

Abstract
In chlorophyll biosynthesis, the magnesium chelatase enzyme complex catalyzes the insertion of a Mg2+ ion into protoporphyrin IX. Prior to this event, two of the three subunits, the AAA+ proteins ChlI and ChlD, form a ChlID–MgATP complex. We used microscale thermophoresis to directly determine dissociation constants for the I-D subunits from Synechocystis, and to show that the formation of a ChlID–MgADP complex, mediated by the arginine finger and the sensor II domain on ChlD, is necessary for the assembly of the catalytically active ChlHID–MgATP complex. The N-terminal AAA+ domain of ChlD is essential for complex formation, but some stability is preserved in the absence of the C-terminal integrin domain of ChlD, particularly if the intervening polyproline linker region is retained. Single molecule force spectroscopy (SMFS) was used to determine the factors that stabilize formation of the ChlID–MgADP complex at the single molecule level; ChlD was attached to an atomic force microscope (AFM) probe in two different orientations, and the ChlI subunits were tethered to a silica surface; the probability of subunits interacting more than doubled in the presence of MgADP, and we show that the N-terminal AAA+ domain of ChlD mediates this process, in agreement with the microscale thermophoresis data. Analysis of the unbinding data revealed a most probable interaction force of around 109 pN for formation of single ChlID–MgADP complexes. These experiments provide a quantitative basis for understanding the assembly and function of the Mg chelatase complex.
Introduction
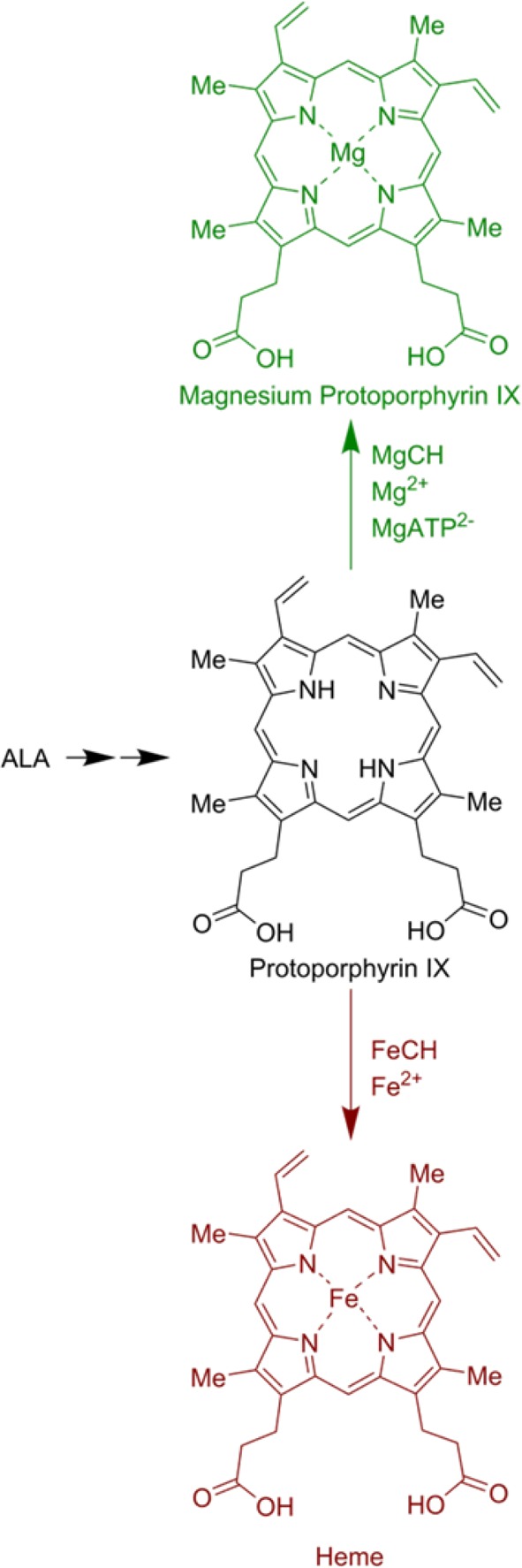
The global-scale biosynthesis of billions of tonnes of chlorophyll forms the basis for photosynthesis, so understanding the mechanism and regulation of chlorophyll biosynthesis is important. Heme and chlorophyll share a common pathway that diverges at metal ion insertion (Scheme 1). A single-subunit enzyme, ferrochelatase (FeCH; E.C. 4.99.11) inserts Fe2+ into protoporphyrin IX, whereas magnesium chelatase (MgCH; E.C. 6.6.1.1) is a large multisubunit enzyme complex that catalyzes the insertion of a Mg2+ ion into protoporphyrin IX in a Mg2+ and MgATP2– dependent manner.1,2 MgCH stands at the branchpoint between heme and chlorophyll biosynthesis in photosynthetic organisms, and it therefore plays pivotal catalytic and regulatory roles in initiating flux through the pathways for biosynthesis of all chlorophyll and bacteriochlorophylls in bacteria and plants.
Scheme 1. Branch point in Tetrapyrrole Biosysnthesis Controlled by Magnesium Chelatase and Ferrochelatase.
ChlI (39 kDa) hydrolyzes ATP in the absence of ChlD.3 ChlD does not hydrolyze ATP in isolation, but decreases the ChlI ATPase rate in the absence of ChlH and porphyrin substrate. ChlD (75 kDa) appears to act as an allosteric regulator of chelatase activity.4 These two subunits form a MgATP–ChlID complex, or a MgATP–BchID complex in Rhodobacter,5 and the subsequent hydrolysis of ATP powers the thermodynamically unfavorable insertion of the Mg2+ into the porphyrin ring in the 150 kDa ChlH subunit.6
Several studies have advanced our understanding of the catalytic cycle of MgCH,2,6−8 but there is currently no structural model for the MgCH complex, nor indeed a settled view on the stoichiometry of the subunits, and the ways in which they interact. The structure of the BchI subunit from Rhodobacter capsulatus was solved some time ago9 revealing the characteristic nucleotide binding motif common to this family of enzymes. Single particle reconstruction of negatively stained protein imaged by electron microscopy revealed the overall architecture of ChlH from Synechocystis,10,11 and recently the ChlH structure has been determined to 2.5 Å by X-ray crystallography.12
ChlI and ChlD are members of the AAA+ (ATPases Associated with various cellular Activities) superfamily of enzymes that display an array of diverse functions such as protein secretion and assembly, proteolysis, cell cycle control, DNA replication, and transcription.13,14 Proteins in the AAA+ superfamily have a structurally conserved region of around 200 amino acids containing the Walker A and B nucleotide binding motifs, as well as structurally important arginine residues. AAA+ proteins link ATP hydrolysis driven conformational changes to chemomechanical motion which is normally transduced throughout a multisubunit complex. This mode of action is likely to be used by MgCH to drive insertion of the Mg2+ ion into protoporphyrin IX. ChlI and the N-terminal half of ChlD share a similar AAA+ domain (approximately 40% sequence identity), and contain the conserved Walker A and B nucleotide binding domains, as well as a sensor II arginine and arginine finger (Figure 1, Supporting Information). The remainder of ChlD has an extended C-terminus comprising central polyproline and acidic regions followed by an integrin I-like C-terminal domain. Integrin I domains are normally involved in cell–cell and cell–matrix interactions and bind to specific complementary motifs.15
A quantitative understanding of the assembly of the ChlID–MgATP complex is essential, since it forms the basis for ATP-dependent insertion of the Mg2+ into the protoporphyrin substrate held within ChlH. Here we report the first directly determined dissociation constants for the subunits of the AAA+ ChlI–ChlD complex. Further, we use SMFS to quantify the nucleotide-dependent binding forces between ChlI and ChlD AAA+ domains that establish the complex that powers Mg chelation.
Results and Discussion
ChlI and ChlD can be co-purified in the presence of nucleotide.4,16 Previously, we have quantified the formation of the ChlID complex by assembly titrations, monitoring the rate of chelation as a function of the concentration of either ChlI or ChlD.4,11 This experiment allowed the determination of an apparent Kd for assembly of a complete, enzymatically active chelatase complex. However, this apparent Kd reflects many assembly and catalysis-related processes, so the present study was designed to yield direct quantitative measurements of the ChlI–ChlD interaction.
The complexities of magnesium and nucleotide binding to ChlI and ChlD make the application of techniques such as differential scanning calorimetry, isothermal calorimetry, and differential scanning fluorescence problematic, so we turned to microscale thermophoresis (MST), a relatively recent technique that provides a readily interpretable signal for binding.17−20 Briefly, protein, labeled with a fluorescent dye, is mixed with different concentrations of binding partner (e.g., protein or small ligand). The movement of a protein in a temperature gradient (thermophoresis) is directly proportional to the Soret coefficient, which takes into account charge, size, and hydration shell. In our work, ChlI was labeled with a fluorescent dye molecule, Alexa Fluor 488 C5 maleimide. After labeling with the Alexa Fluor 488 the site-directed mutant of ChlI, C244S, retains chelatase activity in the presence of nucleotide (Figure 2, Supporting Information). Formation of the ChlID complex alters the intrinsic thermophoresis of ChlI, monitored via the fluorescence signal from ChlI, which yields binding isotherms.
To confirm the requirements of nucleotide to form the ChlID complex, and to ascertain if MST is an appropriate technique for monitoring ChlID complex formation, titrations were performed in the presence and absence of nucleotide (Figure 1). Altering the concentration of ChlD (Figure 1A, black traces) produced no change in thermophoresis of ChlI in the absence of nucleotide, indicating no formation of a ChlID complex. However, there is a clear change in the thermophoresis of ChlI in the presence of 3 mM MgADP– and 10 mM free magnesium (green traces). In the absence of ChlD, ChlI self-assembles into a variety of complexes at concentrations above 1 μM, in the presence or absence of nucleotide.3 In the MST experiment, the concentration of ChlI is held at 20 nM, far below the self-assembly concentration. The formation of the MgADP–ChlID species reaches a steady state, and we calculate a Kd value of 7.6 nM from the curve in Figure 1B.
Figure 1.

Interaction of ChlI C244S Alexa Fluor 488 (hereafter ChlIAF488 C244S) with wild-type (WT) ChlD. Microscale thermophoresis was performed by titrating ChlD WT into a constant concentration (20 nM) of ChlIAF488 C244S. All MST assays were performed in 50 mM Tris/NaOH, 10 mM free Mg2+, 0.1% Pluronic F127, 1 mg mL–1 BSA, pH 7.8 at 20 °C. (A) Raw thermophoresis traces. Black, ChlIAF488 C244S titrated with ChlD in the absence of nucleotide; green, titration performed in the presence of 3 mM MgADP–. Data from the early stages of thermophoresis (red lines) were used for the plots in (B). (B) ○, ChlIAF488 C244S titrated with ChlD WT in the absence of nucleotide; ●, ChlIAF488 C244S titrated with ChlD WT in the presence of 3 mM MgADP–; ChlD WT Kd = 7.57 ± 0.8 nM.
The MST method was used to quantify the differences between AAA+ mutants of ChlD, specifically an arginine finger mutant, R208A, and a R289A mutation in the sensor II domain. The data in Figure 2, summarized in Table 1, show that the high affinity binding of ChlI and WT ChlD in the presence of MgADP– (Kd 7.57 ± 0.8 nM) is weakened significantly by alteration of the arginine finger mutation, R208A (Kd 327.8 ± 67.7 nM), and much more so by the sensor II domain mutation R289A (Kd 2536 ± 219 nM). A previous analysis of these mutants, in which formation of the ChlID–MgATP complex was inferred from measurements of MgCH activity in the presence of ChlH, showed a decreased affinity (Kapp) for ChlI.4 The MST experiments in Figure 2, which directly measure the Kd values of ChlI and ChlD mutants, show that the formation of a ChlID–MgATP complex, mediated by the arginine finger and the sensor II domain, is necessary for the assembly of the catalytically active ChlHID–MgATP complex. The arginine finger is known to act across protomer interfaces to sense the presence of nucleotide in an adjacent AAA+ subunit, and mutating it can impair both oligomerization and ATP hydrolysis, leading to less productive complex formation.21 The sensor II arginine has been shown to discriminate between MgADP– and MgATP2– binding, by interacting with the β-phosphate of MgADP–.22,23 Consistent with these observations for other AAA+ systems, altering the sensor II residues appears to dramatically alter the formation of the ChlID complex, leading to impaired formation of the entire core chelatase.
Figure 2.

Interaction between ChlI and ChlD R208A and R289A. Experimental conditions as in Figure 1. ●, ChlD WT; ▼, ChlD R208A; ■, ChlD R289A. Dissociation constants are listed in Table 1.
Table 1. Comparison of ChlID Apparent Binding Constants and Dissociations Constants.
| protein | Kapp (nM)a | Kd (nM) |
|---|---|---|
| ChlD WT | 0.17 ± 0.9 | 7.6 ± 0.8 |
| ChlD R208A | 14 ± 11 | 327.8 ± 67.7 |
| ChlD R289A | 52 ± 155 | 2536 ± 219 |
Data from Adams and Reid 2013.4
To identify the domains of ChlD involved in binding to ChlI, we produced multiple constructs (Figure 3A) comprising either the N-terminal AAA+ domain (Figure 3A, construct A), the AAA+ domain plus the polyproline region (construct B), the polyproline region and integrin I domain (construct C), or just the integrin I domain (construct D). The new constructs were purified in a similar manner to wild-type protein (Figure 3, Supporting Information). None of these ChlD truncations yielded chelatase activity when reconstituted in vitro with ChlI and ChlH (results not shown), in contrast with results reported for truncations of the recombinant ChlD protein from tobacco, which indicated that ChlD could function without an intact AAA+ domain.24 CD spectroscopy showed that truncations A and B, but not C and D, were both folded (Figure 4, Supporting Information). It is unknown if the C-terminal region of D is inherently disordered, or if the N-terminal truncation of the protein caused it to fold incorrectly.
Figure 3.

Mapping the interactions between ChlI and domains of ChlD using MST. (A) Schematic representation of ChlD, showing the N-terminal AAA+ domain; PP, the polyproline region; and vWD, von Willebrand protein–protein interaction domain. (B) Thermophoretic analysis of ChlI interacting with ChlD WT (●) Kd = 7.57 ± 0.8 nM, truncation A (▲) Kd = 1.36 ± 0.2 μM, and truncation B (△) Kd = 183.29 ± 25.9 nM. Proteins C and D showed no interaction with ChlI, so there are no square symbols in the graph.
These new truncated versions of ChlD were titrated with ChlI under the same conditions as wild-type subunits and analyzed by MST (Figure 3). The dissociation constants determined for AAA+ constructs A and B were 1391 and 178 nM, respectively, and by comparison with the WT value of 7.6 nM, this result shows that the vWD domain is essential for a normal ChlI–D interaction. The data also clearly show that the absence of the polyproline sequence weakens this association 10-fold. The N-terminal truncations C and D, namely mutants with a retained C-terminal domain, completely abolished binding to ChlI.
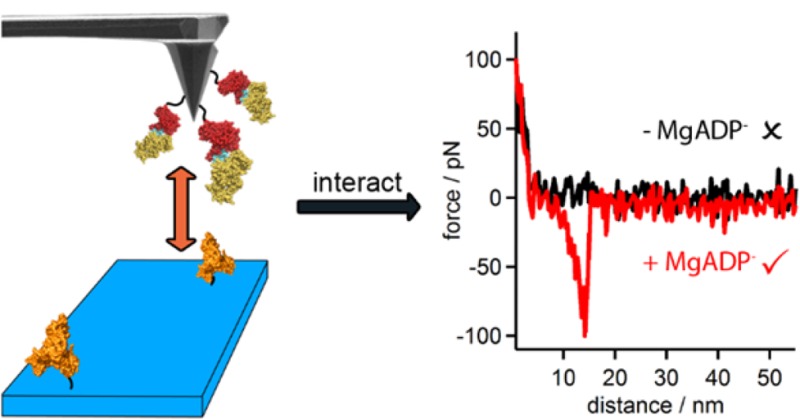
We used SMFS to determine the forces that stabilize the formation of the ChlID–MgADP– complex under physiological conditions25−27 and to control the orientation of ChlI and D molecules as they encounter each other. Instead of obtaining dissociation constants for ensembles of molecules in solution, this single molecule approach yields the most probable unbinding force measured in piconewtons. One of the participants is anchored to a surface, while the other is tethered to the AFM probe; thus, the encounter between the protein molecules is steered and this necessitates the construction of ChlD tagged at either the N- or C-terminus with a His6 sequence. Each of the ChlD variants had WT enzyme activity in assays with ChlI and ChlH (Figure 5, Supporting Information). ChlD subunits were attached to Si AFM probes using silane monolayer chemistry and a polymer linker molecule (SM(PEG)24, ∼10 nm length) terminated with Ni-NTA groups, (Figure 4A). The Ni-NTA-His-tag coupling approach has been demonstrated to provide the appropriate orientation, high mobility, and low coupling density of biological molecules, while at the same time minimizing nonspecific adsorption.28,29 In addition, coupling with a long flexible spacer ensures that the molecule on the AFM tip is free to move and orient, favoring complex formation with its surface-bound partner. In a similar way, we created a monolayer of immobilized ChlI molecules on a SiOx substrate functionalized, again via silane monolayer chemistry, with Ni-NTA (Figure 4A,B). His-Ni2+-NTA bridges remove the need for a covalent chemical linkage, and can achieve long-lasting attachment of proteins (several thousand force–distance cycles, up to several hours), while sustaining significant force stresses.28,30−32
Figure 4.

(A) Schematic representation of AFM experiment. ChlD is attached to the AFM tip; ChlI is attached to surface via their His6 tags. (B) AFM topography image of ChlI surface showing even distribution of protein molecules of the correct height. (C) Example force distance curves showing differences in no, specific, and nonspecific interactions between surface and tip.
The protein domain of ChlD that encounters surface-attached ChlI molecules was controlled by altering the location of the His6 tag. The orientation of the attachment of ChlI to the surface did not alter the results. The experiment is performed by bringing the probe-borne ChlD molecules into contact with the ChlI molecules on the sample surface and then retracting the tip. In the event of a specific interaction, a clear unbinding event is observed (Figure 4C, red trace) where the separation distance and the force of interaction can be measured. The characteristic hyperbolic part of the curve prior to the rupture clearly signifies the stretching of the PEG linker followed by the dissociation of ChlD and ChlI subunits.29 Nonspecific (Figure 4C, blue trace) and no interaction (Figure 4C, black trace) traces were discarded during the data analysis. To verify the specificity of the observed unbinding events, a series of control experiments was performed. The probe-borne ChlD AAA+ domain was brought into contact with a clean, noncoated Si surface, as well as with Si surface coated with bovine serum albumin (BSA) and a lower interaction force of approximately 22 pN was observed (Figure 6, Supporting Information).
We monitored the interaction between the two different orientations of ChlD, with either the N- or C-terminal domain brought into contact with surface-attached ChlI, in the presence and absence of nucleotide. Many data sets (each consisting of 800–1000 force–distance curves) were recorded over different surface locations. Each data set was analyzed to evaluate the interaction probability, as well as the most probable rupture force and most probable separation distance as defined as the maximum of a Gaussian distribution fitted to the histogram (Figure 5).33 There was a relatively low probability (14.5%) for interaction between ChlI and the N-terminal domain of ChlD in the absence of MgADP– (Figure 5A, panel i). We observed a much higher probability for interaction (30.5%) in the presence of nucleotide (Figure 5A, panel ii).
Figure 5.

Analysis of SMFS data for the interaction between ChlI and ChlD. ChlD was attached to the AFM tip, and ChlI to the surface as indicated by the diagrams on the left. (A) C-terminally attached proteins in the absence (i) and presence (ii) of 3 mM MgADP– and 10 mM free Mg2+ when bought together interacted with a higher probability (14.5% in the absence and 30.5% in the presence of MgADP–) compared to N-terminally attached D interacting with C-terminally attached ChlI (B i and ii). Fitting a Gaussian to binned data in panel A(ii) indicated a rupture force of 109 ± 1.5 pN with a rupture distance of 11.9 ± 0.3 nm.
Assuming a Poisson distribution in single molecule interaction events, unbinding probability of about 30% ensures that 80% of binding/unbinding events arise from single molecule interactions, while an unbinding probability of about 14% ensures 90–95% single molecule interactions.34,35
The histogram analysis revealed a most probable interaction force of around 109 pN at a separation distance of around 12 nm, in good agreement with the length of the flexible linker used for attachment of the ChlD. The modal values for interaction forces and separations distances obtained from the histogram were 107.8 pN and 10.1 nm, respectively.
The two lower panels in Figure 4 show that the C-terminal domain of ChlD cannot mediate the interaction with ChlI, whether or not MgADP– is present; the histograms of the rupture forces and the separation distances did not reveal clear peaks and the interaction probability was around 7%, which is comparable to noise level for the probability of interaction.
The model for the MgCH complex from R. capsulatus(36,37) proposes that BchI and BchD form two homohexamers stacked upon each other. The authors predict an interaction between the sensor II arginine in BchI and the BchD integrin I domain,37 instead of the usual function of penetrating a nucleotide binding site of an adjacent subunit to directly interact with the γ-phosphates on ATP.23 We show that with the cyanobacterial enzyme, removal of the integrin I domain of ChlD does not affect the interaction with ChlI, although we have established that the sensor II arginine in ChlD is essential for chelatase activity.
Conclusion
We have used two novel experimental approaches, thermophoresis and SMFS, to analyze the subunit interactions that govern the catalytic mechanism of the MgCH enzyme complex. Apart from its biosynthetic importance, standing at the gateway to chlorophyll biosynthesis, MgCH is also a valuable model for studying AAA+ proteins in general, given the availability of optical signals for monitoring the formation of porphyrin product states. AAA+ proteins are molecular machines that hydrolyze the P–O bond of ATP and transmit the energy to provide chemomechanical motion to power reactions. These proteins are known to form complex homo- or heteroring structures; in the former case, homohexamers display differences in their bound nucleotide state and availability for binding.38 In the case of hetero-AAA+ complexes, in which protein–protein interactions alter as they progress through a reaction cycle, it is important to understand how the force obtained from hydrolysis of ATP is transmitted to adjacent or nonadjacent subunits.
We show that the N-terminal AAA+ domain of ChlD interacts with the AAA+ protein ChlI, and we quantify the dissociation constant, which is 7.6 ± 0.8 nM. Although we cannot be sure that our SMFS monitors the same process, we suggest that this bulk Kd determination reflects an ensemble of nucleotide-dependent unbinding forces between single ChlI and ChlD in solution of 109 ± 1.5 pN. The interaction itself is not unexpected, as AAA+ proteins form complexes where the nucleotide binding domains are at the interfaces between proteins.
It is well established that in SMFS measurements the unbinding forces depend on the loading rate of the bond.34 To put the ChlI–ChlD unbinding force (measured at a loading rate of about 75 nN s–1) in a clearer context, we can compare it with previously published unbinding forces, measured at similar loading rates: 90–100 pN for single cohesin–dockerin unbinding event,39 177 pN for streptavidin–biotin unbinding,40 and 60 pN for the lactose–galectin-3 complex unbinding.41 We can conclude that the ChlI–ChlD unbinding force is consistent with the unbinding forces measured for other biologically relevant (and of comparable size) high-affinity ligand–receptor pairs that form stable complexes.
As AAA+ proteins provide chemomechanical motion to a distal site, the changes in the force of interaction between these proteins while proceeding through an ATP hydrolysis cycle will likely correlate with the force transduced across the enzyme to the active site, in this case the magnesium ion insertion. Performing similar studies on the proteins with nonhydrolyzable analogues of ATP will be useful to explore this link between bound ATP state and protein–protein interaction force.
Abbreviations
AAA+, ATPases Associated with various cellular Activities; AFM, atomic force microscope; MgCH, magnesium chelatase; MST, microscale thermophoresis; SMFS, single molecule force spectroscopy; vWD, von Willebrand domain.
Acknowledgments
N.B.P.A., A.A.B., and C.N.H. gratefully acknowledge financial support from the Biotechnology and Biological Sciences Research Council (BBSRC U.K.), award number BB/M000265/1. C.V. and C.N.H. were supported as part of the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC 0001035. C.N.H. was also supported by an Advanced Award 338895 from the European Research Council.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02827.
Experimental methods, supplementary figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jensen P. E.; Gibson L. C.; Henningsen K. W.; Hunter C. N. J. Biol. Chem. 1996, 271 (28), 16662–16667. 10.1074/jbc.271.28.16662. [DOI] [PubMed] [Google Scholar]
- Jensen P. E.; Gibson L. C.; Hunter C. N. Biochem. J. 1998, 334 (Pt 2), 335–344. 10.1042/bj3340335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J. D.; Siebert C. A.; Bullough P. A.; Hunter C. N. Biochemistry 2003, 42 (22), 6912–6920. 10.1021/bi034082q. [DOI] [PubMed] [Google Scholar]
- Adams N. B. P.; Reid J. D. J. Biol. Chem. 2013, 288, 28727–28732. 10.1074/jbc.M113.477943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson L.; Jensen P. E.; Hunter C. N. Biochem. J. 1999, 337 (Pt 2), 243. 10.1042/bj3370243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid J. D.; Hunter C. N. J. Biol. Chem. 2004, 279 (26), 26893. 10.1074/jbc.M400958200. [DOI] [PubMed] [Google Scholar]
- Adams N. B. P.; Marklew C. J.; Brindley A. A.; Hunter C. N.; Reid J. D. Biochem. J. 2014, 457 (1), 163–170. 10.1042/BJ20130834. [DOI] [PubMed] [Google Scholar]
- Adams N. B. P.; Reid J. D. Biochemistry 2012, 51, 2029–2031. 10.1021/bi300149z. [DOI] [PubMed] [Google Scholar]
- Fodje M. N.; Hansson A.; Hansson M.; Olsen J. G.; Gough S.; Willows R. D.; Al-Karadaghi S. J. Mol. Biol. 2001, 311 (1), 111–122. 10.1006/jmbi.2001.4834. [DOI] [PubMed] [Google Scholar]
- Qian P.; Marklew C. J.; Viney J.; Davison P. A.; Brindley A. A.; Soderberg C.; Al-Karadaghi S.; Bullough P. A.; Grossmann J. G.; Hunter C. N. J. Biol. Chem. 2012, 287 (7), 4946–4956. 10.1074/jbc.M111.308239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams N. B. P.; Marklew C. J.; Qian P.; Brindley A. A.; Davison P. A.; Bullough P. A.; Hunter C. N. Biochem. J. 2014, 464 (3), 315–322. 10.1042/BJ20140463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Pu H.; Fang Y.; Wang X.; Zhao S.; Lin Y.; Zhang M.; Dai H.-E.; Gong W.; Liu L. Nat. Plants 2015, 1 (9), 15125. 10.1038/nplants.2015.125. [DOI] [PubMed] [Google Scholar]
- Erzberger J. P.; Berger J. M. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- Licht S.; Lee I. Biochemistry 2008, 47 (12), 3595–3605. 10.1021/bi800025g. [DOI] [PubMed] [Google Scholar]
- Hynes R. O. Cell 2002, 110 (6), 673–687. 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Jensen P. E.; Gibson L.; Hunter C. N. Biochem. J. 1999, 339 (Pt 1), 127. 10.1042/0264-6021:3390127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken C. J.; Baaske P.; Rothbauer U.; Braun D.; Duhr S. Nat. Commun. 2010, 1 (7), 100. 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- Jerabek-Willemsen M.; André T.; Wanner R.; Roth H. M.; Duhr S.; Baaske P.; Breitsprecher D. J. Mol. Struct. 2014, 1077, 101–113. 10.1016/j.molstruc.2014.03.009. [DOI] [Google Scholar]
- Seidel S. A. I.; Dijkman P. M.; Lea W. A.; van den Bogaart G.; Jerabek-Willemsen M.; Lazic A.; Joseph J. S.; Srinivasan P.; Baaske P.; Simeonov A.; Katritch I.; Melo F. A.; Ladbury J. E.; Schreiber G.; Watts A.; Braun D.; Duhr S. Methods 2013, 59 (3), 301–315. 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhr S.; Braun D. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (52), 19678–19682. 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T.; Whiteheart S. W.; Wilkinson A. J. J. Struct. Biol. 2004, 146 (1–2), 106–112. 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Wendler P.; Ciniawsky S.; Kock M.; Kube S. Biochim. Biophys. Acta, Mol. Cell Res. 2012, 1823 (1), 2–14. 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Hanson P. I.; Whiteheart S. W. Nat. Rev. Mol. Cell Biol. 2005, 6 (7), 519–529. 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Gräfe S.; Saluz H. P.; Grimm B.; Hänel F. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1941–1946. 10.1073/pnas.96.5.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rief M.; Oesterhelt F.; Heymann B.; Gaub H. E. Science 1997, 275, 1295–1297. 10.1126/science.275.5304.1295. [DOI] [PubMed] [Google Scholar]
- Janshoff A.; Neitzert M.; Oberdörfer Y.; Fuchs H. Angew. Chem., Int. Ed. 2000, 39, 3212–3237. . [DOI] [PubMed] [Google Scholar]
- Zlatanova J.; Lindsay S. M.; Leuba S. H. Prog. Biophys. Mol. Biol. 2000, 74 (1–2), 37–61. 10.1016/S0079-6107(00)00014-6. [DOI] [PubMed] [Google Scholar]
- Dupres V.; Menozzi F. D.; Locht C.; Clare B. H.; Abbott N. L.; Cuenot S.; Bompard C.; Raze D.; Dufrêne Y. F. Nat. Methods 2005, 2 (7), 515–520. 10.1038/nmeth769. [DOI] [PubMed] [Google Scholar]
- Verbelen C.; Gruber H. J.; Dufrêne Y. F. J. Mol. Recognit. 2007, 20 (6), 490–494. 10.1002/jmr.833. [DOI] [PubMed] [Google Scholar]
- Nevo R.; Stroh C.; Kienberger F.; Kaftan D.; Brumfeld V.; Elbaum M.; Reich Z.; Hinterdorfer P. Nat. Struct. Biol. 2003, 10 (7), 553–557. 10.1038/nsb940. [DOI] [PubMed] [Google Scholar]
- Riener C. K.; Kienberger F.; Hahn C. D.; Buchinger G. M.; Egwim I. O. C.; Haselgrübler T.; Ebner A.; Romanin C.; Klampfl C.; Lackner B.; Prinz H.; Blaas D.; Hinterdorfer P.; Gruber H. J. Anal. Chim. Acta 2003, 497 (1–2), 101–114. 10.1016/j.aca.2003.08.041. [DOI] [Google Scholar]
- Berquand A.; Xia N.; Castner D. G.; Clare B. H.; Abbott N. L.; Dupres V.; Adriaensen Y.; Dufrêne Y. F. Langmuir 2005, 21 (12), 5517–5523. 10.1021/la050162e. [DOI] [PubMed] [Google Scholar]
- Lebed K.; Lekka M.; Dabrowska A.; Kulik A. J.; Lekki J.; Strachura Z.. Condensed Matter: New Research; Das M. P., Ed.; Nova Science Publishers, Inc.: New York, 2007. [Google Scholar]
- Evans E. Faraday Discuss. 1998, (111), 1–16. [DOI] [PubMed] [Google Scholar]
- Tees D. F.; Waugh R. E.; Hammer D. A. Biophys. J. 2001, 80 (2), 668–682. 10.1016/S0006-3495(01)76047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmlund H.; Lundqvist J.; Al-Karadaghi S.; Hansson M.; Hebert H.; Lindahl M. J. Mol. Biol. 2008, 375 (4), 934–947. 10.1016/j.jmb.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Lundqvist J.; Elmlund H.; Wulff R. P.; Berglund L.; Elmlund D.; Emanuelsson C.; Hebert H.; Willows R. D.; Hansson M.; Lindahl M.; Al-Karadaghi S. Structure 2010, 18 (3), 354–365. 10.1016/j.str.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Schmidt H.; Gleave E. S.; Carter A. P. Nat. Struct. Mol. Biol. 2012, 19 (5), 492–497. 10.1038/nsmb.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl S. W.; Nash M. A.; Fried D. B.; Slutzki M.; Barak Y.; Bayer E. A.; Gaub H. E. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (50), 20431–20436. 10.1073/pnas.1211929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico F.; Moy V. T. J. Mol. Recognit. 2007, 20 (6), 495–501. 10.1002/jmr.841. [DOI] [PubMed] [Google Scholar]
- Bowers C. M.; Carlson D. A.; Rivera M.; Clark R. L.; Toone E. J. J. Phys. Chem. B 2013, 117 (17), 4755–4762. 10.1021/jp309393s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.