

Abstract
The strategic incorporation of the trifluoromethyl (CF3) functionality within therapeutic or agrochemical agents is a proven strategy for altering their associated physicochemical properties (e.g., metabolic stability, lipophilicity, and bioavailability). Electrophilic trifluoromethylation has emerged as an important methodology for installing the CF3 moiety onto an array of molecular architectures, and, in particular, CF3 λ3-iodanes have garnered significant interest because of their unique reactivity and ease of handling. Trifluoromethylations mediated by these hypervalent iodine reagents often require activation through an exogenous Lewis or Brønsted acid; thus, putative intermediates invoked in these transformations are cationic CF3 iodoniums. These iodoniums have, thus far, eluded isolation and investigation of their innate reactivity (which has encouraged speculation that such species cannot be accessed). A more complete understanding of the mechanistic relevance of CF3 iodoniums is paramount for the development of new trifluoromethylative strategies involving λ3-iodanes. Here, we demonstrate that CF3 iodonium salts are readily prepared from common λ3-iodane precursors and exhibit remarkable persistence under ambient conditions. These reagents are competent electrophiles for a variety of trifluoromethylation reactions, and their reactivity is reminiscent of that observed when CF3 iodanes are activated using Lewis acids. As such, our results suggest the mechanistic relevance of CF3 iodonium intermediates in trifluoromethylative processes mediated by λ3-iodanes. The isolation of CF3 iodonium salts also presents the unique opportunity to employ them more generally as mechanistic probes.
Short abstract
The isolation of trifluoromethyl iodonium salts, putative intermediates in common trifluoromethylations, is reported. Probing their reactivity has implications for developing robust routes to therapeutic and agrochemical agents.
Introduction
Synthetic strategies for incorporating the trifluoromethyl (CF3) functionality into molecular scaffolds continue to garner extensive research interest, given that various therapeutic and agrochemical agents are decorated with this moiety (Figure 1).1−4 Apart from serving as a metabolically stable analogue of CH3, the CF3 group is also routinely harnessed as a Cl surrogate (given that the group electronegativity of CF3 is commensurate with that of Cl).1−4 The unique stereoelectronic properties of the CF3 group render it a privileged motif for tailoring pharmacokinetics and pharmacodynamics, and the synthesis of fludelone (a potent antimitotic analogue of epothilone B) is a particularly salient example of how the introduction of a CF3 substituent can enhance a drug’s stability without sacrificing efficacy (Figure 1).5 Of the available trifluoromethylative methodologies, those involving electrophilic reagents (which are typically easier to handle in the laboratory and can often be derived from inexpensive precursors) have risen to the forefront of the field.2 Electrophilic trifluoromethylations involving λ3-iodanes6 {e.g., 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole [1]; 1-trifluoromethyl-1,2-benziodoxol-3-(1H)-one [2]; Figure 2}7−9 have proven to be especially versatile methodologies, and the chemistry of these hypervalent iodine species has been extensively studied.10 A variety of nucleophiles (including examples from groups 14–16) have been reported to undergo trifluoromethylation in the presence of 1 or 2;11−29 however, these transformations typically require the action of a Lewis or Brønsted acid in concert with the iodane. As such, CF3 iodonium intermediates have long been speculated to play a critical role in the aforementioned reactions (Figure 2). Conclusively demonstrating the existence of a CF3 iodonium and investigating its reactivity would be extremely valuable for elucidating mechanistic insights that can inform future reaction discovery and development.
Figure 1.

Representative CF3-containing therapeutic and agrochemical compounds.
Figure 2.

Trifluoromethylations involving hypervalent iodine reagents and proposed reactive intermediates. (A) Conventional λ3-iodanes for electrophilic trifluoromethylation. (B) General mechanism for trifluoromethylation of a nucleophile under Lewis acid catalysis. The activated iodane is highlighted in red. (C) Structural distinctions between proposed intermediates in λ3-iodane mediated trifluoromethylations. (D) Experimental evidence suggests the predominance of iodane character for 1 and 2 under Lewis or Brønsted acid activation.
Although spectroscopic evidence has suggested the transient existence of these species,30 recent reports have rekindled the debate surrounding the relevance of CF3 iodoniums. While exploring the Zn(NTf2)2 [NTf = bis(trifluoromethylsulfonyl)imide] mediated trifluoromethyl etherification of alcohols, Koller et al. isolated and characterized zinc complex 3 (Figure 2).12 A salient feature of 3 is the lengthened I–O bond with respect to parent compound 2 [cf. 2.403(12) Å versus 2.283(2) Å],7 which is suggestive of distortion toward a species with iodonium character. As such, the authors concluded that 3 was the reactive intermediate responsible for the observed etherifications. In a separate report, protonation of 1 with Brookhart’s acid {[H(OEt)2][(3,5-CF3C6H3)4B]} was found to afford dimeric species 4 (Figure 2).31 The solid-state structure of 4 exhibited elongated I–O bonds with respect to parent compound 1 [cf. 2.440(4) Å and 2.257(3) Å versus 2.118(2) Å],14 again suggesting distortion toward a CF3 iodonium. However, interactions involving iodine(III) are known to exhibit weak covalent character, and bond lengths that exceed the sum of the covalent radii of I and O (2.06 Å) are not necessarily indicative of true iodonium character. The experimentally determined structures of 3 and 4 support the hypothesis that activation of CF3 iodanes using Lewis or Brønsted acids does not afford the corresponding iodoniums.10
Despite decades of effort, the successful synthesis and isolation of a genuine CF3 iodonium has remained elusive (Scheme 1). Early efforts by Umemoto and Yagupolskii failed to prepare CF3 iodoniums using the respective protocols each laboratory had developed for the synthesis of perfluoroalkyliodonium salts.32−34 The Togni laboratory also attempted to react (dihaloiodo)arenes and bis(acetoxy)iodoarenes with TMSCF3 in the presence of CsF or tetrabutylammonium difluorotriphenylsilicate, but these synthetic efforts were ultimately unfruitful.7 Given these failures, conventional wisdom has dictated that CF3 iodoniums are inherently unstable and cannot be isolated. Wang and Liu more recently reported the in situ formation of a putative CF3 iodonium (which was detected by mass spectrometry) from the reaction of TMSCF3 with iodobenzenediacetate.30 However, there is currently no direct evidence that CF3 iodoniums are more than transient intermediates generated during ionization in a mass spectrometer.
Scheme 1. Attempted Syntheses of CF3 Iodoniums That Have Previously Been Reported.

We speculated that the inductive destabilization of the trifluoromethyl group should not necessarily preclude the isolation of a CF3 iodonium, at least from a thermodynamic perspective. Literature precedents for the isolation of C3F7 iodonium salts were particularly encouraging, given that the group electronegativity of the C3F7 fragment has been estimated to be similar to that of CF3 (cf. ∼3.3 versus 3.4, respectively, on the Pauling scale).35 As such, we surmised that kinetic stabilization of the nascent ion pair would be paramount for accessing the desired iodoniums. The judicious selection of a mildly iodophilic anion seemed pertinent, given that strongly coordinating anions would generate a tight ion pair and subsequently undergo deleterious reductive eliminations. From a synthetic perspective, we decided to depart from traditional approaches to CF3 iodoniums and discard (dihaloiodo)arenes or bis(alkoxy)iodoarenes as potential precursors (given the established unsuitability of these moieties). Instead, our attention focused on commercially available 1 and 2 as convenient scaffolds for transformation into the corresponding iodoniums. Cleavage of the I–O bond in 1 or 2 under the action of a Brønsted acid presented itself as a plausible route for accessing the target compounds, and we ultimately decided to employ mineral acids (which have not been investigated in this context) to ascertain whether or not halide salts of CF3 iodoniums would exhibit persistence.
Results and Discussion
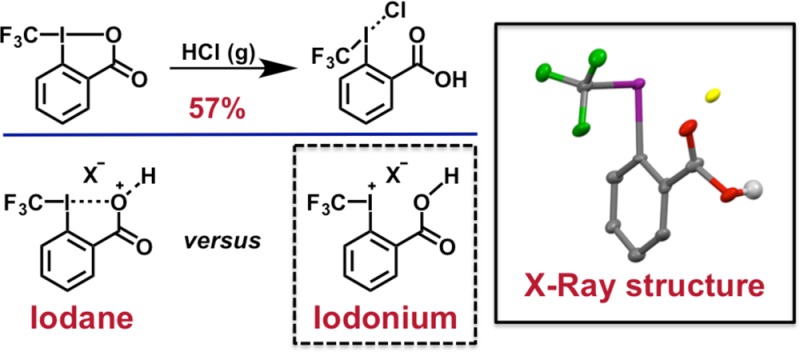
Dissolution of 1 in chloroform followed by sparging with HCl gas for approximately 5 min afforded a white, crystalline solid (1·HCl; 96% yield) upon removal of volatiles (Figures 3 and 4). Analysis by 19F nuclear magnetic resonance (NMR) spectroscopy revealed a singlet that was shifted significantly downfield relative to 1 (Δδ ≈ 13 ppm), which suggested the formation of a species with iodonium character.10 The solubility properties of 1·HCl were consistent with an ionic compound, as the salt exhibited poor solubility in low polarity solvents (e.g., aromatic, halogenated, or ethereal solvents). In contrast, 1·HCl was readily dissolved in polar solvents (e.g., alcohols, acetone, acetonitrile). Single crystals of 1·HCl spontaneously formed in a saturated CDCl3 solution and were analyzed by X-ray crystallography (Figure 4). The solid-state structure of 1·HCl exhibited a distorted T-shape geometry, as evidenced by the 173.37(16)° CCF3–I–Cl bond angle. Importantly, the I–O interatomic distance was found to be 3.080(4) Å, which unambiguously indicates that there is no bonding interaction between the hypervalent iodine center and the pendent alcohol. The cleavage of the I–O bond occurs with concomitant shortening of the CCF3–I bond [cf. 2.217(6) Å for 1·HCl versus 2.267(2) Å for 1].
Figure 3.

Synthesis of (A) 1·HCl and (B) 2·HCl by reacting their parent iodanes with HCl (g). (C) The chloride counterion proved paramount for successful iodonium synthesis, as the remaining halide series proved ineffectual for stabilizing the nascent iodonium. (D) Anion metathesis of 1·HCl with NaBArF24 afforded the corresponding salt, 1·HBArF24. (E) Anion metathesis of 1·HCl with NaOTf affords the unstable salt 1·HOTf, which subsequently decomposes via reductive elimination. One decomposition product was identified as 1·HBF4, and the X-ray structure of 1·HBF4 (thermal ellipsoids at 50% probability) is shown [atom code: green (F); red (O); pink (B); violet (I)]. Only one of the two inequivalent molecules of 1·HBF4 is shown. Hydrogen atoms other than the O–H atom have been omitted for clarity.
Figure 4.

(A) The solid-state structure of 1·HCl (thermal ellipsoids at 50% probability). While the hydroxyl hydrogen atom is shown, all others have been removed for clarity. Atom code: green (F); red (O); violet (I); yellow (Cl). (B) Halide bridging interaction between one molecule of 1·HCl in the asymmetric unit and a symmetry generated partner. (C) The solid-state structure of 11 (thermal ellipsoids at 50% probability). Hydrogen atoms have been removed for clarity. Atom code: green (F); violet (I); yellow (Cl); beige (Si).
The chloride counterion interacts weakly with the iodine center [the I–Cl bond length is 2.8331(14) Å], and, consequently, bridging chloride interactions are observed throughout the structure of 1·HCl (Figure 4). Notably, the observed I–Cl bond length is significantly longer than the distances observed in CF3ICl2 [2.478(2) Å and 2.4572(2) Å]36 and iodobenzene dichloride [2.45(15) Å].37 Furthermore, the I–Cl length of 1·HCl approaches the I–Clbridging interactions observed in diphenyliodonium chloride [3.064(3) Å and 3.105(3) Å].38 We also note that the distorted T-shape geometry of 1·HCl is consistent with that observed for diphenyliodonium chloride. Whereas the CF3–I–Caryl bond angle of 1·HCl is 90.5(2)°, the Caryl–I–Caryl bond angle of diphenyliodonium chloride is 92.6(3)°.38 Crystal packing forces can clearly enforce a coordination environment that favors the T-shape geometry; thus, distortion from a bent geometry (which one might anticipate for an iodonium salt) in the solid state does not necessarily preclude the existence of such a geometry in solution. We also prepared perfluoroalkyl iodonium chloride 11 for additional comparison, and a number of salient features regarding the crystal structure of 11 merit discussion [see the Supporting Information for additional details]. Figure 4 shows the two inequivalent molecules of 11 in the unit cell, which both exhibit similar distorted T-shape geometries (the Calkyl–I–Cl bond angles are 178.14(7)° and 178.78(7)°, respectively). Moreover, the I–Cl bond lengths observed in 11 [2.7546(7) Å and 2.7826(7) Å] are both significantly shorter than that of 1·HCl. Taken together these data suggest that 1·HCl exhibits significant iodonium character.
Consistent with our kinetic stabilization hypothesis (vide supra), the nature of the iodonium counteranion profoundly influenced the overall persistence of the salt (Figure 3). Treatment of 1 with triethylamine trihydrofluoride or salt metathesis of 1·HCl with AgF did not afford 1·HF. In both instances we only observed 1, which suggests that the electronegativity of F precludes a sufficient enthalpic binding affinity to overcome the chelate effect from the pendent oxygen. Although the reaction of 1 with HBr gas afforded a new compound exhibiting a 19F NMR spectrum that was reminiscent of 1·HCl, this species decomposed into a mixture of 1, CF3Br, CF3I, and other minor byproducts after sitting at room temperature. Similarly, anion metathesis of 1·HCl with NaBr led to the rapid formation of CF3I and CF3Br. Finally, salt metathesis of 1·HCl was attempted with NaI, but only CF3I and small amounts of 1 were observed. These results indicate that bromide and iodide are both sufficiently reducing to destabilize 1·HBr and 1·HI. Pseudohalide counteranions also proved deleterious, as 1·HOTf (prepared from 1·HCl using NaOTf) slowly decomposed in solution to afford primarily CF3OTf (Supporting Information). Interestingly, 1·HBF4 was generated during the decomposition of 1·HOTf, as determined by X-ray crystallography (Figure 3). Presumably one of the decomposition products of 1·HOTf abstracted boron from the borosilicate glass vessel in which the salt was generated. The I–O bond lengths observed in the solid state structure of 1·HBF4 [2.514(4) Å and 2.504(3) Å] indicate that this species does not exhibit a meaningful degree of iodonium-like character, which is consistent with poor stabilization of the I(III) center by the BF4 anion. Inspired by this result, we attempted anion metathesis between 1·HCl and other weakly coordinating counterions. For example, reaction of 1·HCl with sodium tetrakis(3,5-trifluoromethyl)phenylborate (NaBArF24) generated 1·HBArF24 in excellent yield (91%; Supporting Information).
1·HCl is remarkably stable as a solid under ambient conditions (i.e., no decomposition was observed by 19F NMR after several months) and appears to be indefinitely stable at 4 °C. No precautions to exclude air or water are required when storing or handling 1·HCl, and solutions of 1·HCl may be heated to 80 °C without detectable decomposition. While we have not observed explosive decomposition of 1·HCl while working with the reagent on larger scales (up to ∼5.5 mmol), it should be noted that 1·HCl decomposes violently when heated above 100 °C [as determined by differential scanning calorimetry (DSC); see Supporting Information]. Intriguingly, 1·HCl may be further elaborated using routine transformations; for example, dissolution of the compound in neat acetyl chloride afforded 1·AcCl in 75% yield (Supporting Information; Scheme 2).
Scheme 2. (Top) Synthesis of 1·AcCl by Dissolution of 1·HCl in Neat Acetyl Chloride at Room Temperature. (Bottom) Synthesis of 2·MeCl under Vilsmeier–Haack Conditions.

The solid-state structure of 1·AcCl is also shown (thermal ellipsoids at 50% probability). Hydrogen atoms are omitted for clarity. Atom code: green (F); red (O); violet (I); yellow (Cl).
Note: The acyl chloride intermediate is directly esterified without isolation. The solid-state structure of 2·MeCl is also shown (thermal ellipsoids at 50% probability). Hydrogen atoms are omitted for clarity. Atom code: green (F); red (O); violet (I); yellow (Cl).
Our synthetic approach could also be extended to prepare CF3 iodoniums from 2 (Figures 3 and 5). Dissolution of 2 in dichloromethane followed by sparging with HCl gas for approximately 30 min induced the precipitation of 2·HCl in 57% yield. As in the case of 1·HCl, we found that 2·HCl was only sparingly soluble in most organic solvents (with the exception of highly polar solvents). Slow crystallization of 2·HCl from a saturated MeCN solution at −30 °C afforded single crystals suitable for X-ray crystallographic analysis (Figure 5). The four inequivalent molecules of 2 in the asymmetric unit all exhibited the expected distorted T-shape geometries [CCF3–I–Cl bond angles of 177.4(2)°, 169.0(2)°, 174.6(2)°, and 172.0(2)°]. The I–O interatomic distances [3.058(6) Å, 2.956(6) Å, 2.963(7) Å, and 3.010(6) Å] are all consistent with the conclusion that no bonding interactions occur between the iodonium center and the ancillary carboxylic acids. As in the case of 1·HCl, the long I–Cl bond lengths [2.996(2) Å, 2.942(2) Å, 3.001(2) Å, and 2.891(2) Å] result in bridging halide interactions within the solid state. The large fluctuation in I–Cl bond lengths is a testament to the ionic nature of 2·HCl, as one would not expect crystal packing forces to have such a pronounced influence on the length of significantly covalent interactions. Furthermore, the extremely long I–Cl bond lengths observed in the solid state (which are commensurate with those observed for diphenyliodonium chloride;38vide supra) unambiguously demonstrate the iodonium nature of 2·HCl.39 As with 1·HCl, we could also prepare derivatives of 2·HCl. For example, methyl ester 2·MeCl could be prepared in 43% yield using a modified synthetic procedure (Supporting Information; Scheme 2).
Figure 5.

Solid-state structure of 2·HCl. (Left) One of the four inequivalent molecules in the asymmetric unit (thermal ellipsoids at 50% probability). While the carboxylic acid hydrogen atom is shown, the remaining hydrogen atoms are omitted for clarity. Cocrystallized MeCN has also been omitted for clarity. Atom code: green (F); red (O); violet (I); yellow (Cl). (Right) The asymmetric unit observed for crystalline 2·HCl (thermal ellipsoids at 50% probability). Hydrogen atoms and MeCN solvent molecules omitted for clarity.
Having established synthetic routes to the targeted iodonium salts, we next turned our attention to evaluating their intrinsic reactivity. Electronic structure calculations were utilized to compare the frontier molecular orbitals of 1·HCl and 2·HCl to their parent iodanes (Supporting Information; Figure 6). All calculations were conducted using density functional theory,40 with the B3LYP exchange-correlation energy functional,41 the 6-311+G(d,p) basis set42 for atoms other than I, and an effective core potential for I. To ensure that any qualitative trends were not basis set dependent, we also performed calculations using the aug-cc-PVTZ basis set for atoms other than I, and an effective core potential for I.10 The lowest unoccupied molecular orbital (LUMO) of 1 is an antibonding orbital aligned along the Caryl–I bond axis. In contrast, the LUMO of 1·HCl exhibits antibonding character that is substantially polarized toward the CCF3–I bond (which suggests significant weakening of the associated 3c–4e interaction).10 Moreover, the LUMO of 1·HCl is substantially lowered with respect to that of 1, which indicates that 1·HCl should be a more potent oxidant. While the highest occupied molecular orbital (HOMO) of 1 exhibits antibonding character along the I–O bond, the HOMO of 1·HCl is predominated by a π-antibonding I–Cl interaction. Qualitatively, the orbital characteristics for the HOMO and LUMO of 2·HCl are analogous to that observed in 1·HCl (the same is true for the HOMO/LUMO characteristics of 2 relative to 1). The LUMO of 2·HCl is significantly lowered with respect to the LUMO of 2, consistent with the conclusion that 2·HCl should exhibit enhanced electrophilicity. As a testament to the oxidizing capability of these iodonium species (which one would expect, given their low-lying LUMOs), we found that various transition metals [e.g., Fe(II) and Au(I)] were susceptible to oxidation by a CF3 iodonium (Scheme 3; Supporting Information; Figure S1).
Figure 6.

Electronic structure calculations showing the frontier molecular orbitals of (A) 1 and 1·HCl and (B) 2 and 2·HCl [DFT; B3LYP; 6-311+G(d,p)].
Scheme 3. Oxidation of Au(I) and Fe(II) Complexes by CF3 Iodoniums.

General conditions: iodonium (1.0–3.0 equiv) and metal complex (1.0 equiv) were combined at room temperature and stirred (2–20 min). See the Supporting Information for complete details.
The isolation of 1·HCl and 2·HCl presented the unique opportunity to also investigate the innate reactivity of CF3 iodoniums experimentally. Within the purview of mechanistic considerations, comparing the reactivity profiles of 1·HCl and 2·HCl to those of Lewis activated 1 and 2 would undoubtedly be informative. For example, Zn activation has been shown to facilitate CF3 ether formation from the reaction of alcohols with 2.12 We found that both 1·HCl and 2·HCl were competent electrophiles for the direct trifluoromethylation of alcohols under solvolytic conditions (Supporting Information); however, the limited solubility of these reagents proved to be an impediment to further reaction optimization. To address this issue, we envisaged leveraging their ionic character under phase transfer conditions. NaBArF24 was selected as a suitable phase transfer catalyst, as 1·HBArF24 was found to have optimal solubility and kinetic persistence (vide supra). Gratifyingly, our strategy afforded CF3 ethers from the corresponding alcohols under mild conditions (55 °C; 3.0 equiv of the alcohol; 10 mol % NaBArF24; 16 h) and in modest yields (Supporting Information; Table 1). Primary and secondary alcohols were suitable substrates, but aryl and tertiary alcohols proved ineffectual (for example, phenol gave poor yields of primarily ring substituted products). The direct use of 1·HBArF24 in a control reaction afforded the desired CF3 ether in modest yield, which supported the validity of the proposed phase transfer pathway (Supporting Information). We should stress that, since both the iodonium and the protonated iodane are competent reagents for alcohol trifluoromethylation (and interconversion between the two species under the reaction conditions cannot be excluded), the reactive intermediate responsible for alcohol trifluoromethylation could plausibly exhibit structural homology to either of these compounds.
Table 1. Trifluoromethylation of Alcohols under Phase Transfer Catalysisa.
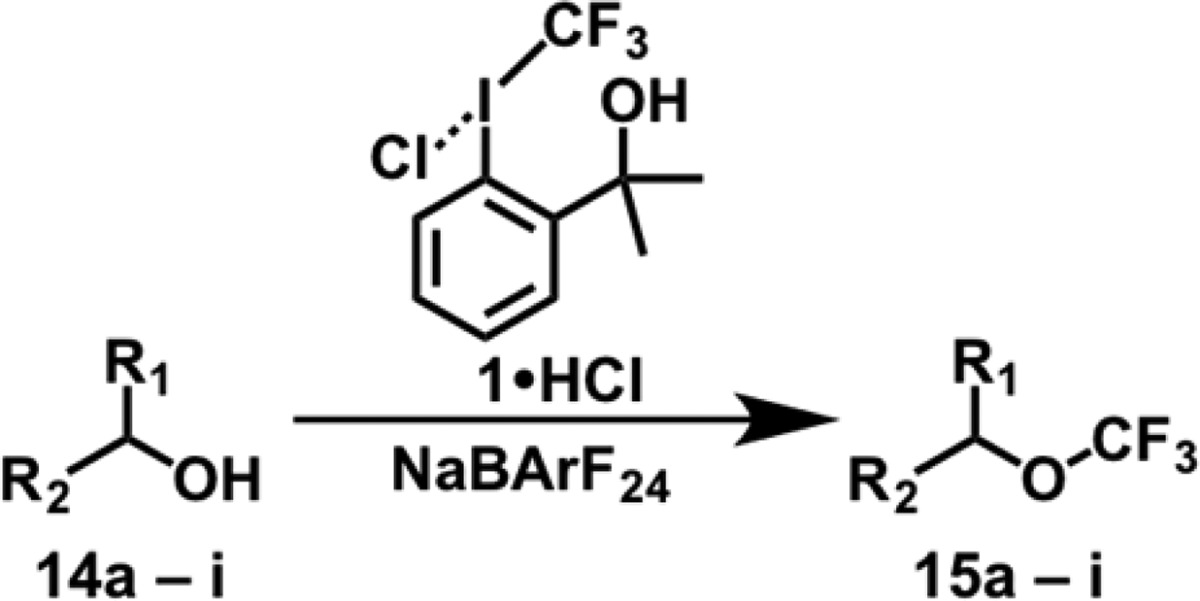
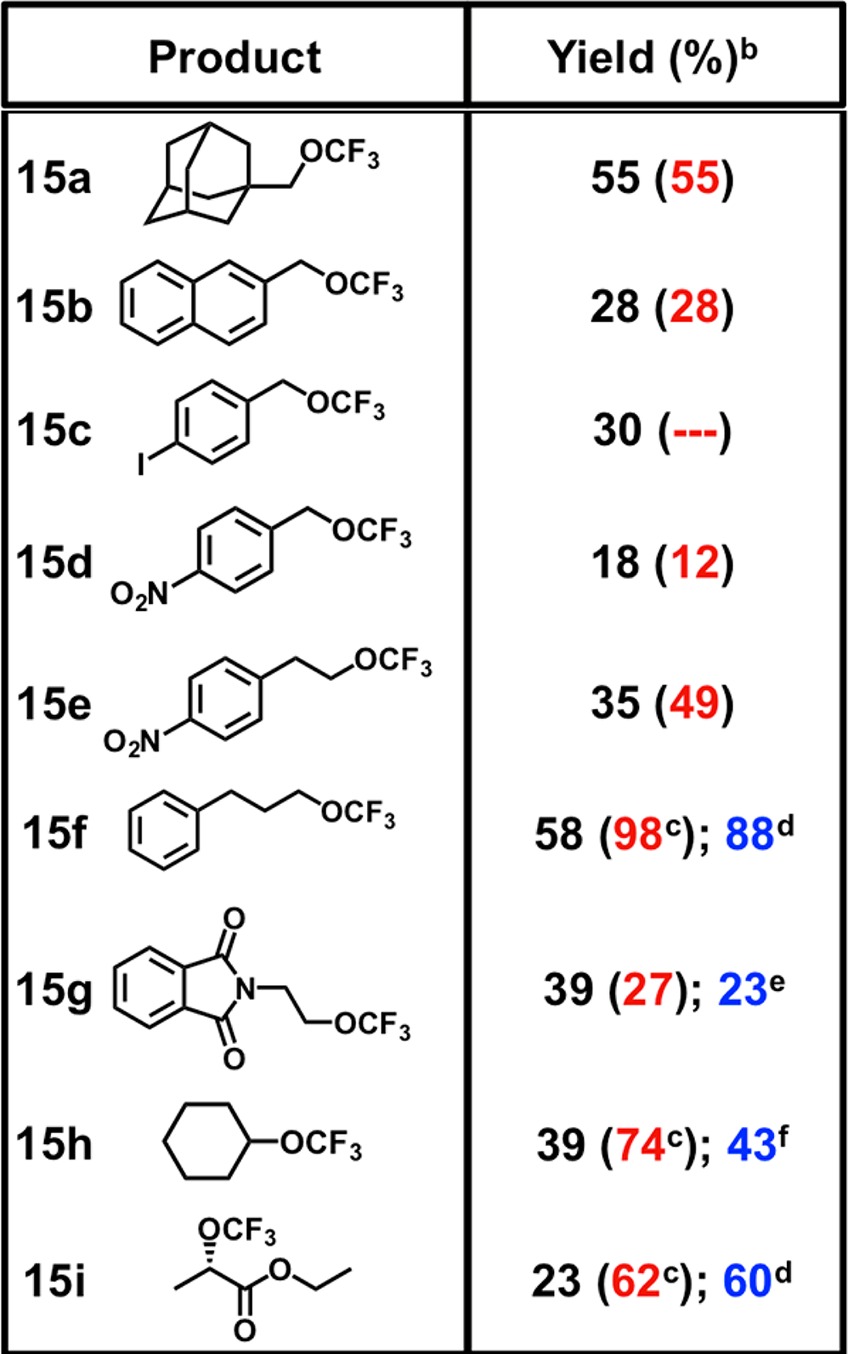
Conditions: 1·HCl (1.0 equiv), alcohol (3.0 equiv), and NaBArF24 (0.1 equiv) are added to MeCN in a conical tube ([1·HCl]0 = 0.5 M) and heated at 55 °C for 16 h.
Determined by 19F NMR spectroscopy using 4-fluorobiphenyl as an internal standard. Parenthetical yields are taken from ref (12) for comparison.
Yield from ref (12) using 75 equiv of alcohol.
NMR yield using 75 equiv of alcohol.
Isolated yield.
NMR yield using 10 equiv of alcohol.
Building on these results, we found that our phase transfer methodology could be extended to benzotriazole, affording a 19:1 mixture of N-trifluoromethylated isomers 17a and 17b in 59% combined yield (Scheme 4). This reactivity is commensurate with previous reports, wherein 17a could be prepared in 58% yield by reacting benzotriazole with 1 in CS2 at 60 °C.19 Benzimidazole could also be trifluoromethylated under phase transfer conditions to afford 19 in 41% yield (Scheme 4). The direct N-trifluoromethylation of benzimidazole is noteworthy, given that previous reports required in situ silylation of the azole to afford good yields (i.e., 71%).19,43 Similar to reactivity observed under Cu activation,28,442·HCl was found to react directly with both carboxybenzyl protected skatole [furnishing 2-(trifluoromethyl) analogue 21 in 49% yield] and 1,1-diphenylethylene (furnishing vinyl product 23 in 37% yield) under relatively mild conditions (Scheme 4). Anionic carbon or heteroatom nucleophiles were also found to react with these iodonium reagents. For example, the potassium salt of 2-ethoxycarbonyl-1-cyclopentanone reacted with 2·MeCl to afford α-trifluoromethylated product 25 in 22% yield (Scheme 4). By comparison, related β-keto esters were found to react directly with Togni-type reagents in similar yields (40–67%).14 The potassium salt of diphenylphosphate and sodium dodecyl sulfide reacted with 2·MeCl to afford the corresponding O- and S-trifluoromethylated products (27 and 29) in 45% and 95% yield, respectively. In contrast, diphenylphosphoric acid and dodecanethiol both gave poor yields of 27 and 29 (6% and 0%, respectively) under identical conditions. Previous work has established that Togni-type reagents react with phosphates45 and sulfides14 to afford congeners of 27 and 29 in similar yields (21–44% and 51%, respectively). The sodium salt of p-toluenesulfonic acid also reacted with 2·MeCl to afford O-trifluoromethyl sulfonate 31 in 70% yield (for comparison, p-toluenesulfonic acid reacted under identical conditions to afford 31 in 78% yield). Again, the reactivity observed here parallels previous reports, which found that 31 could be prepared from 2 in 90% yield.13 In contrast, the salts of hard (i.e., less iodophilic) nucleophiles (such as potassium tert-butoxide, potassium benzotriazolate, potassium phthalimide, and sodium benzoate) failed to give any product upon salt metathesis. This suggests that harder nucleophiles react along a different pathway than the established reaction coordinate for sulfonates and phosphates. Previous kinetic studies regarding the reaction of sulfonic13 and phosphoric acids45 with Togni-type reagents have suggested an operative mechanism involving initial protonation and subsequent reductive elimination to form the trifluoromethylated product, which is consistent with our salt metathesis results. The failure of harder nucleophiles to exhibit analogous reactivity suggests that the mechanism for product formation in these cases is more complex. Regardless, these data show that CF3 iodoniums could play a mechanistic role in reactions involving diverse nucleophiles.
Scheme 4. Reactions of CF3 Iodoniums with Azole (A, B), Indole (C), Olefinic (D), Enolate (E), Phosphate (F), Sulfide (G), and Sulfonate (H) Nucleophiles.

General conditions: iodonium (1.0 equiv), substrate (1.0–3.0 equiv), and NaBArF24 (0.0–0.1 equiv) were combined and stirred at an appropriate temperature (room temperature to 60 °C; 2–16 h). See the Supporting Information for complete details.
In summary, the data reported here unambiguously demonstrate that CF3 iodonium salts are observable species, and, in fact, they can be readily prepared from conventional λ3-iodanes under the action of mineral acids. Chloride salts were found to exhibit optimal persistence, presumably the result of a subtle balance of iodophilicity and electronegativity. More weakly coordinating anions provided insufficient kinetic stabilization (resulting in the formation of species more accurately described as protonated iodanes), and strongly coordinating counterions induced decomposition through reductive elimination pathways. These iodonium salts were capable of trifluoromethylating an array of nucleophiles, suggesting that CF3 iodoniums should not immediately be discarded as relevant intermediates when considering the mechanisms of reactions involving activated CF3 iodanes. Indeed, in some cases the iodonium chloride itself was found to be a competent surrogate for iodane–Lewis acid systems (which suggests the broad potential use of isolated CF3 iodoniums as mechanistic probes). The data presented here will undoubtedly inspire new synthetic strategies for trifluoromethylation, and future studies investigating the complete reactivity of CF3 iodoniums will be reported in due course.
Acknowledgments
We thank National Institute of General Medical Sciences (R01 GM104534) for partial support of this work. J.N.B. was supported by the National Institutes of Health under a Ruth L. Kirschstein National Service Award (F32GM116409). The authors thank William J. Wolf and Cynthia M. Hong (UC Berkeley) for crystallographic assistance, Eva M. Nichols for assistance with cyclic voltammetry, Jeff Mckenna and Vaneet Saini (Novartis Institute of Biomedical Research) for help with DSC and TGA analyses, and Mark D. Levin and Dr. Steven D. Jacob for helpful discussions. X-ray crystallography was performed using the UC Berkeley College of Chemistry CheXray facility supported by the NIH Shared Instrumentation Grant S10-RR027172. Aspira Scientific is gratefully acknowledged for providing reagents 1 and 2.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00119.
Crystallographic data for 1·HBF4 (CIF)
Crystallographic data for 1·HCl (CIF)
Crystallographic data for 2·HCl (CIF)
Crystallographic data for 2·MeCl (CIF)
Crystallographic data for 11 (CIF)
Crystallographic data for 1·AcCl (CIF)
Experimental details including synthetic, oxidation, halide metathesis, and trifluoromethylation procedures, computational methods and data, UV profiles, NMR spectra, crystallographic, DSC, and TGA data, and cyclic voltammograms (PDF)
Author Contributions
† J.N.B. and A.V.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Furuya T.; Kamlet A. S.; Ritter T. Catalysis for fluorination of trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T.; Neumann C. N.; Ritter T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- Egami H.; Sodeoka M. Trifluoromethylation of alkenes with concomitant introduction of additional functional groups. Angew. Chem., Int. Ed. 2014, 53, 8294–8308. 10.1002/anie.201309260. [DOI] [PubMed] [Google Scholar]
- Merino E.; Nevado C. Addition of CF3 across unsaturated moieties: a powerful functionalization tool. Chem. Soc. Rev. 2014, 43, 6598–6608. 10.1039/C4CS00025K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkin A.; Chou T.-C.; Danishefsky S. J. On the remarkable antitumor properties of fludelone: how we got there. Angew. Chem., Int. Ed. 2005, 44, 2838–2850. 10.1002/anie.200461751. [DOI] [PubMed] [Google Scholar]
- Brand J. P.; Fernández González D.; Nicolai S.; Waser J. Benziodoxole-based hypervalent iodine reagents for atom transfer reactions. Chem. Commun. 2011, 47, 102–115. 10.1039/C0CC02265A. [DOI] [PubMed] [Google Scholar]
- Eisenberger P.; Gischig S.; Togni A. Novel 10-I-3 hypervalent iodine-based compounds for electrophilic trifluoromethylation. Chem. - Eur. J. 2006, 12, 2579–2586. 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- Eisenberger P.; Kieltsch I.; Koller R.; Stanek K.; Togni A. Preparation of a trifluoromethyl transfer agent: 1-trifluromethyl-1,3-dihydro-3,3-dimethyl-1,2-benziodoxole. Org. Synth. 2011, 88, 168. 10.1002/0471264229.os088.16. [DOI] [Google Scholar]
- Matoušek V.; Pietrasiak E.; Schwenk R.; Togni A. One-pot synthesis of hypervalent iodine reagents for electrophilic trifluoromethylation. J. Org. Chem. 2013, 78, 6763–6768. 10.1021/jo400774u. [DOI] [PubMed] [Google Scholar]
- Charpentier J.; Früh N.; Togni A. Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 2015, 115, 650–682. 10.1021/cr500223h. [DOI] [PubMed] [Google Scholar]
- Stanek K.; Koller R.; Togni A. Reactivity of a 10-I-3 hypervalent iodine trifluoromethylation reagent with phenols. J. Org. Chem. 2008, 73, 7678–7685. 10.1021/jo8014825. [DOI] [PubMed] [Google Scholar]
- Koller R.; Stanek K.; Stolz D.; Aardoom R.; Niedermann K.; Togni A. Zinc-mediated formation of trifluoromethyl ethers from alcohols and hypervalent iodine trifluoromethylation reagents. Angew. Chem., Int. Ed. 2009, 48, 4332–4336. 10.1002/anie.200900974. [DOI] [PubMed] [Google Scholar]
- Koller R.; Huchet Q.; Battaglia P.; Welch J. M.; Togni A. Acid-mediated formation of trifluoromethyl sulfonates from sulfonic acids and a hypervalent iodine trifluoromethylating agent. Chem. Commun. 2009, 40, 5993–5995. 10.1039/b913962a. [DOI] [PubMed] [Google Scholar]
- Kieltsch I.; Eisenberger P.; Togni A. Mild electrophilic trifluoromethylation of carbon- and sulfur-centered nucleophiles by a hypervalent iodine(III)-CF3 reagent. Angew. Chem., Int. Ed. 2007, 46, 754–757. 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]
- Eisenberger P.; Kieltsch I.; Armanino N.; Togni A. Mild electrophilic trifluoromethylation of secondary and primary aryl- and alkylphosphine using hypervalent iodine(III)-CF3 reagents. Chem. Commun. 2008, 13, 1575–1577. 10.1039/b801424h. [DOI] [PubMed] [Google Scholar]
- Charkoudian L. K.; Liu C. W.; Capone S.; Kapur S.; Cane D. E.; Togni A.; Seebach D.; Khosla C. Probing the interactions of an acyl carrier protein domain from the 6-deoxyerythronolide B synthase. Protein Sci. 2011, 20, 1244–1255. 10.1002/pro.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejía E.; Togni A. Rhenium-catalyzed trifluoromethylation of arenes and heteroarenes by hypervalent iodine reagents. ACS Catal. 2012, 2, 521–527. 10.1021/cs300089y. [DOI] [Google Scholar]
- Santschi N.; Togni A. Electrophilic trifluoromethylation of S-hydrogen phosphorothioates. J. Org. Chem. 2011, 76, 4189–4193. 10.1021/jo200522w. [DOI] [PubMed] [Google Scholar]
- Niedermann K.; Früh N.; Senn R.; Czarniecki B.; Verel R.; Togni A. Direct N-trifluoromethylation of azoles by a hypervalent iodine reagent. Angew. Chem., Int. Ed. 2012, 51, 6511–6515. 10.1002/anie.201201572. [DOI] [PubMed] [Google Scholar]
- Niedermann K.; Früh N.; Vinogradova E.; Wiehn M. S.; Moreno A.; Togni A. A Ritter-type reaction: direct electrophilic trifluoromethylation at nitrogen atoms using hypervalent iodine reagents. Angew. Chem., Int. Ed. 2011, 50, 1059–1063. 10.1002/anie.201006021. [DOI] [PubMed] [Google Scholar]
- Matoušek V.; Pietrasiak E.; Sigrist L.; Czarniecki B.; Togni A. O-trifluoromethylation of N,N-disubstituted hydroxylamines with hypervalent iodine reagents. Eur. J. Org. Chem. 2014, 2014, 3087–3092. 10.1002/ejoc.201402225. [DOI] [Google Scholar]
- Lin X.; Wang G.; Li H.; Huang Y.; He W.; Ye D.; Huang K.-W.; Yuan Y.; Weng Z. Copper-catalyzed trifluoromethylation of aryl sulfinate salts using an electrophilic trifluoromethylation reagent. Tetrahedron 2013, 69, 2628–2632. 10.1016/j.tet.2013.01.041. [DOI] [Google Scholar]
- Fauster K.; Kreutz C.; Micura R. 2′-SCF3 uridine - a powerful label for probing structure and function of RNA by 19F NMR spectroscopy. Angew. Chem., Int. Ed. 2012, 51, 13080–13084. 10.1002/anie.201207128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egami H.; Kawamura S.; Miyazaki A.; Sodeoka M. Trifluoromethylation reactions for the synthesis of β-trifluoromethylamines. Angew. Chem., Int. Ed. 2013, 52, 7841–7844. 10.1002/anie.201303350. [DOI] [PubMed] [Google Scholar]
- Mizuta S.; Engle K. M.; Verhoog S.; Galicia-López O.; O’Duill M.; Médebielle M.; Wheelhouse K.; Rassias G.; Thompson A. L.; Gouverneur V. Trifluoromethylation of allylsilanes under photoredox catalysis. Org. Lett. 2013, 15, 1250–1253. 10.1021/ol400184t. [DOI] [PubMed] [Google Scholar]
- Weng Z.; Li H.; He W.; Yao L.-F.; Tan J.; Chen J.; Yuan Y.; Huang K.-W. Mild copper-catalyzed trifluoromethylation of terminal alkynes using an electrophilic trifluoromethylating reagent. Tetrahedron 2012, 68, 2527–2531. 10.1016/j.tet.2011.12.085. [DOI] [Google Scholar]
- Cai S.; Chen C.; Sun Z.; Xi C. CuCl-catalyzed ortho trifluoromethylation of arenes and heteroarenes with a pivalamido directing group. Chem. Commun. 2013, 49, 4552–4554. 10.1039/c3cc41331d. [DOI] [PubMed] [Google Scholar]
- Shimizu R.; Egami H.; Nagi T.; Chae J.; Hamashima Y.; Sodeoka M. Direct C2-trifluoromethylation of indole derivatives catalyzed by copper acetate. Tetrahedron Lett. 2010, 51, 5947–5949. 10.1016/j.tetlet.2010.09.027. [DOI] [Google Scholar]
- Allen A. E.; Macmillan D. W. C. The productive merger of iodonium salts and organocatalysis: a non-photolytic approach to the enantioselective alpha-trifluoromethylation of aldehydes. J. Am. Chem. Soc. 2010, 132, 4986–4987. 10.1021/ja100748y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Liu J.; Ming W.; Liu Y.; Liu J.; Wang M.; Liu Q. In situ generation of Ph(I)+CF3 and transition-metal-free oxidative sp2 C-H trifluoromethylation. Chem. - Eur. J. 2013, 19, 9104–9109. 10.1002/chem.201301585. [DOI] [PubMed] [Google Scholar]
- Koller R.Taking electrophilic trifluoromethylation chemistry a step further. ETH Diss. 2010, No. (19219), . [Google Scholar]
- Umemoto T.; Kuriu Y.; Shuyama H.; Miyano O.; Nakayama S.-I. Syntheses and properties of (perfluoroalkyl)phenyliodonium triflates (fits reagents) and their analogues. J. Fluorine Chem. 1986, 31, 37–56. 10.1016/S0022-1139(00)85087-3. [DOI] [Google Scholar]
- Umemoto T. Electrophilic perfluoroalkylating agents. Chem. Rev. 1996, 96, 1757–1778. 10.1021/cr941149u. [DOI] [PubMed] [Google Scholar]
- Yagupolskii L. M. Aromatic compounds with new fluorine-containing substituents. J. Fluorine Chem. 1987, 36, 1–28. 10.1016/S0022-1139(00)82050-3. [DOI] [Google Scholar]
- Huheey E. The electronegativity of groups. J. Phys. Chem. 1965, 69, 3284–3291. 10.1021/j100894a011. [DOI] [Google Scholar]
- Minkwitz R.; Berkei M. A new method for preparation and crystal structure of (trifluoromethyl)iodine dichloride. Inorg. Chem. 1999, 38, 5041–5044. 10.1021/ic990441n. [DOI] [PubMed] [Google Scholar]
- Archer E. M.; van Schalkwyk T. G. The crystal structure of benzene iododichloride. Acta Crystallogr. 1953, 6, 88–92. 10.1107/S0365110X53000193. [DOI] [Google Scholar]
- Alcock N. W.; Countryman R. M. Secondary bonding. Part I. Crystal and molecular structures of diphenyliodonium chloride, bromide, and iodide. J. Chem. Soc., Dalton Trans. 1977, 217–219. 10.1039/dt9770000217. [DOI] [Google Scholar]
- The ionic character of iodoniums derived from 1 and 2 was further confirmed using IR spectroscopic analysis and conductivity measurements. For example, the carbonyl stretch of 2·MeCl did not change significantly from the solid state (1727 cm–1) to solutions in either CDCl3 (1729 cm–1) or MeCN (1728 cm–1). This shows that, in solution, the carbonyl is not weakened through interactions with the I(III) center. Conductivity measurements also showed that a MeCN solution of 1·HCl ([1·HCl] = 0.01 M) exhibited a roughly 30-fold increase in conductivity relative to a MeCN solution of 1 ([1] = 0.01 M).
- Parr R. G.; Yang W.. Density Funtional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]
- Becke A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Gordon M. S.; Binkley J. S.; Pople J. A.; Pietro W. J.; Hehre W. J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. 10.1021/ja00374a017. [DOI] [Google Scholar]
- Engl P. S.; Senn R.; Otth E.; Togni A. Synthesis and characterization of N-trifluoromethyl N-heterocyclic carbene ligands and their coplexes. Organometallics 2015, 34, 1384–1395. 10.1021/acs.organomet.5b00137. [DOI] [Google Scholar]
- Parsons A. T.; Buchwald S. L. Copper-catalyzed trifluoromethylation of unactivated olefins. Angew. Chem., Int. Ed. 2011, 50, 9120–9123. 10.1002/anie.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santschi N.; Geissbühler P.; Togni A. Reactivity of an electrophilic hypervalent iodine trifluoromethylation reagent with hydrogen phosphates - a mechanistic study. J. Fluorine Chem. 2012, 135, 83–86. 10.1016/j.jfluchem.2011.08.014. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.