

Abstract
Selective manipulation of protein kinases under living conditions is highly desirable yet extremely challenging, particularly in a gain-of-function fashion. Here we employ our recently developed bioorthogonal cleavage reaction as a general strategy for intracellular activation of individual kinases. Site-specific incorporation of trans-cyclooctene-caged lysine in place of the conserved catalytic lysine, in conjunction with the cleavage partner dimethyl-tetrazine, allowed efficient lysine decaging with the kinase activity chemically rescued in living systems.
Short abstract
Site-specific incorporation of trans-cyclooctene-caged lysine in place of the catalytic lysine, followed by the bioorthogonal cleavage reaction, allowed efficient kinase activation in living systems.
Introduction
As key players in signaling transduction, the more than 500 protein kinases control diverse biological processes that are essential for all aspects of the cell.1,2 The activity of each kinase is precisely regulated by its native physiochemical inputs,1 which are often entangled within the complicated signaling networks, making it exceedingly difficult to manipulate a single kinase with high specificity and/or spatiotemporal resolution. Intensive efforts have therefore been made to specifically inhibit or activate a kinase of interest within a cell. For example, the classical “bump and hole” strategy allows chemical genetic inhibition of engineered inhibitor-sensitized kinase mutants by adenosine derivatives without perturbation to wild-type kinases,3 which provided fruitful insights on kinase-mediated signaling cascades. Meanwhile, multiple protein engineering strategies have been explored for turning on specific kinases,4 including direct chemical rescue of a mutant kinase,5 ligand-gated split kinases,6 the rapamycin-induced allosteric activation of kinases,7 and among others. Because of the gain-of-function nature of these approaches that is advantageous in probing the sufficiency of a specific kinase as opposed to the more widely adopted loss-of-function methods,8 such study greatly enhanced our capability for deciphering and dissecting the intricate kinase networks.9 However, the design and engineering of these kinase variants are often sophisticated and are restricted to certain types of kinases that lack general applicability to the entire kinase families.
Results
Design of Bioorthogonal Chemical Activation Strategy
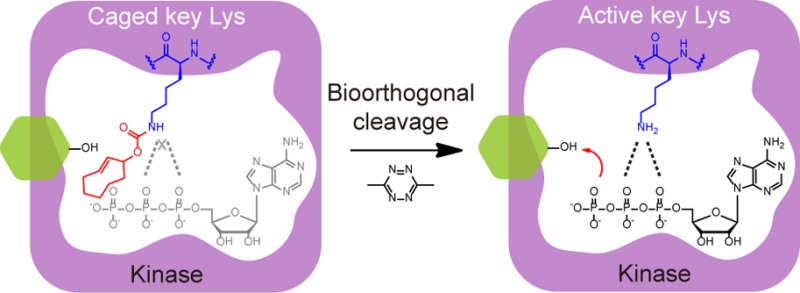
To address these issues, we aim to combine bioorthogonal chemical cleavage reactions10,11 with the genetic code expansion methodology12 to afford a mechanism-based kinase activation strategy with general applicability to virtually all protein kinases. Because lysine is a universally conserved catalytic residue that resides in kinase ATP-binding pocket for ATP docking and phosphoryl transfer, mutation of this lysine residue can abolish the activity of any given kinase.2 Our idea relies on replacing this key lysine residue with a chemical-caged lysine analogue via genetic code expansion for blockage of its enzymatic activity (Figure 1a). The subsequent addition of the bioorthogonal cleavage partner will decage this lysine residue and thus rescue the corresponding wild-type kinase. Although the photocaged version of such lysine-restoration strategy has been previously developed for kinase activation with high temporal resolution and the potential of spatial control,13 the UV light utilized could cause cell damage and perturb the intracellular signaling network.14 In addition, the nitrobenzyl-based photocaging lysine analogue such as o-nitropiperonyl caged lysine (ONPK, 1, Figure S1) employed in the aforementioned study is not fully bioorthogonal and could be decaged in prokaryotic reducing environments (Figure S2), or by enzymes in eukaryotic cells under hypoxic conditions.15 The development of two-photon excitation caging groups circumvented the concern of UV cytotoxicity and could be used to improve the tissue penetration ability.16 Still, a small molecule-based chemical activation strategy has its unique advantages such as better biocompatibility, tunability, and minimal invasiveness, particularly for intact living animals.10 For example, the inverse electron-demand Diels–Alder (invDA) reaction between trans-cyclooctene and tetrazine has been recently employed as a small molecule “click to release” strategy for controlled release of doxorubicin from the antibody-drug conjugates.17 We have further developed the axial isomer of trans-cyclooctene caged lysine (TCOK-a, 2) and 3,6-dimethyl-1,2,4,5-tetrazine (Me2Tz, 3) as a bioorthogonal cleavage pair for invDA-mediated lysine decaging and protein activation inside cells (Figure 1b, Figure S1).18 Therefore, we reasoned that the site-specific incorporation of TCOK-a in place of the catalytic lysine residue can render the kinase inactive, whereas the Me2Tz-triggered invDA reaction and subsequent decaging on TCOK-a may restore the key lysine with the kinase activity chemically rescued (Figure 1c). Herein, we report such a bioorthogonal chemical activation strategy for selective rescue of kinase activity in living cells as well as in living animals.
Figure 1.

Design of bioorthogonal chemical activation strategy for kinases. (a) Chemical decaging of lysine-dependent enzymes. X represents the chemical caging group that renders the enzyme inactive (gray). Upon small molecule-triggered bioorthogonal cleavage reaction, the native lysine residue is restored, which rescues the enzyme activity (orange). (b) Mechanism of the invDA reaction between Me2Tz and the genetically encoded TCOK-a amino acid which subsequently undergoes rearrangement and elimination that ultimately leads to in situ liberation of the ε-amine on lysine. (c) Schematic illustration of invDA-mediated kinase activation. TCOK-a is site-specifically incorporated into a target kinase (cyan) in place of the conserved catalytic lysine residue, which blocks the ATP (green) binding and phosphoryl transfer between kinase and the substrate (orange). The addition of Me2Tz triggers rapid invDA reaction for lysine decaging with the kinase activity chemically rescued (structure based on PDB: 2SRC).
Bioorthogonal Chemical Activation of MEK1 and FAK in Living Cells
We started with the classical Ser/Thr kinase MEK1 in the mitogen-activated protein kinases (MAPK) pathway, one of the fundamental signaling cascades for all eukaryotic systems.19 MEK1 is an essential component in the MAPK pathway and phosphorylates its downstream MAPKs such as extracellular-signal regulated kinases (ERKs) to fulfill diverse essential functions. A constitutively active MEK1 variant with partial N-terminal deletion and S218D/S222D mutation (MEK1-ΔN) was constructed to minimize the interference from endogenous upstream regulation by kinases such as Raf.13 An in vitro kinase assay was first performed between purified MEK1 variants and its substrate ERK to confirm that TCOK-a incorporation in place of the catalytic lysine residue (K97) effectively blocked its kinase activity (Figure S3). In addition, the expression of this caged MEK1 variant (MEK1-ΔN-K97TCOK) did not alter the ERK phosphorylation level inside cells (Figure 2a). We then applied the invDA-mediated chemical activation strategy on MEK1 under living conditions. As expected, the phosphorylation level of ERK and the downstream transcription activity were significantly increased only upon the addition of Me2Tz to cells harboring the MEK1-ΔN-K97TCOK variant (Figure 2a,b). When the noncleavable bicyclo[6.1.0]nonyne caged lysine analogue (BCNK, 4, Figure S1) was used as the control, the caged MEK1 variant (MEK1-ΔN-K97BCNK) was not able to be rescued by Me2Tz under the same condition, which further confirmed that the MEK1 activation was triggered by the designed cleavage reaction instead of the effects from any reagents added. These results demonstrated the capability of TCOK-a/Me2Tz as a bioorthogonal cleavage pair for gain-of-function study of MEK1 within an intracellular context.
Figure 2.

Bioorthogonal chemical activation of kinases of interest in living cells. (a) Cells expressing MEK1-ΔN-K97TCOK showed an increased level of ERK phosphorylation upon Me2Tz addition. (b) ETS domain-containing protein (Elk)-based luciferase reporter showed Elk-related transcription level activation (mean ± s.d., n = 3) of MAPK pathway following chemical cleavage (**P = 0.0095 by two-tailed t test). (c) FAK autophosphorylation level showed activation of hyperactive FAK mutant (FAK-YM) but not the kinase dead mutant (FAK-YM-KD) by the TCOK-a/Me2Tz decaging pair. (d) The phosphorylation level of Src was increased in Me2Tz treated hyperactive FAK (FAK-YM-TCOK) expressing cells but not in the Y397F mutant variant. Me2Tz, 100 μM. HEK293T cells were used in (a-d).
We then moved to another kinase family to prove the general applicability of our chemical activation strategy. Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase (NRTK) central in integrin signaling pathway that is involved in cellular adhesion and spreading.20 To chemically manipulate its activity in living cells, we also constructed the hyperactive FAK mutants according to the literature.7 The Y180A/M183A mutated FAK (FAK-YM) showed higher activity for autophosphorylation as well as the phosphorylation of Src than wild type FAK, while mutation of its catalytic aspartic acid (FAK-YM-KD) or autophosphorylation site (FAK-YM-Y397F) strongly eliminated the phosphorylation activity (Figure S4). TCOK-a caged FAK-YM at the catalytic lysine K454 (FAK-YM-K454TCOK) exhibited a significantly increased autophosphorylation level upon Me2Tz-triggered bioorthogonal cleavage and lysine regeneration, whereas the FAK-YM-KD variant showed no kinase activity after chemical cleavage due to the mutation of the catalytic aspartic acid (Figure 2c and Figure S5a). Similarly, the phosphorylation level of Src was effectively elevated after Me2Tz-triggered decaging of TCOK-a on the FAK-YM-K454TCOK variant, while the Y397F autophosphorylation site mutation eliminated the effects originally caused by binding between the pTyr on FAK and SH2 domain of Src, which led to Src activation (Figure 2d and Figure S5b). The time course study was also conducted, showing an increase of FAK and Src phosphorylation level reached maximum 30 min after Me2Tz treatment (Figure S6). While a basal level of endogenous FAK activity was observed, the hyperactive FAK mutant we rescued exhibited obviously higher activity. In addition, a genetic knockdown or selective kinase inhibitor could be utilized in combination with chemical activation to minimize the perturbation from an endogenous level of kinases, providing an efficient activation strategy with low background.
Bioorthogonal Chemical Activation of Src Oncogenic Variant in Living Cells
Next, considering that many oncogenic forms of kinases are tumor inducing agents with high clinical relevance,1,21 we applied our strategy for in situ rescue of oncogenic kinase mutants which could be used to study its cancer-related cellular responses. The C-terminal tail deletion or Y527F autophosphorylation site mutation forms of Src are its oncogenic variants escaping from C-terminal Src kinase (Csk) regulation with aberrant high activation levels.22 We introduced the Y527F mutation and replaced the catalytic lysine (K295) with TCOK-a, which generated a caged oncogenic Src variant (Src-Y527F-K295TCOK, Figure 3a). Expression of the active oncogenic Src mutant (Src-Y527F) led to its autophosphorylation and downstream tyrosine phosphorylation cascades, while the lysine-caged mutant did not show phosphorylation activity. Notably, upon Me2Tz-mediated lysine decaging on Src-Y527F-K295TCOK, the high kinase activity of Src was restored (Figure 3b). We also investigated the time course and concentration dependence of oncogenic Src activation, which showed that the autophosphorylation of Src was initiated in 5 min with 100 μM Me2Tz, further demonstrating the rapid activation property of this invDA-mediated cleavage reaction (Figure S7a,b). The chemical decaging-mediated protein activation rate was slower but within the same scale as that of photodecaging-mediated protein activation (Figure S7c). Moreover, this activation strategy was verified in a panel of cell lines including 293A, NIH3T3, CHO, and HeLa, which further established its general applicability (Figure S8).
Figure 3.

Bioorthogonal chemical activation of Src oncogenic variant in living cells. (a) Schematic model for bioorthogonal activation of oncogenic Src variant. Substitution of the catalytic lysine (K295) with TCOK-a on the oncogenic Src mutant (Src-Y527F) generated the chemically caged oncogenic Src variant Src-K295TCOK-Y527F (gray). Decaging of TCOK-a via bioorthogonal cleavage reaction could rescue Src activity (orange), leading to phosphorylation of its downstream oncogenic-related substrates. (b) Increase of Src autophosphorylation and downstream phosphorylation level after Me2Tz-mediated decaging of Src-K295TCOK-Y527F in live HEK293T cells. (c) Representative images of 293A cells expressing the oncogenic Src-Y527F variant which led to cell rounding and detaching. 293A cells expressing the “kinase dead” Src mutant (Src-K295R-Y527F) was used as the control. Scale bars, 5 μm. (d) Representative images of phenotypic changes upon chemical rescue of oncogenic Src. Upon Me2Tz mediated bioorthogonal activation, 293A cells expressing Src-K295TCOK-Y527F began to rapidly round up. Me2Tz, 100 μM; Scale bars, 5 μm.
Besides the detection of the phosphorylation level, we also observed that constitutive expression of oncogenic Src variants caused morphological changes such as cell rounding, detaching, and filament disassembly in 293A cells (Figure 3c and Figure S9). These phenotypes resembled the v-src transformed fibroblasts previously reported with increased motility and invasiveness.22 To track this phenotype in a dynamic manner, the bioorthogonal chemical activation was performed for specific and rapid rescue of the oncogenic Src mutant. Cells expressing TCOK-a caged oncogenic Src variant (Src-Y527F-K295TCOK) underwent cell rounding and detaching rapidly after Me2Tz-triggered lysine decaging, while neither the noncleavable benzyloxycarbonyl caged lysine analogue (CbzK, 5, Figure S1) caged Src mutant nor the lysine-mutated Src showed noticeable changes (Figure 3d and Figure S9,S10). Manipulation of oncogenic Src mutant via this bioorthogonal chemical activation strategy may provide a powerful tool in studying other oncogenic kinase variants.
Expanding the Bioorthogonal Chemical Activation Toolkit into Living Animals
Finally, we sought to move our bioorthogonal cleavage strategy into more complicated biological systems such as live animals. Because of the poor penetration capability, many photodecaging strategies are not compatible with deep tissue or animal samples. In contrast, the invDA reaction between trans-cyclooctene and tetrazine derivatives has been established in recent years as the choice of bioorthogonal reactions for applications in intact animals due to their fast reaction rate and high biocompatibility.23,24 To test our invDA-mediated bioorthogonal cleavage reaction in living mice, we first utilized firefly luciferase (fLuc) as the model protein due to the convenience in detecting its enzymatic activity via bioluminescence in vivo.25 A key lysine residue (K529) in fLuc forms hydrogen bonds with ATP to facilitate the production of the bioluminescent oxyluciferin (Figure 4a). Decaging of the premasked lysine residue has been shown previously to restore the blocked luciferase activity via the reformation of hydrogen bonding network between ε-amine on K529 and ATP.18,25 HEK293T cells expressing the TCOK-a caged fLuc variant (fLuc-K529TCOK) were injected into living mice subcutaneously, followed by the treatment of Me2Tz (66 or 6.6 mg/kg body weight) via tail vein injection. The Me2Tz-triggered invDA reaction on TCOK-a led to effective restoration of luciferase activity, as evidenced by the recovered bioluminescence signal in this xenograft mice model (Figure 4b). Toxicity study showed that the Me2Tz molecule did not affect mouse growth up to 20 days with the highest dosage we tested (intravenous injection of 50 μL of 500 mM Me2Tz 3 times a week, equal to 140 mg/kg body weight; Figure S11). We further showed that the activation efficiency was tunable by varying the administered Me2Tz concentration, and the maximum recovery efficiency was comparable to those cells activated by Me2Tz in culture medium (Figure 4c).
Figure 4.

Expanding the bioorthogonal chemical activation strategy into living animals. (a) Schematic flow of bioorthogonal chemical activation strategy in mice by using chemically caged firefly luciferase as the model. HEK293T cells expressing the fLuc-K529TCOK variant were subcutaneously injected into mice, followed by tail vein injection of Me2Tz (50 μL), which led to the regeneration of the active fLuc capable of converting d-luciferin to oxyluciferin in the presence of ATP (structure based on PDB: 2D1Q). (b) Representative images of rescued fLuc activity as measured by restored bioluminescence 60 min after Me2Tz injection. 50 μL of Me2Tz of 300 or 30 mM concentration was injected, equal to 66 or 6.6 mg/kg weight, respectively. HEK293T cells with and without the expression of fLuc-K529TCOK were injected into left legs and right legs, respectively. (c) Statistical analysis (mean ± s.d., n = 3) of bioluminescence values in (b). P = 0.302, 0.097, 0.071, 0.021 in each group by one-tailed t test (*P < 0.05). (d) Activation of Src oncogenic mutant using the invDA-mediated bioorthogonal cleavage reaction in living mice. Cells expressing Src-K295TCOK-Y527F were hypodermically injected into mice, followed by tail vein injection of Me2Tz (50 μL, 300 mM, equal to 66 mg/kg weight). The xenografted cells were extracted to evaluate the Src autophosphorylation level by Western blotting using pY416-Src antibody. The kinase activity of Src was successfully restored with Me2Tz injection. (e) The overexpressed myc-tagged Src was further purified and separated from the endogenous Src kinase by immunoprecipitation on the extracted samples using myc antibody. The Src autophosphorylation level was then analyzed by Western blotting using anti-pY416-Src antibody.
Next, we pursued the activation of Src oncogenic variant in living mice. Similarly, we injected the TCOK-a caged Src oncogenic variant (Src-K295TCOK-Y527F)-expressing cells subcutaneously into living mice, followed by immediate administration of Me2Tz (66 mg/kg body weight) through tail vein injection. These cells were then extracted after 1 h decaging and Src activation was analyzed by detecting its autophosphorylation level on Western blotting gel with pY416-Src antibody. Similar to luciferase activation, the Src autophosphorylation level was obviously elevated in the Me2Tz treated group, with the activation efficiency comparable with the preactivated positive control group (Figure 4d). In addition, the overexpressed myc-tagged Src was purified and separated from the endogenous Src kinase by immunoprecipitation using myc antibody. The detection of Src autophosphorylation by pY416-Src antibody further confirmed efficient activation of Src kinase in living mice (Figure 4e). These results verified the capability of Me2Tz to pass through blood circulation and enter the target cells for in vivo enzyme activation.
Taken together, we demonstrated the compatibility of our bioorthogonal cleavage reaction in living animals, and this chemical activation strategy can be used for gain-of-function study of lysine-dependent enzymes such as kinases within complicated living systems.
Discussion
In summary, we developed a generally applicable strategy to chemically rescue kinase activity in living systems with high efficiency and specificity. Because of the structural and functional similarity among many kinases, general strategies for gain-of-function study of individual kinases are highly desired. Our bioorthogonal chemical rescue approach allowed the activation of a kinase of interest among its closely related family members with very low cytotoxicity or perturbation to cellular processes. This, in conjunction with the capability to differentiate the direct, as opposed to indirect substrates within the downstream signaling cascades, would facilitate the dissection of the intricate and entangled kinase signaling networks in native cellular conditions. Furthermore, our method provides a complementary strategy to the photocaging method for in situ manipulation of kinase activity, which has potential advantages especially in living animals. For example, our study offers an appealing small molecule-based kinase activation strategy within intact animals, which could be used for chemical control of selective cell activation in tissues or animals that are highly desired for cell-based therapy.26,27 In addition, our mechanism-based activation strategy could be readily expanded to a wide range of lysine-dependent enzymes other than kinases, providing a common approach for gain-of-function study of various biological processes under living conditions. Indeed, the emerging of bioorthogonal bond-cleavage reactions significantly expanded our bioorthogonal chemistry toolkit beyond ligation, which may find broad applications, particularly in manipulating biomolecular and/or cellular activities in living systems.10,28,29
Methods
In Vitro Kinase Assay
Purified MEK1 variants (5 ng/μL) and GST-ERK fusion protein (20 ng/μL) were incubated with 1 mM ATP in HMM buffer at 37 °C for indicated time.30 The reaction was then terminated with loading buffer and subjected to SDS-PAGE analysis followed by Western blotting. HMM buffer: 10 mM HEPES, 10 mM MgCl2, 10 mM MnCl2, pH = 7.4.
Expression and Activation of Chemically Caged Kinases in Living Cells
Cells were plated in multiwell plates and grown to 60–80% confluency ready for transfection. Plasmids encoding the target kinases (MEK1, FAK, or oncogenic Src variant) bearing an in-frame amber codon were cotransfected with the plasmid encoding MmPylRS-306A/384F-tRNACUAPyl pair into cells in serum-free DMEM via X-tremeGENE HP (Roche) or lipofectamine LTX (Invitrogen). After 6 h, the medium was changed to DMEM/10% FBS supplemented with and without 1 mM UAA, and cells were further grown for another 18 h before being replaced with fresh DMEM. Me2Tz (100 μM, or specially indicated concentration) was added for 1 h (or specially noted time) to enable the invDA-mediated cleavage reaction to proceed inside live cells before collecting the cell samples for further analysis. For the phosphorylation assays, cells were lysed with lysis buffer and subjected to SDS-PAGE and Western blotting analysis. Lysis buffer: 20 mM HEPES-KOH, pH 7.8, 50 mM KCl, 100 mM NaCl, 1 mM EGTA, 1% NP-40, 1 mM NaF, 0.1 mM Na3VO4.
Dual Luciferase Assay
Cells were plated in 96-well plate and grown to 60–80% confluency ready for transfection. pcDNA4-MEK1-ΔN-K97TAG, pCMV-MmPylRS-306A/384F, Elk-Gal4, Gal4-fLuc, and pRL-TK were cotransfected into cells in serum-free DMEM via X-tremeGENE HP (Roche). After 6 h, medium was changed to DMEM/10% FBS supplemented with 1 mM TCOK or BCNK, and cells were grown for another 18 h before replacing the culture medium with fresh DMEM containing 100 μM Me2Tz. After 8 h, medium was removed and bioluminescence was detected according to the protocol for dual luciferase assay kit (Vigorous) on a Synergy H4 microplate reader (Bio-Tek). Dual luciferase activity was calculated according to the following formula: dual luciferase activity = fLuc bioluminescence/rLuc bioluminescence.
Fluorescence Imaging
For live cell imaging, cells were plated into a four-well chamber and cotransfected with indicated plasmids. Upon confocal imaging, culture medium was removed and replaced with fresh live cell imaging solution (Gibco). Images were captured on LSM700 laser scanning confocal microscope (Zeiss). GFP channel was used for detection of Src-EGFP fluorescence and Texas Red channel was used for detection of LifeAct-mRFP fluorescence. For time lapse images, frames were captured with 2 min intervals for 40 min (10 min before and 30 min after 100 μM Me2Tz addition). For fixed cell imaging, cells were transfected and treated before fixation with 4% PFA, permeabilized with 0.1% Triton-X100, and blocked with 5% FBS, before immunofluorescence staining or phalloidin staining. DAPI was used to stain nucleus in the last step. Images of stained samples were also visualized on LSM 700 laser scanning confocal microscope (Zeiss).
Luciferase and Kinase Activation in Mice
Balb/c nude mice (male, 6–8 weeks) were purchased from Vital River Laboratories, China. All protocols were approved by the Institutional Animal Care and Use Committee of Peking University accredited by AAALAC International. HEK293T cells were cultured in dishes, cotransfected with pcDNA3.1-fLuc-K529TAG (or pcDNA4-Src-K295TAG-Y527F-mycHis) and pCMV-MmPylRS-306A/384F in serum-free DMEM via X-tremeGENE HP (Roche). After 6 h, medium was changed to DMEM/10% FBS supplemented with and without 1 mM TCOK (or with 1 mM CbzK as the negative control in the case of Src) and cells were grown for another 18 h. Cells were then digested, centrifuged, and resuspended in DMEM before being placed on ice or preactivated with 500 μM Me2Tz at R.T. for 20 min. These cells (∼2 × 107 cells/50 μL) were then subcutaneously injected into the mice (left legs, cells with fLuc-K529TCOK/Src-K295TCOK-Y527F expression; right legs, cells without fLuc-K529TCOK/with Src-K295CbzK-Y527F expression). 50 μL Me2Tz of 300 or 30 mM concentration (equal to 66 or 6.6 mg/kg body weight for an ∼25 g mouse) was then intravenously injected into mice via tail vein immediately following the cell injection, allowing bioorthogonal activation of target protein in vivo. For the case of luciferase, after 40 min, 150 μL of luciferin (15 mg/mL) was administered into mice (equal to 90 mg/kg body weight) by intraperitoneal injection, and after another 20 min, the in vivo bioluminescence images of mice were captured on IVIS Lumina II imaging system (PerkinElmer). For the case of Src, cells were extracted 1 h after tetrazine administration, followed by immunoprecipitation by myc antibody and analysis of autophosphorylation level by Western blotting. Immunoprecipitation was intended to distinguish overexpressing Src mutant with endogenous Src protein to make a clear background.
Statistical Analysis
Statistical values in graphs are presented as mean ± s.d. from three independent experiments. The t test or ANOVA is used for a significance test.
Acknowledgments
We thank Prof. Feng Shao (NIBS) and Prof. Li Yu (Tsinghua University) for providing plasmids. This work was supported by the National Natural Science Foundation of China (21225206, 21521003, 21432002, and 91313301), the National Basic Research Program of China (2012CB917301).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00024.
Supplementary experimental procedures; Figure S1. Chemical structures of unnatural amino acids and chemical reagents used in this study. Figure S2. LC-MS analysis of GFP-N149ONPK. Figure S3. In vitro kinase assay on MEK1. Figure S4. Phosphorylation analysis of several FAK mutants. Figure S5. Bioorthogonal cleavage mediated FAK activation. Figure S6. Activation of FAK at different time points after Me2Tz treatment. Figure S7. Time and concentration dependence of chemical decaging mediated oncogenic Src activation and the comparison with UV-mediated decaging. Figure S8. Activation of oncogenic Src variant in different cell lines. Figure S9. Phenotypes of 293A cells expressing Src variants. Figure S10. Phenotype change of 293A cells expressing caged oncogenic Src mutant with or without Me2Tz-mediated activation. Figure S11. Toxicity study on Me2Tz (PDF)
Author Contributions
§ G.Z. and J.L.: equal contribution.
The authors declare no competing financial interest.
Supplementary Material
References
- Blume-Jensen P.; Hunter T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E.; Johnson L. N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 2012, 81, 587–613. 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Bishop A. C.; Ubersax J. A.; Petsch D. T.; Matheos D. P.; Gray N. S.; Blethrow J.; Shimizu E.; Tsien J. Z.; Schultz P. G.; Rose M. D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Rakhit R.; Navarro R.; Wandless T. J. Chemical biology strategies for posttranslational control of protein function. Chem. Biol. 2014, 21, 1238–1252. 10.1016/j.chembiol.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y.; Molina H.; Pandey A.; Zhang J.; Cole P. A. Chemical rescue of a mutant enzyme in living cells. Science 2006, 311, 1293–1297. 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- Camacho-Soto K.; Castillo-Montoya J.; Tye B.; Ghosh I. Ligand-gated split-kinases. J. Am. Chem. Soc. 2014, 136, 3995–4002. 10.1021/ja4130803. [DOI] [PubMed] [Google Scholar]
- Karginov A. V.; Ding F.; Kota P.; Dokholyan N. V.; Hahn K. M. Engineered allosteric activation of kinases in living cells. Nat. Biotechnol. 2010, 28, 743–747. 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn J. A.; Wells J. A. Turning enzymes ON with small molecules. Nat. Chem. Biol. 2010, 6, 179–188. 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- Chu P. H.; Tsygankov D.; Berginski M. E.; Dagliyan O.; Gomez S. M.; Elston T. C.; Karginov A. V.; Hahn K. M. Engineered kinase activation reveals unique morphodynamic phenotypes and associated trafficking for Src family isoforms. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12420–12425. 10.1073/pnas.1404487111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Chen P. R. Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol. 2016, 12, 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Bielski R.; Witczak Z. Strategies for coupling molecular units if subsequent decoupling is required. Chem. Rev. 2013, 113, 2205–2243. 10.1021/cr200338q. [DOI] [PubMed] [Google Scholar]
- Wang L.; Schultz P. G. Expanding the genetic code. Angew. Chem., Int. Ed. 2005, 44, 34–66. 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- Gautier A.; Deiters A.; Chin J. W. Light-activated kinases enable temporal dissection of signaling networks in living cells. J. Am. Chem. Soc. 2011, 133, 2124–2127. 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldchen S.; Lehmann J.; Klein T.; van de Linde S.; Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 2015, 5, 15348. 10.1038/srep15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W.; Ji A.; Wang M. X.; Ai H. W. Expanding the Genetic Code for a Dinitrophenyl Hapten. ChemBioChem 2015, 16, 2007–2010. 10.1002/cbic.201500204. [DOI] [PubMed] [Google Scholar]
- Luo J.; Uprety R.; Naro Y.; Chou C.; Nguyen D. P.; Chin J. W.; Deiters A. Genetically encoded optochemical probes for simultaneous fluorescence reporting and light activation of protein function with two-photon excitation. J. Am. Chem. Soc. 2014, 136, 15551–15558. 10.1021/ja5055862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 2013, 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Li J.; Jia S.; Chen P. R. Diels-Alder reaction-triggered bioorthogonal protein decaging in living cells. Nat. Chem. Biol. 2014, 10, 1003–1005. 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- Seger R.; Krebs E. G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [PubMed] [Google Scholar]
- Mitra S. K.; Hanson D. A.; Schlaepfer D. D. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeatman T. J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckute J.; Devaraj N. K. Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol. 2013, 17, 761–767. 10.1016/j.cbpa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Lin S.; Huang Y.; Zhao J.; Chen P. R. Mechanism-based design of a photoactivatable firefly luciferase. J. Am. Chem. Soc. 2013, 135, 7410–7413. 10.1021/ja4013535. [DOI] [PubMed] [Google Scholar]
- Pawlak J. B.; Gential G. P.; Ruckwardt T. J.; Bremmers J. S.; Meeuwenoord N. J.; Ossendorp F. A.; Overkleeft H. S.; Filippov D. V.; van Kasteren S. I. Bioorthogonal deprotection on the dendritic cell surface for chemical control of antigen cross-presentation. Angew. Chem., Int. Ed. 2015, 54, 5628–5631. 10.1002/anie.201500301. [DOI] [PubMed] [Google Scholar]
- Roybal K. T.; Rupp L. J.; Morsut L.; Walker W. J.; McNally K. A.; Park J. S.; Lim W. A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Bertozzi C. R. A Bioorthogonal Reaction of N-Oxide and Boron Reagents. Angew. Chem., Int. Ed. 2015, 54, 15777–15781. 10.1002/anie.201508861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Li H.; Xu H.; Zhou Y.; Zhang J.; Long C.; Li S.; Chen S.; Zhou J.-M.; Shao F. The Phosphothreonine Lyase Activity of a Bacterial Type III Effector Family. Science 2007, 315, 1000–1003. 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.