

Abstract

While preparative electrolysis of organic molecules has been an active area of research over the past century, modern synthetic chemists have generally been reluctant to adopt this technology. In fact, electrochemical methods possess many benefits over traditional reagent-based transformations, such as high functional group tolerance, mild conditions, and innate scalability and sustainability. In this Outlook we highlight illustrative examples of electrochemical reactions in the context of the synthesis of complex molecules, showcasing the intrinsic benefits of electrochemical reactions versus traditional reagent-based approaches. Our hope is that this field will soon see widespread adoption in the synthetic community.
Short abstract
The use of innately scalable yet underutilized electrochemical methods for the synthesis and functionalization of complex organic molecules is discussed. Selected examples are shown to highlight the benefits of this enabling tool.
Introduction
The field of synthetic organic chemistry is under constant and extreme selection pressure. It is challenged not only to create ever-increasingly complex molecules but also to do so in a timely, atom-economic, and sustainable fashion.1−3 Due in part to these external stimuli, classic technologies such as photochemistry4 and electrochemistry5−9 have reemerged, providing tools that enable chemists to do more with less. As early as the late 19th century, preparative electrolysis began to be used as an industrial process to prepare bulk chemicals on ton-scale. Classic examples include the chloralkali process,10 wherein aqueous sodium chloride is electrolyzed to give chlorine gas and sodium hydroxide, and the Hall–Héroult process,11 which provides elemental aluminum by electrolysis of Al2O3. These profoundly enabling uses of electrochemistry persist to this day, producing millions of metric tons of these valuable chemicals. Yet, examples of electrochemistry for use in organic synthesis and the fine chemicals industry remain scarce. This is perplexing given the fact that this technique generally features relatively mild conditions, good functional group tolerance, and high chemoselectivity. Furthermore, the ease with which many electrochemical reactions can be scaled up, as well as the intrinsic “greenness” of the reactions (because electric current is used in place of stoichiometric oxidants or reductants), make this chemistry attractive in a process chemistry setting. It is, in fact, an innately practical technique.
Those advantages notwithstanding, practicing organic chemists have generally been reluctant to adopt this technology into their own laboratories. In our experience, electrochemistry is widely feared by practicing organic chemists due to the perceived complex reaction setup (potentiostat, divided/undivided cell, electrode composition, experiment type, etc.), the seemingly endless number of reaction variables (electrolyte, electrode composition, cell type, etc.), and the common misconception that only aqueous solvents may be employed and that product separation is difficult. The barrier to adoption becomes higher when one discovers that a “standard” instrument for preparative electrolysis does not exist, and many of the recent elegant literature examples utilize home-built rather than commercially available equipment. This daunting situation certainly discouraged us from exploring electrochemical transformations—indeed it was the difficulty in accessing the dimeric natural product dixiamycin B (1, Figure 1) that brought us to embrace the technology.12
Figure 1.

Synthesis of dixiamycin B by Baran and co-workers.
The most simple retrosynthetic analysis of 1 triggered an N–N bond-forming reaction to couple two xiamycin monomers (2). Despite extensive screening, no reagent-based oxidant was capable of forging the necessary N–N bond. It was only after these exhaustive evaluations that we began to consider the possibility of utilizing an electrochemical oxidation for this key dehydrodimerization step. A literature search revealed studies from Ambrose and co-workers on the reactivity of carbazolium radical cations.13 By substantially modifying the reaction conditions, we found that treating carbazole 2 under a constant potential of 1.15 V vs Ag/AgCl using graphite rod electrodes in a tetraalkylammonium electrolytic solution of 19:1 DMF/MeOH led to formation of dixiamycin B (1) in 28%. The ability to “dial-in” the oxidative strength of the reaction and accomplish what no chemical reagent could was, to us, a convincing demonstration of the power of electrochemistry in organic synthesis, particularly in complex settings that require exquisite chemoselectivity.
It is the goal of this Outlook to allay the aforementioned fears by presenting a forward-looking perspective of electrochemical transformations specifically in complex settings. Although our entry into this area was guided by necessity, we hope that electrochemistry will soon become a routinely employed technique in modern organic chemistry laboratories in order to simplify and enable synthetic pathways.
Electrochemistry Vignettes in Synthesis
Though reports of electrochemical transformations in the synthesis of complex molecules are scarce, there are nonetheless several applications illustrative of its complexity-generating power. An early example is the application of a Kolbe decarboxylative dimerization in Corey’s 1958 synthesis (Figure 2) of pentacyclosqualene (6), α-onoeradiene (not shown), and β-onoceradiene (7).14 Subjecting the ammonium carboxylate salt 4, which was prepared in three steps from sclareolide, to electrolysis at high current density in refluxing methanol resulted in decarboxylation followed by radical dimerization to yield 5 in 17% (R = H) or 34% (R = Ac) yield. Subsequent treatment of the diol with perchloric acid resulted in cyclization to pentacyclosqualene (6), while elimination using POCl3 in pyridine led to β-onoceradiene (7). It is particularly noteworthy that, even today, the invention of reagents to accomplish this type of Csp3–Csp3 coupling is still the subject of ongoing research in many groups.15
Figure 2.

Synthesis of pentacyclosqualene and β-onoceradiene by Corey and co-workers using Kolbe electrolysis.
Some of the most substantial advancements in electrochemical oxidative coupling reactions of the past 20 years have been developed by the Moeller16−33 and Wright34−39 groups. In this way, anodic oxidation has been shown to enable the coupling of two nucleophilic functional groups, thereby leading to new broadly useful umpolung disconnections. An elegant example of this is Moeller’s synthesis of alliacol A (8, Figure 3),28,33 whereby the enoxysilane and furan nucleophiles present in 9 can be coupled together under electrochemical conditions. This reaction proceeds via selective oxidation of the enoxysilane to give radical cation intermediate 10, which undergoes cyclization by attack of the furan to provide the radical oxonium ion 11. Subsequent single electron oxidation at the anode, trapping of the resulting carbocation with methanol, and elimination with TsOH provides furan 13 in 88% yield. At first glance, it may not be clear what the origins of this remarkable selectivity would be. Since oxidation of the functional group with the lowest oxidation potential takes place first, the enoxysilane (Ep1/2 ∼ 0.9 V vs Ag/AgCl) is predictably and selectively oxidized in the presence of a furan (Ep1/2 ∼ 1.3 V vs Ag/AgCl). The oxidation potentials of the individual functional groups in a molecule can be easily approximated by analyzing a cyclic voltammogram of a simple model substrate that contains only the functional group in question. As evidenced by the conversion of 9 to 13, a unique advantage of electrochemistry is the selectivity and tunability of the reaction based on the redox potentials of the functional groups present in the molecule. It is therefore trivial to sequence anodic coupling reactions (and many other electrochemical transformations) since, unlike reagent-based oxidants, the selectivity of the oxidation can be known at the outset and precise control of the potential can be essentially “dialed-in”. In addition, these reactions are tolerant to a wide array of functional groups, as long as the oxidation potential of the group is higher than that of the group that is to be oxidized. The power of this particular transformation has been recognized on numerous occasions, arguably most impressively in Trauner’s synthesis of guanacastepene E (14),40 where stereoselective formation of the C1–C2 bond is accomplished using a similar anodic oxidative coupling.
Figure 3.

Moeller’s synthesis of alliacol A via an intermolecular anodic coupling.
The scalable synthesis of DZ-2384 (15) by Harran and co-workers further enumerates the remarkable functional group compatibility of many electrochemical reaction conditions (Figure 4).41 DZ-2384, a diazonamide-inspired preclinical candidate for oncology, was recently prepared using an intramolecular electrochemical oxidative coupling of 16 between the phenol and indole motifs to give macrocycle 17. Previously, this transformation was accomplished on similar substrates using oxidants such as PhI(OAc)2;42 unfortunately, this reagent-based system also led to considerable byproduct formation, such that this reaction became the most problematic bottleneck in material throughput, hampering access to desperately needed material for downstream studies. The electrochemical conditions for accomplishing this transformation were a marked improvement in terms of selectivity, as well as lower cost and environmental footprint, enabling the reaction to be easily carried out on 60 g of indole substrate 16. It is clear from the successful execution of this transformation that electrochemical reactions can solve not only problems with respect to reactivity but practical challenges in the context of scale-up and process chemistry.
Figure 4.

Synthesis of diazonamide-inspired drug development candidate DZ-2384 by Harran and co-workers.
A striking testament to the utility of electrochemistry in an industrial process setting stems from a recent collaboration between the Waldvogel group and Novartis involving the electrochemical reduction of a geminal dihalide (Figure 5).43,44 In this case, reduction of dibromocyclopropane 18 to cyclopropane 19, an important intermediate for HCV NS5A inhibitors, was accomplished in a separated cell using a leaded bronze cathode and [Et3NMe]O3SOMe as supporting electrolyte. This method ameliorated several problems that plagued alternative routes to 19 including ring-opened products and racemization. Equally important from a process chemistry perspective, the electrochemical method also proved to be significantly more cost-efficient and resulted in considerably less waste generation, making this a “green” process. Highlighting the functional group tolerance of this transformation in a complex setting, the reaction conditions were also applied to the reduction of cyclosporin A analogue 20 to give the reduced product in 98% yield.
Figure 5.

Synthesis of NS5A inhibitor intermediate and reduction of cyclosporin A analogue by Waldvogel and co-workers.
Extensive studies in the synthesis of complex terpenes by us and others led to the realization that no practical, sustainable method for allylic C–H oxidation existed. This fact, combined with the knowledge that an ongoing project at Bristol-Myers Squibb (BMS) required such an oxidation, inspired a collaborative exploration into the use of electrochemistry as a potential solution (Figure 6).45 While this type of transformation can be readily accomplished through a variety of reagent-based systems (e.g., chromium, palladium, rhodium, ruthenium, etc.), most of these systems are unsuitable in an industrial process setting due to toxicity or cost associated with these reagents. Conditions developed in our laboratory employ an N-hydroxyphthalimide catalyst that undergoes anodic oxidation to a highly reactive oxygen-centered radical and engages a substrate through selective allylic C–H atom abstraction. Using this protocol, more than a dozen natural product scaffolds were selectively oxidized, as exemplified by the 100 g scale oxidation of dehydroepiandrosterone derivative 21 to the corresponding enone 22 (performed by Asymchem Life Sciences literally in a bucket). To verify the improved environmental footprint and efficiency of this reaction, the conditions for the conversion of 21 to 22 were compared against commonly employed Cr-promoted and Ru-catalyzed methods in the literature, the latter of which was developed by Schering process chemists. Using the Process Greenness Score (PGS), a metric used at BMS to evaluate greenness of a process, the electrochemical method was found to be a nearly 50% improvement over the previously mentioned methods.
Figure 6.

(a) Electrochemical allylic oxidation by Baran and co-workers. (b) Process greenness score (PGS) for Cr, Ru, and electrochemistry. (c) 100 g scale allylic oxidation conducted in a bucket. Panels b and c reprinted with permission from ref (45). Copyright 2016 Nature Publishing Group.
Selective arene C–H functionalization has seen considerable interest throughout the synthetic community over the past several years,46−54 in large part due to demands in the context of drug discovery. In particular, the ability to functionalize arenes in a selective and predictable fashion in the presence of multiple other functional groups has immediate applications toward the diversification of late stage drug intermediates. A noteworthy example of addressing this challenge has been recently reported by Yoshida and co-workers to accomplish arene C–H amination in a stunning series of reports (Figure 7).55−57 Electrolysis of electron-rich arenes in the presence of electron-deficient nitrogen heterocycles (e.g., pyridine, N-methanesulfonylimidazole, etc.) leads to selective oxidation of the arene to an electrophilic radical cation and trapping by the heterocycle to forge a new C–N bond. Further treatment of the crude reaction mixture under mild conditions provides one of several nitrogen-functionalized arenes, including anilines (e.g., 23 and 24), heterocycles (25), and secondary N-aryl amines (26). The ability to rapidly, selectively, and predictably conduct this C–H amination reaction led to an improved synthesis of a key intermediate in the synthesis of VLA-4 antagonist 27, dramatically reducing the overall step count and increasing the overall yield.
Figure 7.

Electrochemical arene amination by Yoshida.
Radical-based C–H functionalization methods using sulfinate-reagents have emerged as a useful means to modify both simple and complex heterocycles in a medicinally relevant way.58−63 Although this technique utilizes TBHP, a cheap industrial oxidant, the superstoichiometric quantities required can be a deterrent for large-scale applications. In addition, certain types of heterocycles gave consistently lower yields of functionalized product. In collaboration with the Blackmond group, anodic oxidation of sulfinate salts enabled an increase in the yield of the process and eliminated the use of a chemical oxidant altogether (Figure 8).64 Furthermore, anodic oxidation allowed for precise control over the rate of radical formation, slowing the decomposition of the sulfinate salt and dramatically improving the overall reaction.
Figure 8.
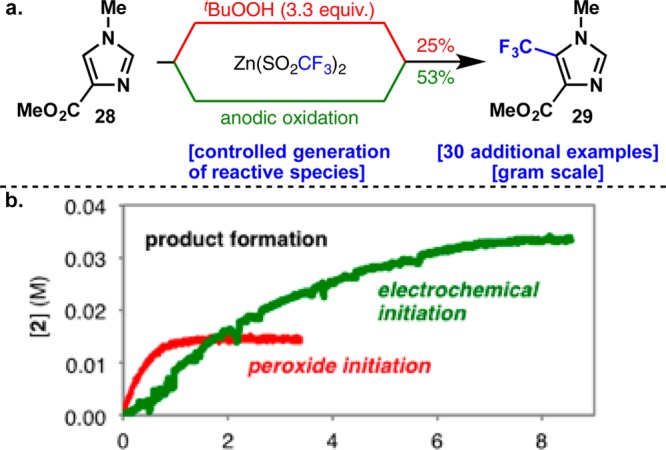
(a) Electrochemical fluoroalkylation of heterocycles by Baran and Blackmond and co-workers. (b) Formation of 29 using tBuOOH and electrochemical protocols. Reprinted with permission from ref (64). Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA.
It is evident from the above examples that anodic oxidations and cathodic reductions are enabling tools for the construction of natural products and small molecule medicines. Nonetheless, concerns about the practical aspects of the reaction setup still act as a barrier to entry for many practicing bench chemists. The assumption that specialized equipment is required to try electrochemical experiments could not, in fact, be further from reality. Recently, Aubé and Moeller have demonstrated a remarkably practical setup to promote the C–H oxidation of complex polycyclic lactams such as 30 utilizing a repurposed cell phone charger as the power supply, and #7 mechanical pencil lead as electrodes (Figure 9).65 The methoxy amide products such as 31 can be further diversified to a variety of useful functionalized products. This elegant, readily accessible experimental setup should serve to lower the barrier to entry for chemists contemplating using organoelectrochemistry, since no specialized equipment (potentiostat or unusual electrodes) is needed. In a similar vein, Moeller previously demonstrated that many electrochemical transformations, including that shown in Figure 3, can be carried out using a 6 V lantern battery, easily obtained from any neighborhood hardware store, as an even simpler power supply.33 Obviously, these simple configurations are not without limitations, because potential and current are not as easily controlled; however, because these setups are so accessible, we hope this information will encourage the average synthetic chemist to incorporate electrochemical transformations into their synthetic toolkit.
Figure 9.

Synthesis of functionalized polycyclic lactams by Aubé and co-workers using a repurposed mobile phone charger. Reprinted with permission from ref (65). Copyright 2015 WILEY-VCH Verlag GmbH & Co. KGaA.
Summary and Future Directions
This outlook is not meant to be an exhaustive review of this ever-expanding field but rather a brief introduction to the area that will inspire others to try electrochemical reactions or use the platform to invent new transformations. For those interested in learning more, several extensive reviews and monographs have been published in this area.3 In addition, Figure 10 showcases a number of exciting contributions that are representative of the vibrant and creative directions that are being explored using electrochemistry.
Figure 10.

Suggested topics for further reading. Reprinted with permission from refs (66−75). Copyright 2002, 2010, and 2014 WILEY-VCH Verlag GmbH & Co. KGaA. Copyright 1983, 2012, and 2015 American Chemical Society. Copyright 2014 Royal Society of Chemistry. Copyright 1988 Elsevier B.V. Copyright 1996 Springer.
The examples outlined herein represent only a tip of the iceberg in terms of the types of major problems that electrochemistry can solve. As sustainability becomes a prime directive for organic synthesis, one could argue that there should be no reason to use superstoichiometric reagents to accomplish simple redox manipulations of functional groups (e.g., alcohol to ketone or ester to alcohol) when the same transformations can be efficiently achieved using electrochemistry. The unique tunability and chemoselectivity of electrochemistry holds great potential for reaction invention in areas such as C–H functionalization, catalysis, and total synthesis. Finally, we believe that for electrochemistry to really take off in all areas of synthetic organic chemistry, more standardized and simplified instrumentation needs to be developed specifically for this community. Although it is great that certain reactions can be run using crude, homemade equipment such as a lantern battery or a cell phone charger, this can lead to reproducibility concerns (battery type, electrode material, etc.). Furthermore, the lack of “out of the box”, standardized equipment for preparative electrolysis certainly discourages widespread adoption of this technique.
Acknowledgments
We are grateful to our industrial collaborators (BMS, Asymchem, Pfizer, LEO Pharma, and Aldrich) for funding portions of this work and NIGMS (GM-097444).
The authors declare no competing financial interest.
References
- Bryan M. C.; Dillon B.; Hamann L. G.; Hughes G. J.; Kopach M. E.; Peterson E. A.; Pourashraf M.; Raheem I.; Richardson P.; Richter D.; Sneddon H. F. Sustainable practices in medicinal chemistry: Current state and future directions. J. Med. Chem. 2013, 56, 6007–6021. 10.1021/jm400250p. [DOI] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Kuttruff C. A.; Eastgate M. D.; Baran P. S. Natural product synthesis in the age of scalability. Nat. Prod. Rep. 2014, 31, 419–432. 10.1039/C3NP70090A. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller K. D. Synthetic applications of anodic electrochemistry. Tetrahedron 2000, 56, 9527–9554. 10.1016/S0040-4020(00)00840-1. [DOI] [Google Scholar]
- Sperry J. B.; Wright D. L. The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 2006, 35, 605–621. 10.1039/b512308a. [DOI] [PubMed] [Google Scholar]
- Sequeira C. A. C.; Santos D. M. F. Electrochemical routes for industrial synthesis. J. Braz. Chem. Soc. 2009, 20, 387–406. 10.1590/S0103-50532009000300002. [DOI] [Google Scholar]
- Yoshida J.; Kataoka K.; Horcajada R.; Nagaki A. Modern strategies in electroorganic synthesis. Chem. Rev. 2008, 108, 2265–2299. 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]
- Ogibin Y. N.; Elinson M. N.; Nikishin G. I. Mediator oxidation systems in organic electrosynthesis. Russ. Chem. Rev. 2009, 78, 89–140. 10.1070/RC2009v078n02ABEH003886. [DOI] [Google Scholar]
- O’Brien T. F.; Bommoraju T. V.; Hine F.. Handbook of Chlor-Alkali Technology; Springer: Dordecht, Netherlands, 2005. [Google Scholar]
- American Chemical Society National Historic Chemical Landmarks. Hall Process: Production and Commercialization of Aluminum. http://www.acs.org/content/acs/en/education/whatischemistry/landmarks/aluminumprocess.html (accessed April 27, 2016).
- Rosen B. R.; Werner E. W.; O’Brien A. G.; Baran P. S. Total synthesis of dixiamycin B by electrochemical oxidation. J. Am. Chem. Soc. 2014, 136, 5571–5574. 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose J. F.; Carpenter L. L.; Nelson R. F. Electrochemical and spectroscopic properties of cation radicals III. Reaction pathways of carbazolium radical ions. J. Electrochem. Soc. 1975, 122, 876–894. 10.1149/1.2134365. [DOI] [Google Scholar]
- Corey E. J.; Sauers R. R. The Synthesis of pentacyclosqualene (8,8′-cycloönocerene) and the α- and β-onoceradienes. J. Am. Chem. Soc. 1959, 81, 1739–1743. 10.1021/ja01516a054. [DOI] [Google Scholar]
- For a review of recent applications of Csp3–Csp3 cross coupling in natural product synthesi, see:Geist E.; Kirschning A.; Schmidt T. sp3-sp3 Coupling reactions in the synthesis of natural products and biologically active molecules. Nat. Prod. Rep. 2014, 31, 441–448. 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- Redden A.; Perkins R. J.; Moeller K. D. Oxidative cyclization reactions: Controlling the course of a radical cation-derived reaction with the use of a second nucleophile. Angew. Chem., Int. Ed. 2013, 52, 12865–12868. 10.1002/anie.201308739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A.; Moeller K. D. Oxidative cyclizations, the synthesis of aryl-substituted C-glycosides, and the role of the second electron transfer step. Org. Lett. 2013, 15, 5818–5821. 10.1021/ol402826z. [DOI] [PubMed] [Google Scholar]
- Redden A.; Moeller K. D. Anodic coupling reactions: Exploring the generality of Curtin–Hammett controlled reactions. Org. Lett. 2011, 13, 1678–1681. 10.1021/ol200182f. [DOI] [PubMed] [Google Scholar]
- Xu H.-C.; Moeller K. D. Intramolecular anodic olefin coupling reactions: Use of the reaction rate to control substrate/product selectivity. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. 10.1002/anie.201003924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Moeller K. D. Anodic coupling reactions and the synthesis of C-glycosides. Org. Lett. 2010, 12, 2590–2593. 10.1021/ol100800u. [DOI] [PubMed] [Google Scholar]
- Xu H.-C.; Moeller K. D. Intramolecular anodic olefin coupling reactions: Using competition studies to probe the mechanism of oxidative cyclization reactions. Org. Lett. 2010, 12, 1720–1723. 10.1021/ol100317t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.-C.; Moeller K. D. Intramolecular anodic olefin coupling reactions and the synthesis of cyclic amines. J. Am. Chem. Soc. 2010, 132, 2839–2844. 10.1021/ja910586v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.-C.; Moeller K. D. Intramolecular anodic olefin coupling reactions: The use of a nitrogen trapping group. J. Am. Chem. Soc. 2008, 130, 13542–13543. 10.1021/ja806259z. [DOI] [PubMed] [Google Scholar]
- Wu H.; Moeller K. D. Anodic coupling reactions: A sequential cyclization route to the arteannuin ring skeleton. Org. Lett. 2007, 9, 4599–4602. 10.1021/ol702118n. [DOI] [PubMed] [Google Scholar]
- Tang F.; Moeller K. D. Intramolecular anodic olefin coupling reactions: The effect of polarization on carbon–carbon bond formation. J. Am. Chem. Soc. 2007, 129, 12414–12415. 10.1021/ja076172e. [DOI] [PubMed] [Google Scholar]
- Brandt J. D.; Moeller K. D. Oxidative cyclization reactions: Amide trapping groups and the synthesis of furanones. Org. Lett. 2005, 7, 3553–3556. 10.1021/ol051296m. [DOI] [PubMed] [Google Scholar]
- Huang Y. T.; Moeller K. D. Anodic coupling reactions: The use of N,O-ketene acetal coupling partners. Org. Lett. 2004, 6, 4199–4202. 10.1021/ol048450+. [DOI] [PubMed] [Google Scholar]
- Mihelcic J.; Moeller K. D. Oxidative cyclizations: The asymmetric synthesis of (−)-alliacol A. J. Am. Chem. Soc. 2004, 126, 9106–9111. 10.1021/ja048085h. [DOI] [PubMed] [Google Scholar]
- Mihelcic J.; Moeller K. D. Anodic cyclization reactions: The total synthesis of alliacol A. J. Am. Chem. Soc. 2003, 125, 36–37. 10.1021/ja029064v. [DOI] [PubMed] [Google Scholar]
- Duan S. Q.; Moeller K. D. Anodic cyclization reactions: Capitalizing on an intramolecular electron transfer to trigger the synthesis of a key tetrahydropyran building block. J. Am. Chem. Soc. 2002, 124, 9368–9369. 10.1021/ja027227+. [DOI] [PubMed] [Google Scholar]
- Sutterer A.; Moeller K. D. Reversing the polarity of enol ethers: An anodic route to tetrahydrofuran and tetrahydropyran rings. J. Am. Chem. Soc. 2000, 122, 5636–5637. 10.1021/ja001063k. [DOI] [Google Scholar]
- Frey D. A.; Krishna Reddy S. H.; Moeller K. D. Intramolecular anodic olefin coupling reactions: The use of allylsilane coupling partners with allylic alkoxy groups. J. Org. Chem. 1999, 64, 2805–2813. 10.1021/jo982280v. [DOI] [PubMed] [Google Scholar]
- Frey D. A.; Wu N.; Moeller K. D. Anodic electrochemistry and the use of a 6-V lantern battery: A simple method for attempting electrochemically based synthetic transformations. Tetrahedron Lett. 1996, 37, 8317–8320. 10.1016/0040-4039(96)01946-6. [DOI] [Google Scholar]
- Sperry J. B.; Ghiviriga I.; Wright D. L. Electrochemical annulation of five-membered rings through dearomatization of furans and thiophenes. Chem. Commun. 2006, 194–196. 10.1039/B513532J. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Wright D. L. The gem-dialkyl effect in electron transfer reactions: Rapid synthesis of seven-membered rings through an electrochemical annulation. J. Am. Chem. Soc. 2005, 127, 8034–8035. 10.1021/ja051826+. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Wright D. L. Annulated heterocycles through a radical-cation cyclization: synthetic and mechanistic studies. Tetrahedron 2006, 62, 6551–6557. 10.1016/j.tet.2006.03.058. [DOI] [Google Scholar]
- Sperry J. B.; Whitehead C. R.; Ghiviriga I.; Walczak R. M.; Wright D. L. Electrooxidative coupling of furans and silyl enol ethers: Application to the synthesis of annulated furans. J. Org. Chem. 2004, 69, 3726–3734. 10.1021/jo049889i. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Wright D. L. Synthesis of the hamigeran skeleton through an electro-oxidative coupling reaction. Tetrahedron Lett. 2005, 46, 411–414. 10.1016/j.tetlet.2004.11.108. [DOI] [Google Scholar]
- Whitehead C. R.; Sessions E. H.; Ghiviriga I.; Wright D. L. Two-step electrochemical annulation for the assembly of polycyclic systems. Org. Lett. 2002, 4, 3763–3765. 10.1021/ol026771k. [DOI] [PubMed] [Google Scholar]
- Miller A. K.; Hughes C. C.; Kennedy-Smith J. J.; Gradl S. N.; Trauner D. Total synthesis of (−)-heptemerone B and (−)-guanacastepene E. J. Am. Chem. Soc. 2006, 128, 17057–17062. 10.1021/ja0660507. [DOI] [PubMed] [Google Scholar]
- Ding H.; DeRoy P. L.; Perreault C.; Larivée A.; Siddiqui A.; Caldwell C. G.; Harran S.; Harran P. G. Electrolytic macrocyclizations: Scalable synthesis of a diazonamide-based drug development candidate. Angew. Chem., Int. Ed. 2015, 54, 4818–4822. 10.1002/anie.201411663. [DOI] [PubMed] [Google Scholar]
- Burgett A. W. G.; Li Q.; Wei Q.; Harran P. G. A concise and flexible total synthesis of (−)-diazonamide A. Angew. Chem., Int. Ed. 2003, 42, 4961–4966. 10.1002/anie.200352577. [DOI] [PubMed] [Google Scholar]
- Gütz C.; Selt M.; Bänziger M.; Bucher C.; Römelt C.; Hecken N.; Gallou F.; Galvão T. R.; Waldvogel S. R. A novel cathode material for cathodic dehalogenation of 1,1-dibromo cyclopropane derivatives. Chem. - Eur. J. 2015, 21, 13878–13882. 10.1002/chem.201502064. [DOI] [PubMed] [Google Scholar]
- Gütz C.; Bänziger M.; Bucher C.; Galvão T. R.; Waldvogel S. R. Development and scale-up of the electrochemical dehalogenation for the synthesis of a key intermediate for NS5A inhibitors. Org. Process Res. Dev. 2015, 19, 1428–1433. 10.1021/acs.oprd.5b00272. [DOI] [Google Scholar]
- Horn E. J.; Rosen B. R.; Chen Y.; Tang J.; Chen K.; Eastgate M. D.; Baran P. S. Scalable and sustainable electrochemical allylic C–H oxidation. Nature 2016, 533, 77–81. 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle K. M.; Mei T.-S.; Wasa M.; Yu J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby D. A.; Bergman R. G.; Ellman J. A. Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation. Chem. Rev. 2010, 110, 624–655. 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons T. W.; Sanford M. S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann L. Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: Mechanism and scope. Chem. Rev. 2011, 111, 1315–1345. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- Arockiam P. B.; Bruneau C.; Dixneuf P. H. Ruthenium(II)-catalyzed C–H bond activation and functionalization. Chem. Rev. 2012, 112, 5879–5918. 10.1021/cr300153j. [DOI] [PubMed] [Google Scholar]
- Seregin I. V.; Gevorgyan V. Direct transition metal-catalyzed functionalization of heteroaromatic compounds. Chem. Soc. Rev. 2007, 36, 1173–1193. 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: Versatility and practicality. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlacken G. P.; Bateman L. M. Recent advances in aryl–aryl bond formation by direct arylation. Chem. Soc. Rev. 2009, 38, 2447–2464. 10.1039/b805701j. [DOI] [PubMed] [Google Scholar]
- Brueckl T.; Baxter R. D.; Ishihara Y.; Baran P. S. Innate and guided C–H functionalization logic. Acc. Chem. Res. 2012, 45, 826–839. 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Electrochemical C–H amination: Synthesis of aromatic primary amines via N-arylpyridinium ions. J. Am. Chem. Soc. 2013, 135, 5000–5003. 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Direct C–N coupling of imidazoles with aromatic and benzylic compounds via electrooxidative C–H functionalization. J. Am. Chem. Soc. 2014, 136, 4496–4499. 10.1021/ja501093m. [DOI] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Heterocyclization approach for electrooxidative coupling of functional primary alkylamines with aromatics. J. Am. Chem. Soc. 2015, 137, 9816–9819. 10.1021/jacs.5b06526. [DOI] [PubMed] [Google Scholar]
- Ji Y.; Brueckl T.; Baxter R. D.; Fujiwara Y.; Seiple I. B.; Su S.; Blackmond D. G.; Baran P. S. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411–14415. 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y.; Dixon J. A.; Rodriguez R. A.; Baxter R. D.; Dixon D. D.; Collins M. R.; Blackmond D. G.; Baran P. S. A new reagent for direct difluoromethylation. J. Am. Chem. Soc. 2012, 134, 1494–1497. 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y.; Dixon J. A.; O’Hara F.; Funder E. D.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herlé B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Practical and innate carbon–hydrogen functionalization of heterocycles. Nature 2012, 492, 95–99. 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Ruffoni A.; Gianatassio R.; Fujiwara Y.; Sella E.; Shabat D.; Baran P. S. Direct synthesis of fluorinated heteroarylether bioisosteres. Angew. Chem., Int. Ed. 2013, 52, 3949–3952. 10.1002/anie.201300763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara F.; Blackmond D. G.; Baran P. S. Radical-based regioselective C–H functionalization of electron-deficient heteroarenes: Scope, tunability, and predictability. J. Am. Chem. Soc. 2013, 135, 12122–12134. 10.1021/ja406223k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianatassio R.; Kawamura S.; Eprile C. L.; Foo K.; Burns A. C.; Collins M. R.; Baran P. S. Simple sulfinate synthesis enables C–H trifluoromethylcyclopropanation. Angew. Chem., Int. Ed. 2014, 53, 9851–9855. 10.1002/anie.201406622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien A. G.; Maruyama A.; Inokuma Y.; Fujita M.; Baran P. S.; Blackmond D. G. Radical C–H functionalization of heteroarenes under electrochemical control. Angew. Chem., Int. Ed. 2014, 53, 11868–11871. 10.1002/anie.201407948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowski K. J.; Liu R.; Milligan G. L.; Moeller K. D.; Aubé J. Practical Electrochemical Anodic Oxidation of Polycyclic Lactams for Late Stage Functionalization. Angew. Chem., Int. Ed. 2015, 54, 10555–10558. 10.1002/anie.201504775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsler B.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Metal- and reagent-free highly selective anodic cross-coupling reaction of phenols. Angew. Chem., Int. Ed. 2014, 53, 5210–5213. 10.1002/anie.201400627. [DOI] [PubMed] [Google Scholar]
- Kirste A.; Elsler B.; Schnakenburg G.; Waldvogel S. R. Efficient anodic and direct phenol-arene C–C cross-coupling: The benign role of water or methanol. J. Am. Chem. Soc. 2012, 134, 3571–3576. 10.1021/ja211005g. [DOI] [PubMed] [Google Scholar]
- Kirste A.; Schnakenburg G.; Stecker F.; Fischer A.; Waldvogel S. R. Anodic phenol–arene cross-coupling reaction on boron-doped diamond electrodes. Angew. Chem., Int. Ed. 2010, 49, 971–975. 10.1002/anie.200904763. [DOI] [PubMed] [Google Scholar]
- For a comprehensive review, see:Francke R.; Little R. D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- For an overview of the cation pool method, see:Yoshida J.; Suga S. Basic concepts of “Cation Pool” and “Cation Flow” methods and their applications in conventional and combinatorial organic synthesis. Chem. - Eur. J. 2002, 8, 2650–2658. 10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rafiee M.; Miles K. C.; Stahl S. S. Electrocatalytic alcohol oxidation with TEMPO and bicyclic nitroxyl derivatives: Driving force trumps steric effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. 10.1021/jacs.5b09672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey D. P.; Milton R. D.; Chen D.; Sigman M. S.; Minteer M. S. TEMPO-modified linear poly(ethylenimine) for immobilization-enhanced electrocatalytic oxidation of alcohols. ACS Catal. 2015, 5, 5519–5524. 10.1021/acscatal.5b01668. [DOI] [Google Scholar]
- Semmelhack M. F.; Schmid C. R. Nitroxyl-mediated electro-oxidation of amines to nitriles and carbonyl compounds. J. Am. Chem. Soc. 1983, 105, 6732–6734. 10.1021/ja00360a042. [DOI] [Google Scholar]
- Becking L.; Schäfer H. J. Pyrrolidines by intramolecular addition of Kolbe radicals generated from β-allylaminoalkanoates. Tetrahedron Lett. 1988, 29, 2797–2800. For other examples of a variety of electrochemically mediated radical cascades, see ref (5). 10.1016/0040-4039(88)85212-2. [DOI] [Google Scholar]
- For a review on electrochemical reductive cyclizations, see:Little R. D.; Schwaebe M. K. Electrochemical Cyclization at the Cathode. Top. Curr. Chem. 1997, 185, 1–48. 10.1007/3-540-61454-0_69. [DOI] [Google Scholar]