata K.C., Nagase R., Horikawa S., Hayashi Y., Saika M., Fukuyama T., Izawa K., Oki T., Nakahara F., Kitaura J. (2014) The molecular basis of myeloid malignancies. Proc. Jpn. Acad. Ser. B 90, 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly L.M., Kutok J.L., Williams I.R., Boulton C.L., Amaral S.M., Curley D.P., Ley T.J., Gilliland D.G. (2002) PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc. Natl Acad. Sci. U. S. A. 99, 8283–8288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grisolano J.L., Wesselschmidt R.L., Pelicci P.G., Ley T.J. (1997) Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 89, 376–387 [PubMed] [Google Scholar]
- 9.Ono R., Nakajima H., Ozaki K., Kumagai H., Kawashima T., Taki T., Kitamura T., Hayashi Y., Nosaka T. (2005) Dimerization of MLL fusion proteins and FLT3 activation synergize to induce multiple-lineage leukemogenesis. J. Clin. Invest. 115, 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stubbs M.C., Kim Y.M., Krivtsov A.V., Wright R.D., Feng Z., Agarwal J., Kung A.L., Armstrong S.A. (2008) MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia 22, 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ono R., Kumagai H., Nakajima H., Hishiya A., Taki T., Horikawa K., Takatsu K., Satoh T., Hayashi Y., Kitamura T., Nosaka T. (2009) Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia 23, 2197–2209 [DOI] [PubMed] [Google Scholar]
- 12.Nick H.J., Kim H.G., Chang C.W., Harris K.W., Reddy V., Klug C.A. (2012) Distinct classes of c-Kit-activating mutations differ in their ability to promote RUNX1-ETO-associated acute myeloid leukemia. Blood 119, 1522–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine R.L., Wadleigh M., Cools J., Ebert B.L., Wernig G., Huntly B.J., Boggon T.J., Wlodarska I., Clark J.J., Moore S., Adelsperger J., Koo S., Lee J.C., Gabriel S., Mercher T., D'Andrea A., Frohling S., Dohner K., Marynen P., Vandenberghe P., Mesa R.A., Tefferi A., Griffin J.D., Eck M.J., Sellers W.R., Meyerson M., Golub T.R., Lee S.J., Gilliland D.G. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 [DOI] [PubMed] [Google Scholar]
- 14.James C., Ugo V., Le Couedic J.P., Staerk J., Delhommeau F., Lacout C., Garcon L., Raslova H., Berger R., Bennaceur-Griscelli A., Villeval J.L., Constantinescu S.N., Casadevall N., Vainchenker W. (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 [DOI] [PubMed] [Google Scholar]
- 15.Baxter E.J., Scott L.M., Campbell P.J., East C., Fourouclas N., Swanton S., Vassiliou G.S., Bench A.J., Boyd E.M., Curtin N., Scott M.A., Erber W.N., Green A.R. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061 [DOI] [PubMed] [Google Scholar]
- 16.Kralovics R., Passamonti F., Buser A.S., Teo S.S., Tiedt R., Passweg J.R., Tichelli A., Cazzola M., Skoda R.C. (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 [DOI] [PubMed] [Google Scholar]
- 17.Pikman Y., Lee B.H., Mercher T., McDowell E., Ebert B.L., Gozo M., Cuker A., Wernig G., Moore S., Galinsky I., DeAngelo D.J., Clark J.J., Lee S.J., Golub T.R., Wadleigh M., Gilliland D.G., Levine R.L. (2006) MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3, e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nangalia J., Massie C.E., Baxter E.J., Nice F.L., Gundem G., Wedge D.C., Avezov E., Li J., Kollmann K., Kent D.G., Aziz A., Godfrey A.L., Hinton J., Martincorena I., Van Loo P., Jones A.V., Guglielmelli P., Tarpey P., Harding H.P., Fitzpatrick J.D., Goudie C.T., Ortmann C.A., Loughran S.J., Raine K., Jones D.R., Butler A.P., Teague J.W., O'Meara S., McLaren S., Bianchi M., Silber Y., Dimitropoulou D., Bloxham D., Mudie L., Maddison M., Robinson B., Keohane C., Maclean C., Hill K., Orchard K., Tauro S., Du M.Q., Greaves M., Bowen D., Huntly B.J., Harrison C.N., Cross N.C., Ron D., Vannucchi A.M., Papaemmanuil E., Campbell P.J., Green A.R. (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klampfl T., Gisslinger H., Harutyunyan A.S., Nivarthi H., Rumi E., Milosevic J.D., Them N.C., Berg T., Gisslinger B., Pietra D., Chen D., Vladimer G.I., Bagienski K., Milanesi C., Casetti I.C., Sant'Antonio E., Ferretti V., Elena C., Schischlik F., Cleary C., Six M., Schalling M., Schonegger A., Bock C., Malcovati L., Pascutto C., Superti-Furga G., Cazzola M., Kralovics R. (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390 [DOI] [PubMed] [Google Scholar]
- 20.MacKenzie K.L., Dolnikov A., Millington M., Shounan Y., Symonds G. (1999) Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated mice. Blood 93, 2043–2056 [PubMed] [Google Scholar]
- 21.Cools J., Stover E.H., Boulton C.L., Gotlib J., Legare R.D., Amaral S.M., Curley D.P., Duclos N., Rowan R., Kutok J.L., Lee B.H., Williams I.R., Coutre S.E., Stone R.M., DeAngelo D.J., Marynen P., Manley P.W., Meyer T., Fabbro D., Neuberg D., Weisberg E., Griffin J.D., Gilliland D.G. (2003) PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRalpha-induced myeloproliferative disease. Cancer Cell 3, 459–469 [DOI] [PubMed] [Google Scholar]
- 22.Imai Y., Kurokawa M., Izutsu K., Hangaishi A., Takeuchi K., Maki K., Ogawa S., Chiba S., Mitani K., Hirai H. (2000) Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood 96, 3154–3160 [PubMed] [Google Scholar]
- 23.Harada H., Harada Y., Niimi H., Kyo T., Kimura A., Inaba T. (2004) High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 103, 2316–2324 [DOI] [PubMed] [Google Scholar]
- 24.Gombart A.F., Hofmann W.K., Kawano S., Takeuchi S., Krug U., Kwok S.H., Larsen R.J., Asou H., Miller C.W., Hoelzer D., Koeffler H.P. (2002) Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood 99, 1332–1340 [DOI] [PubMed] [Google Scholar]
- 25.Raza A., Galili N. (2012) The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat. Rev. Cancer 12, 849–859 [DOI] [PubMed] [Google Scholar]
- 26.Haferlach T., Nagata Y., Grossmann V., Okuno Y., Bacher U., Nagae G., Schnittger S., Sanada M., Kon A., Alpermann T., Yoshida K., Roller A., Nadarajah N., Shiraishi Y., Shiozawa Y., Chiba K., Tanaka H., Koeffler H.P., Klein H.U., Dugas M., Aburatani H., Kohlmann A., Miyano S., Haferlach C., Kern W., Ogawa S. (2014) Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Y.W., Slape C., Zhang Z., Aplan P.D. (2005) NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 106, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenblatt S., Li L., Slape C., Nguyen B., Novak R., Duffield A., Huso D., Desiderio S., Borowitz M.J., Aplan P., Small D. (2012) Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 119, 2883–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe-Okochi N., Kitaura J., Ono R., Harada H., Harada Y., Komeno Y., Nakajima H., Nosaka T., Inaba T., Kitamura T. (2008) AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 111, 4297–4308 [DOI] [PubMed] [Google Scholar]
- 30.Harada Y., Inoue D., Ding Y., Imagawa J., Doki N., Matsui H., Yahata T., Matsushita H., Ando K., Sashida G., Iwama A., Kitamura T., Harada H. (2013) RUNX1/AML1 mutant collaborates with BMI1 overexpression in the development of human and murine myelodysplastic syndromes. Blood 121, 3434–3446 [DOI] [PubMed] [Google Scholar]
- 31.Nerlov C. (2004) C/EBPalpha mutations in acute myeloid leukaemias. Nat. Rev. Cancer 4, 394–400 [DOI] [PubMed] [Google Scholar]
- 32.Bereshchenko O., Mancini E., Moore S., Bilbao D., Mansson R., Luc S., Grover A., Jacobsen S.E., Bryder D., Nerlov C. (2009) Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 16, 390–400 [DOI] [PubMed] [Google Scholar]
- 33.Kato N., Kitaura J., Doki N., Komeno Y., Watanabe-Okochi N., Togami K., Nakahara F., Oki T., Enomoto Y., Fukuchi Y., Nakajima H., Harada Y., Harada H., Kitamura T. (2011) Two types of C/EBPalpha mutations play distinct but collaborative roles in leukemogenesis: lessons from clinical data and BMT models. Blood 117, 221–233 [DOI] [PubMed] [Google Scholar]
- 34.Togami K., Kitaura J., Uchida T., Inoue D., Nishimura K., Kawabata K.C., Nagase R., Horikawa S., Izawa K., Fukuyama T., Nakahara F., Oki T., Harada Y., Harada H., Aburatani H., Kitamura T. (2015) A C-terminal mutant of CCAAT-enhancer-binding protein alpha (C/EBPalpha-Cm) downregulates Csf1r, a potent accelerator in the progression of acute myeloid leukemia with C/EBPalpha-Cm. Exp. Hematol. 43, 300–308, e301 [DOI] [PubMed] [Google Scholar]
- 35.Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M., Chalkidis G., Suzuki Y., Shiosaka M., Kawahata R., Yamaguchi T., Otsu M., Obara N., Sakata-Yanagimoto M., Ishiyama K., Mori H., Nolte F., Hofmann W.K., Miyawaki S., Sugano S., Haferlach C., Koeffler H.P., Shih L.Y., Haferlach T., Chiba S., Nakauchi H., Miyano S., Ogawa S. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 [DOI] [PubMed] [Google Scholar]
- 36.Papaemmanuil E., Cazzola M., Boultwood J., Malcovati L., Vyas P., Bowen D., Pellagatti A., Wainscoat J.S., Hellstrom-Lindberg E., Gambacorti-Passerini C., Godfrey A.L., Rapado I., Cvejic A., Rance R., McGee C., Ellis P., Mudie L.J., Stephens P.J., McLaren S., Massie C.E., Tarpey P.S., Varela I., Nik-Zainal S., Davies H.R., Shlien A., Jones D., Raine K., Hinton J., Butler A.P., Teague J.W., Baxter E.J., Score J., Galli A., Della Porta M.G., Travaglino E., Groves M., Tauro S., Munshi N.C., Anderson K.C., El-Naggar A., Fischer A., Mustonen V., Warren A.J., Cross N.C., Green A.R., Futreal P.A., Stratton M.R., Campbell P.J. (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 365, 1384–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papaemmanuil E., Gerstung M., Malcovati L., Tauro S., Gundem G., Van Loo P., Yoon C.J., Ellis P., Wedge D.C., Pellagatti A., Shlien A., Groves M.J., Forbes S.A., Raine K., Hinton J., Mudie L.J., McLaren S., Hardy C., Latimer C., Della Porta M.G., O'Meara S., Ambaglio I., Galli A., Butler A.P., Walldin G., Teague J.W., Quek L., Sternberg A., Gambacorti-Passerini C., Cross N.C., Green A.R., Boultwood J., Vyas P., Hellstrom-Lindberg E., Bowen D., Cazzola M., Stratton M.R., Campbell P.J. (2013) Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627; quiz 3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gelsi-Boyer V., Trouplin V., Adelaide J., Bonansea J., Cervera N., Carbuccia N., Lagarde A., Prebet T., Nezri M., Sainty D., Olschwang S., Xerri L., Chaffanet M., Mozziconacci M.J., Vey N., Birnbaum D. (2009) Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 145, 788–800 [DOI] [PubMed] [Google Scholar]
- 39.Thol F., Friesen I., Damm F., Yun H., Weissinger E.M., Krauter J., Wagner K., Chaturvedi A., Sharma A., Wichmann M., Gohring G., Schumann C., Bug G., Ottmann O., Hofmann W.K., Schlegelberger B., Heuser M., Ganser A. (2011) Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. 29, 2499–2506 [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Wahab O., Gao J., Adli M., Dey A., Trimarchi T., Chung Y.R., Kuscu C., Hricik T., Ndiaye-Lobry D., Lafave L.M., Koche R., Shih A.H., Guryanova O.A., Kim E., Li S., Pandey S., Shin J.Y., Telis L., Liu J., Bhatt P.K., Monette S., Zhao X., Mason C.E., Park C.Y., Bernstein B.E., Aifantis I., Levine R.L. (2013) Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 210, 2641–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J., Li Z., He Y., Pan F., Chen S., Rhodes S., Nguyen L., Yuan J., Jiang L., Yang X., Weeks O., Liu Z., Zhou J., Ni H., Cai C.L., Xu M., Yang F.C. (2014) Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue D., Kitaura J., Togami K., Nishimura K., Enomoto Y., Uchida T., Kagiyama Y., Kawabata K.C., Nakahara F., Izawa K., Oki T., Maehara A., Isobe M., Tsuchiya A., Harada Y., Harada H., Ochiya T., Aburatani H., Kimura H., Thol F., Heuser M., Levine R.L., Abdel-Wahab O., Kitamura T. (2013) Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J. Clin. Invest. 123, 4627–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue D., Kitaura J., Matui H., Hou H.-A., Chou W.-C., Nagamachi A., Kawabata K.C., Togami K., Nagase R., Horikawa K., Saika M., Micol J.-B., Hayashi Y., Harada Y., Harada H., Inaba T., Tien H.-F., Abdel-Wahab O., Kitamura T. (2015) SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia 29, 847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quivoron C., Couronne L., Della Valle V., Lopez C.K., Plo I., Wagner-Ballon O., Do Cruzeiro M., Delhommeau F., Arnulf B., Stern M.H., Godley L., Opolon P., Tilly H., Solary E., Duffourd Y., Dessen P., Merle-Beral H., Nguyen-Khac F., Fontenay M., Vainchenker W., Bastard C., Mercher T., Bernard O.A. (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 [DOI] [PubMed] [Google Scholar]
- 45.Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M.E., Vasanthakumar A., Patel J., Zhao X., Perna F., Pandey S., Madzo J., Song C., Dai Q., He C., Ibrahim S., Beran M., Zavadil J., Nimer S.D., Melnick A., Godley L.A., Aifantis I., Levine R.L. (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Figueroa M.E., Abdel-Wahab O., Lu C., Ward P.S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H.F., Tallman M.S., Sun Z., Wolniak K., Peeters J.K., Liu W., Choe S.E., Fantin V.R., Paietta E., Lowenberg B., Licht J.D., Godley L.A., Delwel R., Valk P.J., Thompson C.B., Levine R.L., Melnick A. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T., Liu L.X., Jiang W.Q., Liu J., Zhang J.Y., Wang B., Frye S., Zhang Y., Xu Y.H., Lei Q.Y., Guan K.L., Zhao S.M., Xiong Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaturvedi A., Araujo Cruz M.M., Jyotsana N., Sharma A., Yun H., Gorlich K., Wichmann M., Schwarzer A., Preller M., Thol F., Meyer J., Haemmerle R., Struys E.A., Jansen E.E., Modlich U., Li Z., Sly L.M., Geffers R., Lindner R., Manstein D.J., Lehmann U., Krauter J., Ganser A., Heuser M. (2013) Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122, 2877–2887 [DOI] [PubMed] [Google Scholar]
- 49.Kats L.M., Reschke M., Taulli R., Pozdnyakova O., Burgess K., Bhargava P., Straley K., Karnik R., Meissner A., Small D., Su S.M., Yen K., Zhang J., Pandolfi P.P. (2014) Proto-Oncogenic Role of Mutant IDH2 in Leukemia Initiation and Maintenance. Cell Stem Cell 14, 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J., Wang Y.Y., Dai Y.J., Zhang W., Zhang W.N., Xiong S.M., Gu Z.H., Wang K.K., Zeng R., Chen Z., Chen S.J. (2014) DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl Acad. Sci. U. S. A. 111, 2620–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S.J., Zhao H., Hardikar S., Singh A.K., Goodell M.A., Chen T. (2013) A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 122, 4086–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cancer Genome Alras Research Network (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med . 368, 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe-Okochi N., Yoshimi A., Sato T., Ikeda T., Kumano K., Taoka K., Satoh Y., Shinohara A., Tsuruta T., Masuda A., Yokota H., Yatomi Y., Takahashi K., Kitaura J., Kitamura T., Kurokawa M. (2013) The shortest isoform of C/EBPbeta, liver inhibitory protein (LIP), collaborates with Evi1 to induce AML in a mouse BMT model. Blood 121, 4142–4155 [DOI] [PubMed] [Google Scholar]
- 54.Foley S.B., Hildenbrand Z.L., Soyombo A.A., Magee J.A., Wu Y., Oravecz-Wilson K.I., Ross T.S. (2013) Expression of BCR/ABL p210 from a knockin allele enhances bone marrow engraftment without inducing neoplasia. Cell Rep. 5, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rocquain J., Carbuccia N., Trouplin V., Raynaud S., Murati A., Nezri M., Tadrist Z., Olschwang S., Vey N., Birnbaum D., Gelsi-Boyer V., Mozziconacci M.J. (2010) Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 10, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y., Ahn J., Abdel-Wahab O., Shih A., Lu C., Ward P.S., Tsai J.J., Hricik T., Tosello V., Tallman J.E., Zhao X., Daniels D., Dai Q., Ciminio L., Aifantis I., He C., Fuks F., Tallman M.S., Ferrando A., Nimer S., Paietta E., Thompson C.B., Licht J.D., Mason C.E., Godley L.A., Melnick A., Figueroa M.E., Levine R.L. (2014) DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thiede C. (2012) Mutant DNMT3A: teaming up to transform. Blood 119, 5615–5617 [DOI] [PubMed] [Google Scholar]
- 58.Ribeiro A.F., Pratcorona M., Erpelinck-Verschueren C., Rockova V., Sanders M., Abbas S., Figueroa M.E., Zeilemaker A., Melnick A., Lowenberg B., Valk P.J., Delwel R. (2012) Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood 119, 5824–5831 [DOI] [PubMed] [Google Scholar]