

Abstract
The use of NMR chiral solvating agents (CSAs) for the analysis of enantiopurity has been known for decades, but has been supplanted in recent years by chromatographic enantioseparation technology. While chromatographic methods for the analysis of enantiopurity are now commonplace and easy to implement, there are still individual compounds and entire classes of analytes where enantioseparation can prove extremely difficult, notably, compounds that are chiral by virtue of very subtle differences such as isotopic substitution or small differences in alkyl chain length. NMR analysis using CSAs can often be useful for such problems, but the traditional approach to selection of an appropriate CSA and the development of an NMR-based analysis method often involves a trial-and-error approach that can be relatively slow and tedious. In this study we describe a high-throughput experimentation approach to the selection of NMR CSAs that employs automation-enabled screening of prepared libraries of CSAs in a systematic fashion. This approach affords excellent results for a standard set of enantioenriched compounds, providing a valuable comparative data set for the effectiveness of CSAs for different classes of compounds. In addition, the technique has been successfully applied to challenging pharmaceutical development problems that are not amenable to chromatographic solutions. Overall, this methodology provides a rapid and powerful approach for investigating enantiopurity that compliments and augments conventional chromatographic approaches.
Short abstract
We have developed a high-throughput experimental approach to facilitate the selection of NMR CSAs that employs automation-enabled screening of prepared libraries of CSAs in a systematic fashion. It provides a powerful, orthogonal method to conventional chromatographic enantioseparation for enantiopurity analysis.
Introduction
The NMR analysis of enantiopurity using chiral solvating agents (CSAs) has a nearly 50 year history of utility.1−3 However, the emergence of fast and convenient chromatographic methods of enantiopurity determination has diminished the use of NMR approaches with CSAs. Despite the convenience of modern chromatographic methods for the separation of enantiomers, the technique is by no means universal. Chromatographic enantioseparation relies on the differential adsorption of the two enantiomers of the compound of interest by an enantiopure adsorbent—a sometimes difficult task for compounds that are chiral by virtue of very subtle differences such as isotopic substitution or variations in alkyl chain length.4 In contrast, while differentiation in the NMR spectrum may occur because of differential binding of the two enantiomers to the CSA, this is not mandatory. Enantiomeric differentiation in the NMR spectrum often depends on different geometries for the two diastereomeric complexes formed between the enantiomers of the analyte of interest and the enantiopure CSA. Specifically, isoenergetic diastereomeric adsorbates cannot lead to separation in chromatography, but can lead to distinct NMR signals. Consequently, NMR analysis using CSAs can often complement enantioselective chromatography, performing well for those compounds that are the most difficult to resolve chromatographically.
Despite these advantages, the use of NMR CSAs is limited by several factors including the lack of predictive rules or simple to use method development strategies that ensure rapid success in the selection of a CSA and development of an NMR based approach for analysis of enantiopurity. Factors influencing the selection of the best CSA for a given analyte are also poorly understood. Unlike most reports of new chromatographic phases, almost all reports of new NMR CSAs examine a rather limited group of analytes. A more significant problem is that these reports seldom provide any comparison with known CSAs, presumably stemming from the challenge of preparing and analyzing large numbers of samples. The lack of data comparing the effectiveness of different CSAs makes choice of the optimal CSA for a given analyte a challenging task. To our knowledge, this report represents the first time where direct comparison of a wide range of CSAs (32 total) on analytes from a variety of functional group classes has been undertaken. A historical lack of automation in many NMR laboratories has, until recently, made CSA identification and method development a one at a time ad hoc undertaking that is still reminiscent of the early days of enantioselective chromatography. Given the inherent power of the CSA approach, the extensive body of literature accumulated over the past half century,2,3 and the orthogonality to conventional enantioselective chromatographic methods, we have endeavored to create an automated high-throughput experimental approach to the rapid selection and optimization of NMR-based approaches to the analysis of enantiopurity using CSAs.
Results and Discussion
Selection of CSA Library and Analytes
We conducted the initial exploratory study in chloroform-d, although the method is easily extended to other NMR solvents. Commercial availability of CSAs known to have broad applicability was an important criterion used in selecting our test library of 32 CSAs (Figure 1). Additionally, we shaped our library to contain mostly CSAs that were commercially available (C1–C18, C20, C23–C27, C29–C31). Unfortunately, many of the large number of reported NMR CSAs are not commercially available at this time, presumably because it remains unknown, at least in part, whether these reagents exhibit improved performance and broader applicability than those that can already be purchased from a commercial vendor. Many of these literature-reported CSAs also require multistep syntheses, constituting an additional deterrent to their use. We did include several CSAs in the test library that are not commercially available, most of which have been developed in our laboratories (C19, C21, C22, C28). A rhodium reagent (C32) possessing effectiveness for use with soft Lewis base analytes was also studied. All of the noncommercial CSAs included in the library are readily prepared from commercially available reagents, most in a single step. Clearly, additional CSAs could be added to future iterations of the library.
Figure 1.

CSA library exploited in NMR high-throughput screening. CSAs with both enantiomeric and multiple diastereomeric forms that have been screened are labeled with *.
For the initial study, a group of analytes were selected that span the range of functionalities and structural variability commonly found in drug candidates (A1–A16, Figure 2). Analytes available in enantiopure form were chosen so that solutions could be enriched in one of the enantiomers, thereby aiding understanding of how each enantiomer undergoes chemical shift perturbation upon complexation with a given CSA. In addition, both enantiomers of each CSA were used when available, facilitating the determination of whether or not enantiomeric differentiation is observed in the NMR spectrum.
Figure 2.

Analytes used in CSA NMR high-throughput screening.
Preparation and Use of CSA Screening Kits
A streamlined approach for rapid selection of the optimal CSA for analysis of enantiopurity is analogous to platform screening approaches previously developed in these laboratories for chromatographic separation, or choices of crystallization or catalysis conditions.5 (Figure 3) Using automated liquid handling, an aliquot of a stock solution of each CSA in acetonitrile was dispensed into a glass vial insert of a 96-well tray. Evaporation of solvent afforded CSA kits containing predosed amounts of the 32 different CSAs, which were sealed under plastic cap mats and stored in a refrigerator under nitrogen until needed. When performing an NMR screening, 80 μL of a 10 mM stock solution of the analyte in chloroform-d was dispensed into each of the 32 inserts in the CSA screening kit. The plate is slowly shaken to dissolve the CSAs, and then 35 μL of each solution is transferred, using an automated liquid handler, to a 1.7 mm NMR tube in a tube rack having the footprint of a 96-well tray. The rack of 1.7 mm NMR tubes is then placed into an automatic sample changer, and the NMR spectrum for each tube is collected using automated acquisition and data processing protocols.
Figure 3.

Overview of the preparation and use of CSA screening kits.
An important question with such a screening protocol involves the concentration of CSA and analyte. Since NMR enantiodifferentiation with CSAs can occur because of either differential association or the diastereomeric nature of the complexes, the optimal ratio of CSA to analyte depends on which mechanism of intermolecular association predominates. If the diastereomeric nature of the complexes is important, using excess CSA relative to analyte is often warranted. Based on previous experience, we opted to use a CSA concentration of 20 mM and analyte concentration of 10 mM for initial screening. These concentrations are common6−8in NMR studies of other CSAs and facilitate sufficient complexation to promote formation of the diastereomeric complexes. Assuming that at least one CSA produces enantiomeric differentiation in the NMR spectrum of the analyte, the conditions for using the best combination can then be optimized in subsequent studies.
Effectiveness of CSAs
A summary of the results obtained for all the CSA–analyte combinations is provided in Table 1. Gray cells in Table 1 indicate that the CSA caused no enantiodifferentiation for any resonances of the analyte, yellow indicates partial enantiodifferentiation of one or more resonance, and green indicates complete enantiodifferentiation for one or more resonances.
Table 1. Summary of CSA High-Throughput Screening Resultsa.
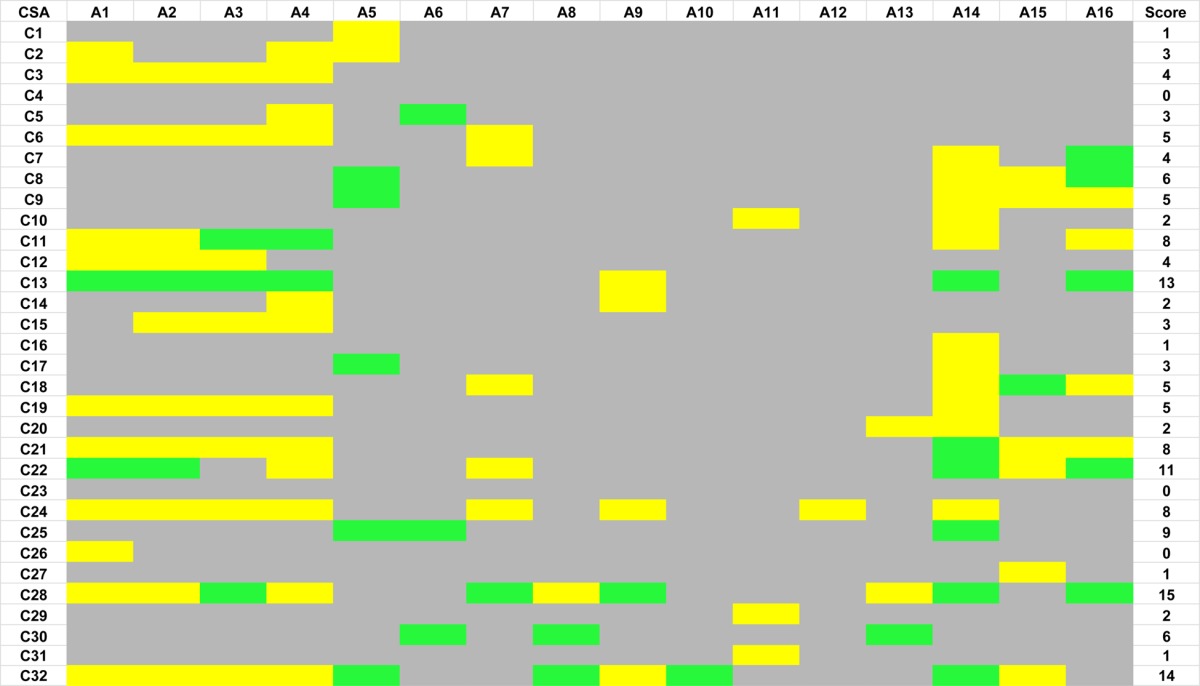
32 CSAs are screened by NMR spectroscopy on 16 enantiomerically enriched analytes. Green, yellow, and gray cells respectively indicate full, partial, and no enantiodifferentiation. The score is based on whether complete (2 points) or partial (1 point) enantiodifferentation is observed for each analyte.
As evidenced by the large number of gray cells in Table 1, most CSA–analyte combinations resulted in no enantiodifferentiation in the NMR spectrum. Only a very few combinations (green cells) resulted in complete enantiodifferentiation of one or more resonances. Nevertheless, every analyte in the study did show partial or complete enantiodifferentiation of at least one resonance with one or more CSAs in the library, including the challenging analytes A8 and A11, which are chiral by virtue of isotopic substitution. In no case would it have been possible to predict a priori which CSA would perform best for which analyte, but broad screening of CSAs facilitates the rapid identification of a suitable lead, from which optimization of CSA–analyte ratio, solvent, temperature, and NMR conditions can proceed. The data in Table 1 also provided a rare opportunity to compare the general effectiveness of a wide range of NMR CSAs. Included in Table 1 is a weighted score based on the number of analytes where a particular CSA causes complete (2 points) or partial (1 point) enantiodifferentation in the NMR spectrum. Whelk-O (C28), Rh2(MTPA)4 (C32), quinine (C13), the 1-(1-naphthyl)ethyl urea derivatives of tert-leucine (C21) and valine (C22), and dibenzoyltartaric acid (C24) were the most broadly applicable CSAs in the study group. The Whelk-O reagent is an analogue of a broadly applicable chiral discriminator from liquid chromatographic studies9,10 and has received some limited attention as an NMR CSA.11−14 The rhodium reagent, which might generally be thought of as applicable to soft Lewis bases,15 but which has also been shown to be effective for some hard Lewis bases,16 was observed to be effective for a number of analytes in our study with hard Lewis base functionalities. Quinine2,3,17 and the naphthyl urea derivatives of valine and tert-leucine18 have been the focus of only limited studies, but our results indicate that they are applicable to a broad range of analytes. Dibenzoyltartaric acid has not been widely studied19,20 and, while it only results in partial enantiodifferentiation in most cases, was shown to be effective for a broad range of analytes. 1-(1-Naphthyl)ethylamine (C11)2,21,22 also performed very well, which, to a large degree, was the result of its applicability for carboxylic acids. Our study suggests that commercial availability of CSAs such as Whelk-O, Rh2(MTPA)4, and naphthylurea derivatives of valine and tert-leucine may be warranted.
Figure 4 shows an example of the complete enantiodifferentiation of the methyl doublet of 2-amino-3,3-dimethylbutane (A5) with 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (C25). The mixture is enriched in the (R)-enantiomer of A5, and the reversal in the order of the two doublets of the two enantiomers of A5 with (R)- and (S)-C25 is apparent.
Figure 4.

1H NMR (500 MHz, CDCl3, 298 K) spectrum of the methyl resonance of (a) 2-amino-3,3-dimethylbutane (A5, R/S 3:2, 10 mM) with (b) 1,1′-binaphthyl-2,2′diyl hydrogen phosphate ((S)-C25, 20 mM) and (c) (R)-C25 (20 mM).
The spectra shown in Figure 5 illustrate the complete enantiomeric differentiation of the H5 and H7 aryl resonances of an (S)-enriched mixture of naproxen (A3) with quinine (C13). Either the H5 or H7 resonances in this mixture are suitable for determining enantiopurity. Having more than one enantiodifferentiated resonance improves the chances that a usable resonance will be in a region of the spectrum free of overlap with other analyte or CSA resonances.
Figure 5.

1H NMR (500 MHz, CDCl3, 298 K) spectrum of a portion of the aryl region of (a) naproxen (A3, R/S 4:7, 10 mM) with (b) quinine (9R-C13, 20 mM).
Figure 6 shows the aryl region of an (S)-enriched mixture of omeprazole (A16) with quinine (C13). H13 appears as a singlet at about 8.23 ppm and H7 as a doublet at about 6.90 ppm. Both H13 and H7 exhibit complete enantiomeric differentiation in the presence of C13 as seen in Figure 6b. H5 appears as a doublet of doublets at about 6.95 ppm and is only partially differentiated in the presence of C13. In many cases, partial enantiomeric differentiation with CSAs is still sufficient to estimate enantiopurity, although this is not the case for the H5 resonance in Figure 6b. Alternatively, it is possible to use a pure shift NMR pulse sequence in which the overlapped multiplets are collapsed into singlets leading to significantly improved resolution in the NMR spectrum.23−25Figure 6c shows the corresponding pure shift spectrum for the mixture of A16 with C13. Pure shift spectra were obtained for the entire CSA library under automated conditions, so no attempt was made to optimize the signal-to-noise ratio for the rapid screening study. Two enantiodifferentiated singlets are seen in the pure shift spectrum for both H7 and H13. Of particular note are the two singlets seen for the H5 resonance, clearly showing that this resonance is also enantiodifferentiated. Based on these initial results, subsequent studies could be performed to optimize the signal-to-noise of the pure shift spectrum for a more accurate determination of enantiopurity.
Figure 6.

1H NMR (500 MHz, CDCl3, 298 K) spectrum of the aryl region of (a) omeprazole (A16, R/S 3:7, 10 mM) with (b) quinine (9R-C13, 20 mM). Spectrum c has the same concentrations as spectrum b but is obtained in a pure shift mode.
Two other CSAs deserve mention for specialized applications. 1,1′-Binaphthyl-2,2′-diyl hydrogen phosphate (C25)2,26 is noteworthy for its enantiodifferentiation of amines, and the europium(III) tris-β-diketonate of (3-heptafluorobutyryl)-camphorate (C30, Eu(hfc)3)2,27 is effective for some analytes where very few of the other CSAs afforded any enantiodifferentiation. Chiral lanthanide reagents were commonly used when investigators only had access to NMR spectrometers of lower field strength (200 MHz or less).28 On higher field instruments, line broadening with these CSAs is often a problem and the spectra that we obtained with C30 were often broadened to such a degree as to render the spectra useless. However, in certain instances, there was obvious enantiodifferentiation of a magnitude (e.g., 0.25 and 0.22 ppm for the diastereotopic H4 methylene atoms of 2-amino-4-phenylbutane) far greater than that observed with other reagents. The use of the (+)- and (−)-Eu(hfc)3 with an analyte enriched in one enantiomer is especially useful in determining whether the highly shifted resonances exhibit enantiodifferentiation. Decreasing the concentration of the europium CSAs from 20 mM to 5 mM or even lower would likely be warranted in future studies.
Interestingly, the widely used CSA, 2,2,2-trifluoro-1-(9-anthryl)ethanol (C7), commonly known as Pirkle’s alcohol, did not perform well in this study. While care should be taken in generalizing based on a small analyte sample set, this result does help to emphasize the value of screening a diverse set of CSAs. The 3,5-dinitrobenzoyl (DNB) derivatives of phenylglycine (C17), 1-phenylethylamine (C18), and l-leucine (C19) did cause enantiodifferentiation in the NMR spectra of some analytes, but were not as broadly applicable as the Whelk-O CSAs (C28). Similar results are observed in liquid chromatographic studies in which the Whelk-O chiral stationary phase shows enhanced effectiveness relative to the DNB-amino acid phases.29
Analysis of the screening outcomes can be used to remove certain CSAs from the library because either they are ineffective or their effectiveness is duplicated by other CSAs. However, it is important to recall that some of the CSAs work well for certain types of compounds (e.g., TRISPHAT (C26) for cationic metal complexes30) that are not well represented within the set of analytes chosen for this study. Further research will be required to develop a complete understanding, but at this juncture it would seem that CSAs C1–C4, C6, C10, C12, C14–C16, C19, C20, C23, C26, C27, and C31 could be removed from the library without substantial decrease in overall performance.
Another outcome of analyzing so many CSAs is that it allowed us to determine whether or not particular functionalities were amenable for study by many or only a few CSAs. Carboxylic acid containing analytes had by far the most CSAs that produced some degree of enantiomeric differentiation in the NMR spectrum, suggesting that the development of additional CSAs for carboxylic acids should be a fairly low priority. A number of CSAs afforded enantiomeric differentiation of the amines, with 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (C25) being most noteworthy. Consequently, in future reports of new CSAs for amines, the degree of enantiodifferentiation should be compared with C25. Similarly, several CSAs afforded enantiodifferentiation of sulfoxides, particularly the Whelk-O reagent (C28). The alcohols in our study were the least effectively differentiated, suggesting a potential focus for new CSA development. For three of the alcohols, only one or two CSAs (A10 with C32; A12 with C24; and A13 with C28 and C30) afford enantiodifferentiation in the NMR spectrum. Terpinen-4-ol (A10) is a highly hindered tertiary alcohol, and it may be that Rh2(MTPA)4 (C32) is effective because of association with the olefin instead of the hydroxyl group.31 The one diol in our test group was enantiodifferentiated by a number of CSAs.
Many CSAs contain aryl rings, which can afford significant shielding and upfield shifts of the resonances of analyte hydrogen atoms that are positioned over the ring, thereby leading to enantiodifferentiation in the NMR spectrum when this shielding is selective for one of the complexed enantiomers. For example, the shielding of the H5 and H7 resonances of naproxen with quinine is apparent in Figure 5b. This shielding is often ascribed to a face-centered pi–pi stacking of the aryl rings of the analyte and CSA, but recent studies have shown that edge-to-face interactions are also important, particularly when the interaction of two electron-rich rings is involved.32−34 All of the aryl rings in our analytes are electron rich; however, of the CSAs containing electron-deficient dinitrobenzoyl aromatic rings (C17, C18, C19, and C28), only C28 showed a general ability to afford enantiodifferentiation for a large number of analytes. It should be noted that C28 possesses an electron rich naphthyl ring in addition to the electron deficient dinitrobenzyl ring. For the combinations in Table 1 that led to enantiodifferentiation, the degree of shielding of aryl resonances of the analytes varied with different CSAs.
A few of the analytes (A14–A16) have more than one functional group capable of interacting with CSAs, and in each case a number of the CSAs caused enantiodifferentiation in the NMR spectrum. This is not surprising since different CSAs can be expected to associate at different types of functionalities.
Many compounds of pharmaceutical interest contain fluorine atoms, allowing for the possibility of enantiodifferentiation in the 19F NMR spectrum. The observation of the 19F NMR nonequivalence in diastereomeric complexes with CSAs was one of the first examples of NMR nonequivalence reported.119F NMR spectra were recorded for A2 with the CSA library. Even though the fluorine atom in A2 is relatively remote from the stereocenter, C32 caused complete enantiodifferentiation and C28 caused partial enantiodifferentiation of the fluorine singlet (Figure 7). Note that the spectrum shown in Figure 7b has two sets of enantiodifferentiated fluorine signals, each set of which has two peaks with areas equal to the proportions of R- and S-enantiomers in the mixture. It is known that analytes can form 1:1 or 2:1 complexes with C32(15) and the two sets of signals presumably result from the presence of both complexes in solution under conditions of slow exchange. The unexpectedly complicated nature of the CF3 resonance of the MTPA group in the rhodium complex provides further evidence for the formation of the 1:1 and 2:1 complexes. In no cases was there any evidence for the formation of multiple complexes in the 1H NMR spectral results with C32 reported in Table 1. The high degree of spectral dispersion, the observation that the 19F NMR spectrum for many compounds consists of a singlet, and the lack of obscuring signals from the CSA are significant advantages that suggest great potential for this approach for fluorine-containing molecules.
Figure 7.

19F NMR (471 MHz, CDCl3, 298 K) spectrum of flurbiprofen (A2, R/S 7:3, 10 mM) with (a) (S,S)-Whelk-O (C28, 20 mM) and (b) Rh2(MTPA)4 (C32, 20 mM).
The high-throughput CSA screening kit approach has proven useful for the rapid development of NMR methods for quantifying the enantiopurity of enantiomeric mixtures that are not amenable to chromatographic enantioseparation methods. Attempts to resolve the enantiomers of amino-lipid A17(35) with standard methods for chromatographic enantioseparation proved completely ineffective, perhaps not surprising given that the asymmetry of the molecule only becomes apparent at a distance eight bonds from the stereocenter. CSA library screening revealed hits for several possible CSAs, with 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (C25) showing the best results. Figure 8a shows the N-methyl singlet, olefinic multiplets, and allylic methylenes at about 2.20, 5.35, and 2.05 ppm, respectively. In Figure 8b, the N-methyl resonance splits into two distinct doublets at a 1:1 ratio in the presence of C25, which suggests that the amino-lipid is a racemic mixture. The olefinic (H-16) and allylic protons (H-21) also exhibit partial enantiodifferentiation that is clearly observed in the pure shift spectrum by the 1:1 singlets at 5.38 and 2.04 ppm, respectively (Figure 8d). A question that arises from the interpretation of Figure 8d is whether the chemical shift nonequivalence of the two N-methyl singlets results from slow chemical exchange caused by N-protonation when A17 binds with C25, thereby rendering the two methyl groups diastereotopic. In other words, are two singlets observed in Figure 8d because of diastereotopic resolution or because the two N-methyl groups have the same chemical shift and are enantiodifferentiated? The 1H–1H COSY correlations between the exchangeable proton at 11.75 ppm, the N-methyl doublets at 2.65 and 2.69 ppm, and the adjacent methylene resonances provide evidence for the protonation of A17 nitrogen by C25, which is corroborated by the 4.2 Hz J-coupling between the N-methyl and the downfield proton. However, the strong NOE between the N-methyl groups and the flanking methylene groups and the absence of observable NOE between the N-methyl groups (Figure 9) indicates that the two singlets for the N-methyl groups in Figure 8d are the result of enantiodifferentiation rather than diastereotopic resolution. The NOE between the N-methyl and C25 resonances at 7.55 and 7.89 ppm further suggests that the CSA associates with the racemic amino-lipid via hydrogen bonding.
Figure 8.

Application of high-throughput screening of CSAs for NMR enantiodifferentiation of a chiral lipid that is resistant to chromatographic resolution. 1H NMR (500 MHz, CDCl3, 298 K) spectrum of (a) amino-lipid A17 (10 mM) with (b) 1,1′-binaphthyl-2,2′diyl hydrogen phosphate ((S)-C25, 20 mM). Spectra c and d have the same concentrations as spectra a and b but are obtained in a pure shift mode.
Figure 9.

2D 500 MHz ROESY spectrum expansion of amino-lipid A17 with 1,1′-binaphthyl-2,2′diyl hydrogen phosphate (S)-C25.
These results clearly show the value of the CSA screening approach to provide a powerful, orthogonal method to conventional chiral chromatography for rapid method development and enantiopurity analysis. Elimination of nonproductive CSAs from the library and addition of new CSAs can be expected to lead to further improvements in performance, speed and generality. While this study has focused on NMR automation using a robotic tube-changing autosampler, alternative approaches using flow chemistry with a flow-through NMR probe could be conceived, potentially offering further speed advantages.
Experimental Section
Preparation of CSA Kits
Volumetric dispensing and aspiration of CSA mixtures was carried out on a Chemspeed SWING XL robot equipped with a 4-needle head dispenser and tumble stirrer, under positive nitrogen flow. CSA mixtures were prepared at 32 mM concentration in MeCN-d3 or varying mixtures MeCN-d3/CDCl3. The solvent choice for each CSA was driven by the ability to form a uniform solution or slurry, with the goal of achieving uniform dispensing. The resulting mixtures were dispensed to a 96-well polypropylene tray with 4 × 21 mm (200 μL) glass vial inserts (Analytical-Sales Cat. No. 96342) at 50 μL volume. The solvents were evaporated to leave behind 1.6 μmol of CSA in each well, and the kits were bagged under nitrogen to minimize exposure to moisture upon storage.
Preparation of NMR Samples
Judiciously chosen analytes were precisely weighed as mixtures of enantiomers at a concentration of 10 mM. These were typically prepared as enantiomeric mixtures enriched in one enantiomer (i.e., 7 mM/3 mM) and prepared as stock solutions in CDCl3. 80 μL of the substrate solution (0.8 μmol) was manually transferred with a multichannel pipet to an insert containing 1.6 μmol of preloaded CSA on a 96-well reaction plate. The resulting mixture of CSA and substrate in CDCl3 was sealed with a top cover and well mixed on a shaker for 0.5 h. Transfer of 35 μL of CSA/substrate complex from the insets to each 1.7 mm NMR tube (96-tube rack) was facilitated by a PrepGilsonST liquid handler. Subsequently, the 96-tube rack was loaded onto a SampleJet NMR sample changer on a 500 MHz NMR spectrometer equipped with 1.7 mm MicroCryoProbe. All NMR experiments were acquired in automation using IconNMR software.
Conclusions
In this study we have demonstrated the value of a high-throughput experimentation approach to the selection of enantioselective NMR CSAs that employs automation-enabled screening of prepared libraries of CSAs in a systematic fashion. This approach is helpful in providing a comparative data set for the effectiveness of CSAs for different classes of compounds. Good results were obtained for a standard set of enantioenriched compounds, with the technique proving especially valuable for rapid development of analytical methods for studying the enantiomers of compounds that proved challenging or impossible to resolve using conventional chromatographic enantioseparation approaches.
Acknowledgments
We are grateful to Merck Creative Studios for assistance with Figure 3, to the MRL Department of Process & Analytical Chemistry for providing funding for this collaboration, and to Melissa Lin for help with the NMR sample preparation and data acquisition. We also thank Gary Martin for insightful suggestions on pure shift NMR experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00062.
NMR spectra of all screening results and summary of HTE screening results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pirkle W. H. The nonequivalence of physical properties of enantiomers in optically active solvents. differences in nuclear magnetic resonance spectra. I. J. Am. Chem. Soc. 1966, 88, 1837. 10.1021/ja00960a060.5942993 [DOI] [Google Scholar]
- Wenzel T. J.Discrimination of Chiral Compounds Using NMR Spectroscopy; Wiley: Hoboken, 2007. [Google Scholar]
- Uccello-Barretta G.; Balzano F. Chiral NMR solvating additives for differentiation of enantiomers. Top. Curr. Chem. 2013, 341, 69. 10.1007/128_2013_445. [DOI] [PubMed] [Google Scholar]
- Pirkle W. H.; Gan K. Z. A direct chromatographic separation of enantiomers chiral by virtue of isotopic substitution. Tetrahedron: Asymmetry 1997, 8, 811–814. 10.1016/S0957-4166(97)00037-2. [DOI] [Google Scholar]
- Davies I. W.; Welch C. J. Looking forward in pharmaceutical process chemistry. Science 2009, 325, 701–704. 10.1126/science.1174501. [DOI] [PubMed] [Google Scholar]
- Pham N. H.; Wenzel T. J. A water-soluble calix[4]resorcinarene with α-methyl-l-prolinylmethyl groups as a chiral NMR solvating agent. J. Org. Chem. 2011, 76, 986–989. 10.1021/jo102197w. [DOI] [PubMed] [Google Scholar]
- Chisholm C. D.; Wenzel T. J. Enantiomeric discrimination of aromatic-containing anionic substrates using cationic cyclodextrins. Tetrahedron: Asymmetry 2011, 22, 62–68. 10.1016/j.tetasy.2010.12.001. [DOI] [Google Scholar]
- Chisholm C. D.; Fülöp F.; Forró E.; Wenzel T. J. Enantiomeric discrimination of cyclic β-amino acids using (18-crown-6)-2,3,11,12-tetracarboxylic acid as a chiral NMR solvating agent. Tetrahedron: Asymmetry 2010, 21, 2289–2294. 10.1016/j.tetasy.2010.07.031. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J.; Lamm B. Design, synthesis, and evaluation of an improved enantioselective naproxen selector. J. Org. Chem. 1992, 57, 3854–3860. 10.1021/jo00040a026. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J. An improved chiral stationary phase for the chromatographic separation of underivatized naproxen enantiomers. J. Liq. Chromatogr. 1992, 15, 1947–1955. 10.1080/10826079208020869. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J.; Wilson S. R. Assignment of absolute configuration to an improved enantioselective naproxen selector. Chirality 1994, 6, 615–622. 10.1002/chir.530060803. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J. Chromtographic and 1H NMR support for a proposed chiral recognition model. J. Chromatogr. 1994, 683, 347–353. 10.1016/0021-9673(94)00477-3. [DOI] [Google Scholar]
- Welch C. J.; Kress M. H.; Beconi M.; Mathre D. J. Studies on the racemization of a stereolabile 5-aryl-thiazolidinedione. Chirality 2003, 15, 143–147. 10.1002/chir.10180. [DOI] [PubMed] [Google Scholar]
- Koscho M. E.; Pirkle W. H. Investigation of a broadly applicable chiral selector used in enantioselective chromatography (Whelk-O 1) as a chiral solvating agent for NMR determination of enantiomeric composition. Tetrahedron: Asymmetry 2005, 16, 3345–3351. 10.1016/j.tetasy.2005.08.032. [DOI] [Google Scholar]
- Duddeck H. Rh2[MTPA]4, a dirhodium complex as NMR auxiliary for chiral recognition. Chem. Rec. 2005, 5, 396–409. 10.1002/tcr.20059. [DOI] [PubMed] [Google Scholar]
- Mattiza J. T.; Meyer V.; Ozuduru G.; Heine T.; Fohrer J.; Duddeck H. Competition of ester, amide, ether, carbonate, alcohol, and epoxide ligands in the dirhodium experiment (chiral discrimination by NMR spectroscopy). Nat. Prod. Commun. 2012, 7, 359–362. [PubMed] [Google Scholar]
- Rosini C.; Uccello-Barretta G.; Pini D.; Abete C.; Salvadori P. Quinine: an inexpensive chiral solvating agent for the determination of enantiomeric composition of binaphthyl derivatives and alkylarylcarbinols by NMR spectroscopy. J. Org. Chem. 1988, 53, 4579–4581. 10.1021/jo00254a031. [DOI] [Google Scholar]
- Wenzel T. J.; Miles R. D.; Weinstein S. E. Chiral nuclear magnetic resonance shift reagents: lanthanide mixtures with 1-(1-naphthyl) ethylurea derivatives of amino acids. Chirality 1997, 9, 1–9. 10.1002/(SICI)1520-636X(1997)9:1<1::AID-CHIR1>3.0.CO;2-M. [DOI] [Google Scholar]
- Ohki A.; Miyashita R.; Naka K.; Maeda S. Enantioselective extraction of di-O-benzoyltartrate anion by ion-pair extractant having binaphthyl-unit. Bull. Chem. Soc. Jpn. 1991, 64, 2714–2719. 10.1246/bcsj.64.2714. [DOI] [Google Scholar]
- Toşa M.; Paizs C.; Majdik C.; Novak L.; Kolonits P.; Irimie F.; Poppe L. Optically active 3-substituted-10-alkyl-10H-phenothiazine-5-oxides by enantiomer selective biotransformations. Tetrahedron: Asymmetry 2002, 13, 211–221. 10.1016/S0957-4166(02)00077-0. [DOI] [Google Scholar]
- Burlingame T. G.; Pirkle W. H. The nonequivalence of physical properties of enantiomers in optically active solvents. differences in proton magnetic resonance spectra. II. J. Am. Chem. Soc. 1966, 88, 4294. 10.1021/ja00970a055. [DOI] [Google Scholar]
- Horeau A.; Guette J. P. Absolute configuration of α-isopropylphenylacetic acid, and a nuclear magnetic resonance determination of its optical purity. C. R. Seances Acad. Sci. C 1968, 267, 257–259. [Google Scholar]
- Foroozandeh M.; Adams R. W.; Meharry N. J.; Jeannerat D.; Nilsson M.; Morris G. A. Ultrahigh-resolution NMR spectroscopy. Angew. Chem., Int. Ed. 2014, 53, 6990–6992. 10.1002/anie.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroozandeh M.; Adams R. W.; Kiraly P.; Nilsson M.; Morris G. A. Measuring couplings in crowded NMR spectra: pure shift NMR with multiplet analysis. Chem. Commun. 2015, 51, 15410–15413. 10.1039/C5CC06293D. [DOI] [PubMed] [Google Scholar]
- Castañar L.; Pérez-Trujillo M.; Nolis P.; Monteagudo E.; Virgili A.; Parella T. Enantiodifferentiation through frequency-selective pure-shift (1)H nuclear magnetic resonance spectroscopy. ChemPhysChem 2014, 15, 854–857. 10.1002/cphc.201301130. [DOI] [PubMed] [Google Scholar]
- Shapiro M. J.; Archinal A. E.; Jarema M. A. Chiral purity determination by 1H NMR spectroscopy. Novel use of l,l′-binaphthyl-2,2′-diylphosphoric acid. J. Org. Chem. 1989, 54, 5826–5828. 10.1021/jo00285a036. [DOI] [Google Scholar]
- Fraser R. R.; Petit M. A.; Saunders J. K. Determination of enantiomeric purity by an optically active nuclear magnetic resonance shift reagent of wide applicability. J. Chem. Soc. D 1971, 1450–1451. 10.1039/c29710001450. [DOI] [Google Scholar]
- Wenzel T. J.NMR Shift Reagents; CRC Press: Boca Raton, FL, 1987. [Google Scholar]
- Welch C. J. Evolution of chiral stationary phase design in the Pirkle laboratories. J. Chromatogr. A 1994, 666, 3–26. 10.1016/0021-9673(94)80367-6. [DOI] [Google Scholar]
- Lacour J. Chiral hexacoordinated phosphates: From pioneering studies to modern uses in stereochemistry. C. R. Chim. 2010, 13, 985–997. 10.1016/j.crci.2010.02.009. [DOI] [Google Scholar]
- Wypchlo K.; Duddeck H. Chiral recognition of olefins by 1H NMR spectroscopy in the presence of a chiral dirhodium complex. Tetrahedron: Asymmetry 1994, 5, 27–30. 10.1016/S0957-4166(00)80477-2. [DOI] [Google Scholar]
- Martinez C. R.; Iverson B. L. Rethinking the term ″pi-stacking. Chem. Sci. 2012, 3, 2191–2201. 10.1039/c2sc20045g. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J.; Hyun M. H. Concerning the role of face-to-edge π-π interactions in chiral recognition. J. Chromatogr. A 1992, 607, 126–30. 10.1016/0021-9673(92)87062-D. [DOI] [Google Scholar]
- Pirkle W. H.; Welch C. J. Use of simultaneous face to face and face to edge π-π interactions to facilitate chiral recognition. Tetrahedron: Asymmetry 1994, 5, 777–780. 10.1016/S0957-4166(00)86225-4. [DOI] [Google Scholar]
- Gindy M. E.; Feuston B.; Glass A.; Arrington L.; Haas R. M.; Schariter J.; Stirdivant S. M. Stabilization of ostwald ripening in low molecular weight amino lipid nanoparticles for systemic delivery of siRNA therapeutics. Mol. Pharmaceutics 2014, 11, 4143–4153. 10.1021/mp500367k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.