

Abstract
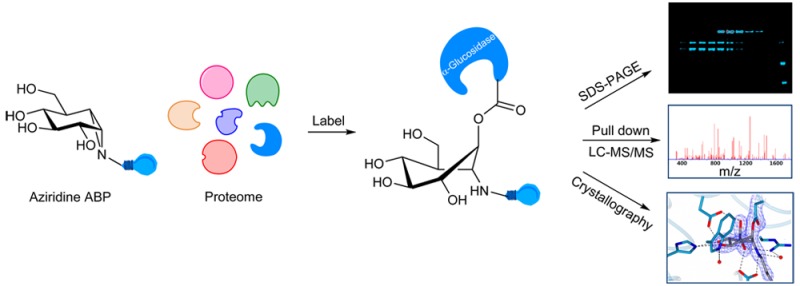
The development of small molecule activity-based probes (ABPs) is an evolving and powerful area of chemistry. There is a major need for synthetically accessible and specific ABPs to advance our understanding of enzymes in health and disease. α-Glucosidases are involved in diverse physiological processes including carbohydrate assimilation in the gastrointestinal tract, glycoprotein processing in the endoplasmic reticulum (ER), and intralysosomal glycogen catabolism. Inherited deficiency of the lysosomal acid α-glucosidase (GAA) causes the lysosomal glycogen storage disorder, Pompe disease. Here, we design a synthetic route for fluorescent and biotin-modified ABPs for in vitro and in situ monitoring of α-glucosidases. We show, through mass spectrometry, gel electrophoresis, and X-ray crystallography, that α-glucopyranose configured cyclophellitol aziridines label distinct retaining α-glucosidases including GAA and ER α-glucosidase II, and that this labeling can be tuned by pH. We illustrate a direct diagnostic application in Pompe disease patient cells, and discuss how the probes may be further exploited for diverse applications.
Short abstract
α-Glucosidases control food digestion, protein folding, and glycogen turnover. Designer probes report on tissue- and pH-specific α-glucosidase activity and reveal loss of α-glucosidase activity in Pompe disease patient material.
Introduction
Lysosomal α-glucosidase (GAA, α-1,4-glucosidase, acid maltase) (E.C. 3.2.1.20) is a retaining α-glucosidase, which has been classified into CAZy family GH31.1−4 Following processing within endoplasmic reticulum the 110 kDa (952 AA) precursor is transported to the lysosomes, where it is modified to active 76 and 70 kDa isoforms.5,6 Within the lysosomes, GAA catalyzes the degradation of glycogen via a general acid/base catalyzed double displacement mechanism,7,8 releasing a molecule of α-glucose with net retention of stereochemistry at its anomeric center (Figure 1a). Deficiency in GAA leads to the glycogen storage disease type II, known as Pompe disease.9 In Pompe patients, intralysosomal glycogen accumulation causes progressive muscle weakness in heart and skeletal muscles and also affects the liver and nervous system.10 Different clinical forms of Pompe disease are usually discerned based on age of onset. The infantile-onset form manifests at 4 to 8 months and, when untreated, results in death in the first years of life.11 Later onset forms generally progress more slowly and are characterized by progressive decrease in muscle strength in the legs followed by smaller muscles in the trunk and arms and ultimately to fatality through respiratory failure.12 The severity of the Pompe disease and its progress correlates with the extent of enzyme loss. Pompe disease is currently treated by chronic intravenous administration of recombinant human GAA (rGAA, alglucosidase alfa; Myozyme). Impressive delay of fatal symptoms by enzyme replacement therapy (ERT) is observed in infantile Pompe disease patients,13 but treatment of late onset disease requires very large amounts of therapeutic enzyme due to poor correction of muscle cell pathology.14
Figure 1.

The mechanism of action of retaining α-glucosidases allows the development of activity-based probes. (a) Koshland double-displacement mechanism employed by retaining α-glucosidases. (b) α-Glucose-configured N-alkyl cyclophellitol aziridines as mechanism-based irreversible retaining α-glucosidase inhibitors (1, 2) and probes (3–6).
The identification of GAA activity in living cells or tissues is challenging. We have previously shown that activity-based protein profiling (ABPP) is an effective methodology to quantify β-glucosidases responsible for the lysosomal storage disorder, Gaucher disease.15,16 Linking “reporter moieties” to the aziridine nitrogen in the retaining β-glucosidase inhibitor, cyclophellitol aziridine, yielded in-class broad-spectrum retaining β-glucosidase probes. These probes label, besides lysosomal glucosylceramidase (GBA-the enzyme deficient in Gaucher disease), also neutral glucosylceramidase (GBA2), broad-specificity β-glucosidase (GBA3), and lactase phloridzin hydrolase (LPH).16−21 Based on these findings we hypothesized that the corresponding α-glucopyranose configured, nitrogen-substituted cyclophellitol aziridines (Figure 1b) would be effective ABPs for specific labeling of GH31 retaining α-glucosidases, besides GAA also ER α-glucosidase II (GANAB, a crucial enzyme in the quality control of newly formed glycoproteins in the ER) and intestinal α-glucosidases involved in food processing. Here we report the successful development of such in-class GH31 α-glucosidase ABPs. We demonstrate covalent labeling by mass spectrometry and X-ray crystallography of a representative bacterial GH31 enzyme homologous to GAA. We reveal that α-glucosidases are labeled in situ by our ABPs and that the absence of GAA in Pompe disease cells is readily demonstrated in a diagnostic manner.
Results
Synthesis of Mechanism-Based Inhibitors and Activity-Based Probes
As the first research objective we set out to develop an efficient route of synthesis for the preparation of α-configured cyclophellitol aziridine 1 (Figure 2). During our previous work on retaining β-glucosidase probes we developed an optimized route toward cyclohexene 8, the precursor in the total synthesis, by Madsen and co-workers, of the natural product and retaining β-glucosidase inhibitor, cyclophellitol.22 We reasoned that an iodocyclization scheme, with an appropriate nitrogen nucleophile delivered to the alkene-derived iodonium ion from the allylic alcohol position, would yield an appropriately configured 2-amino-1-iodo species for subsequent intramolecular cyclization in a stereospecific fashion. For this purpose, the benzyl protective groups in 8 were removed by Birch reduction, after which the 4,6-benzylidene (glucopyranose numbering) was installed to give 9. The allylic alcohol in 9 proved the most reactive of the two secondary alcohols toward trichloroacetonitrile in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the catalytic base, providing imidate 10 as the major product in the presence of small amounts of the bis-imidate. Subsequent key iodocyclization afforded in a stereospecific fashion cyclic imidate 11. Both cyclic imidate and benzylidene acetal were hydrolyzed under acidic conditions, and the resulting crude primary amine 12 was treated with sodium bicarbonate giving aziridine 1. The aziridine nitrogen in 1 was alkylated with 1-iodo-7-azidoheptane to yield 2, onto which and by means of copper(I)-catalyzed azide–alkyne [2 + 3] cycloaddition either a BODIPY-green dye, a BODIPY-red dye, a Cy5 dye, or a biotin was conjugated, to give ABPs 3, 4, 5 and 6, respectively (Figure 2). 1,6-epi-Cyclophellitol 7 was prepared by stereoselective epoxidation of 9 with 3-chloroperbenzoic acid (mCPBA) and deprotection of benzylidene acetal group of the resulting 17 by Pearlman’s catalyst hydrogenation. As reference competitive inhibitors, we used deoxynojirimycin derivatives 18–22 and maltose 23 (Figure S1).23
Figure 2.

Synthesis of the cyclophellitol aziridine inhibitors 1, 2, probes 3–6 and 1,6-epi-cyclophellitol 7. Reagents and conditions: (a) (i) Li, NH3, THF, −60 °C, 57%; (ii) PhCH(OMe)2, CSA, DMF, 61%. (b) CCl3CN, DBU, DCM, 0 °C. (c) NaHCO3, I2, H2O, two step yield 41%. (d) 37% HCl aq, dioxane. (e) NaHCO3, MeOH, two step yield 63%. (f) 1-Azido-8-iodooctane, K2CO3, DMF, 80 °C, 39%. (g) 13, 14, 15, or 16, CuSO4, sodium ascorbate, DMF, 38% 3, 11% 4, 24% 5, 23% 6. (h) mCPBA, DCM, 40 °C, 44%. (i) Pd(OH)2/C, H2, MeOH, 68%.
In Vitro Inhibition and Labeling of Recombinant Human GAA
We first tested the inhibitory properties of compounds 1–7 (Figure 2) toward rGAA (Myozyme, Genzyme) at pH 4.0 using 4-methylumbelliferyl-α-glucose as substrate (Figure 3a). All compounds proved to be potent rGAA inhibitors with apparent IC50 values in the nanomolar range. The most potent inhibitor of the series is cyclophellitol aziridine 1, with epoxide 7 and N-alkyl aziridine 2 within the same range, albeit slightly weaker (Figure 3a). Fluorescent ABPs 3–5 all showed about 10-fold increased apparent IC50 values compared to 1, and biotin-conjugated ABP 6 a further 2-fold increase. In situ enzyme inhibition within human fibroblasts exhibited similar potency, except for biotin compound 6 (IC50 > 10 μM), which apparently is the least cell-permeable of the series. The pH optimum of 5.0 found for labeling of 110 kDa rGAA differed slightly from that of enzymatic activity toward 4MU-α-glucose pH 4.0 (Figure 3b). When rGAA was preincubated for 30 min with 10 μM 1, 2, and 4–7, followed by labeling with compound 3, green fluorescent labeling of rGAA by compound 3 was abrogated (Figure 3c). Likewise, the presence of high concentrations of the substrates 4MU-α-glucose and maltose reduced labeling by compound 3 as does prior denaturation of rGAA by incubation with 2% SDS. Inhibitors 1–7 reacted too fast for kinetics analysis: for instance, time dependent labeling of rGAA by 100 nM ABP 3 at 4 and 37 °C demonstrated that most of the enzyme could be labeled within 2 min (Figure S2a).
Figure 3.

In vitro inhibition and labeling of α-glucosidases. (a) Inhibition of recombinant GAA. Apparent IC50 values (extrapolated with one phase exponential association) are the mean of three separate experiments. Error ranges depict standard deviation. (b) Labeling of rGAA with compound 3 at various pH values as compared to activity toward 4-MU-α-d-glucopyranose. Error bars represent standard deviation. (c) ABP labeling of rGAA with 3 competed with compounds 1, 2, and 4–7. (d) Stereoscopic view of the CjAgd31B active site in complex with compound 2, showing covalent link to CjAgd31B enzymatic nucleophile Asp412, and H-bonding interactions to neighboring residues. Electron density is REFMAC maximum-likelihood/σA-weighted 2 Fo – Fc synthesis contoured at 0.49 electrons per Å3.
To establish the mechanism-based mode of action of our probes, we took advantage of a bacterial homologue of GAA from CAZy family GH31, the α-glucosidase from Cellvibrio japonicus, CjAgd31B, which is readily amenable to structure studies of GH31 ligand complexes and displays 27% identity to the human GAA enzyme over 615 amino acids including absolute conservation of the active center “-1” glucosyl site.24 The 1.95 Å resolution structure of CjAgd31B (Figure 3d) soaked with cyclophellitol aziridine 2 revealed unambiguous electron density showing covalent binding of the ring opened cyclophellitol aziridine β-linked to the active site nucleophile residue (Asp412; equating to Asp518 of GAA and Asp542 of GANAB). Apart from demonstrating irrevocably the mechanism-based mode of action of our probes, the GH31 complex with 2 is consistent with the reaction pathway employed by GH31 in processing α-glucosidic linkages. Substrates are proposed to bind in a 4C1 conformation with catalysis occurring via a 4H3 oxacarbenium ion like transition state to a covalent adduct in 1S3 skew-boat conformation.25,26 This 1S3 conformation of the enzyme–inhibitor adduct is well captured in the complex shown in Figure 3d. Similarly, the 1S3 conformation of the enzyme–inhibitor 1 adduct was also well captured in 1.85 Å resolution (Figure S2b).
ABP Labeling of Multiple α-Glucosidases in Homogenates of Fibroblasts
Having established the expected covalent inactivation we next examined ABP labeling of α-glucosidases in a more complex and physiologically relevant biological mixture of proteins. For this, we prepared homogenates of cultured fibroblasts and exposed these to 1 μM ABP 5 at various pH values. Incubation with ABP 5 at pH 4.0 gave clean labeling of what appeared to be the mature lysosomal forms of GAA (two bands at around 70 kDa). At higher pH values an additional protein with a molecular weight of around 100 kDa was observed (Figure 4a). GAA precursor amounts are low in cells and are—as with the mature form—optimally active at low pH. We therefore thought it unlikely that the protein labeled at 100 kDa would be the GAA pro-form. Rather, we envisaged that this band would correspond to another α-glucosidase, possibly ER α-glucosidase II. To establish, unambiguously, the nature of the 70 kDa proteins identified at acidic pH as well as the 100 kDa protein seen at neutral pH, we used affinity purification and chemical proteomics of fibroblast homogenates treated with biotin-conjugated ABP 6. A lysate of fibroblasts was incubated at pH 4.0 and pH 7.0 with ABP 6, in either the presence or absence of pretreated ABP 3. Next, biotinylated proteins were enriched by pull-down using streptavidin-coated magnetic beads. Loaded streptavidin beads were split for in-gel digestion and on-bead digestion. Captured proteins for subsequent in-gel digestion were released in Laemmli buffer, resolved on SDS–PAGE and visualized by silver staining (Figure 4b). Three distinct bands were obtained that all could be competed for by inclusion of α-cyclophellitol aziridine 3. Two bands with apparent molecular masses of around 70 kDa are most prominent in the pull-down at pH 4.0, and one band at approximately 100 kDa was the predominant species labeled at pH 7.0. Tryptic digestion was performed on these three protein bands, and the resulting tryptic peptides were analyzed by nanoscale liquid chromatography coupled to tandem mass spectrometry (nano-LCMS/MS). The proteins were identified via matching of the obtained peptide sequences against the Mascot database. In this manner, the ∼70 kDa proteins were identified as the two mature forms (70 and 76 kDa) of GAA (Figure 4c). The labeled 100 kDa protein proved to be GANAB and isoform 2 of GANAB, the known retaining ER α-glucosidase II (Figure 4c).
Figure 4.

Labeling of multiple α-glucosidases and their identification by proteomics. (a) Labeling of proteins in fibroblast lysate with compound 5. (b) Workflow of pull-down proteomics experiments. (c) In-gel digestion silver staining and identification of target proteins modified by biotin-ABP 6. (d) Glycosidase identification with Mascot score and emPAI values after on-bead pull-down and processing of fibroblast lysate with biotin-ABP 6.
Using on-bead trypsin digestion very similar results were obtained. GAA in the pH 4.0 incubation and GANAB in the pH 7.0 incubation topped the exponentially modified protein abundance index (emPAI), a relative quantitation of the proteins in a mixture based on protein coverage by the peptide matches in a database search result, and protein scores from Mascot search report (Figure 4d and Figure S3). Comparable experiments with mouse liver lysates (Figures S4, S5) resulted in identification of GAA and GANAB as the major targets of biotin-aziridine ABP 6, matching the fluorescent bands on SDS–PAGE. Using a final concentration of 10 μM of biotin-aziridine 6 in the affinity purification experiments, we also observed enrichment of a protein with apparent molecular mass of 60–65 kDa (Figure S4). Proteomics analysis identified this protein as GBA, the lysosomal acid β-glucosidase deficient in Gaucher disease. To corroborate this observation we determined possible inhibition of enzymatic activity of pure GBA by compounds 1–7. Indeed, GBA is also inhibited by all of the prepared compounds with apparent IC50 values in the range of 592.8 nM to 155.3 μM (Table S1). For comparison, the corresponding β-configured cyclophellitol aziridine JJB34315 was a far more potent inhibitor toward GBA with 500-fold lower apparent IC50 values than α-aziridine 4 JJB347 in the same test.
Labeling of α-Glucosidases Present in Murine Gastrointestinal Tracts
Given the observed high affinity labeling with our ABPs of both GAA and GANAB, we next examined labeling of proteins in extracts from intestines, tissue known to contain additional retaining GH31 α-glucosidases. For this purpose, mouse intestine was freshly collected, food remains and bacteria were removed by rinsing, and the tissue was lysed in KPi-Triton buffer using sonication. The lysate was incubated with biotinylated compound 6, and bound proteins were analyzed by gel electrophoresis and chemical proteomics as before (Figures S6, S7). In this way we observed, in addition to biotin-ABP 6 labeling of GAA and GANAB, labeling of sucrase-isomaltase (Sis) and maltase-glycoamylase (MGAM), two retaining α-glucosidases expressed specifically in intestinal tissue. In addition, the retaining β-glucosidases, lactase (Lct) and GBA, were identified. The findings were recapitulated by labeling lysates of pull-down and supernatant samples with ABPs 3 and 6, protein separation by gel electrophoresis, and fluorescence imaging of the wet gel slabs, followed by transferring protein to polyvinylidene fluoride (PVDF) membrane for Western blot analysis with HRP-streptavidin. Fluorescent bands, chemiluminescence bands, and silver stain protein bands matched well, and the expected molecular masses for GAA, GANAB, Sis, MGAM, Lct, and GBA were observed (Figure S6).
Labeling and Concomitant Inhibition of α-Glucosidases in Intact Cells
Prompted by the effective in vitro labeling of GH31 α-glucosidases in cell and tissue extracts, we studied the labeling of enzymes in intact cultured fibroblasts with ABPs 3 and 5. Fibroblasts were exposed to 100 nM ABP 5 for various times (from 10 min to 4 h) by including these in the culture medium. Subsequently, cells were extensively washed and harvested, and lysates were prepared at 4 °C and then separated by SDS–PAGE followed by fluorescence scanning of the wet gel slabs (Figure 5a left). Fluorescent labeling of GAA and GANAB was prominent in blue bands. Of note, no GBA was concomitantly labeled in situ, illustrating the far lower affinity of the α-glucopyranose ABP to label this β-glucosidase, but GBA was labeled by excess of ABP 3 under in vitro conditions (Figure 5a right). We added during the procedure an excess of ABP 3 to exclude artificial labeling of enzymes by nonreacted free ABP 5 during the process of cell lysis. This addition did not change the result, demonstrating that labeling of GAA and GANAB truly occurs in situ.
Figure 5.

In situ labeling of GAA and GANAB in living fibroblasts. (a) Time-dependent labeling of GAA and GANAB in fibroblasts by ABP 5in situ and ABP 3in vitro. (b) Chloroquine blocks in situ GAA labeling.
GAA requires an acid pH for labeling by the α-glucopyranose ABP (Figure 3a). We therefore investigated the effect of chloroquine, an agent that raises intralysosomal pH.27 Fibroblasts were incubated with either increasing amount of chloroquine (10, 25, and 50 μM) for 3 h or increasing treatment time (1, 2, and 3 h), after which ABP 5 was added to the medium. After the time points, cells were washed, harvested, and analyzed on labeled protein. As shown in Figure 5b, the presence of chloroquine abolished in situ labeling of GAA but not GANAB.
Diagnostic Application: Probing for GAA Activity in Pompe Disease Patient Material
Finally, we studied the value of our ABPs for diagnosis of Pompe disease. Fibroblasts of normal individuals and Pompe disease patients suffering from the infantile and adult variants of disease were cultured. Cell lysates were labeled with 0.5 μM of ABP 3 for 30 min at pH 4.0 and pH 7.0. Gel electrophoresis of the denatured protein content and subsequent fluorescence imaging and immunoblots revealed a prominent distinction between material from normal persons and Pompe disease patients (Figure 6 and Figure S8). Marked absence of fluorescently labeled mature 70/76 kDa GAA with concomitant normal levels of GANAB was only observed for the patient fibroblasts. This finding demonstrates the potential of our α-glucosidase ABPs in laboratory diagnosis of Pompe disease.
Figure 6.

In vitro ABP labeling and Western blot detection of α-glucosidases in wild type and Pompe fibroblasts. (a). In vitro GAA labeling at pH 4.0 with 3, followed by Western blot detection of GAA in various fibroblast lysates, containing wild type or mutant (Pompe) GAA. (b). In vitro GANAB labeling at pH 7.0 with 3 followed by Western blot detection of GAA in various fibroblast lysates, containing wild type or mutant (Pompe) GAA.
Discussion
Activity-based protein profiling (ABPP) has emerged in the past 15 years as a powerful technique to identify enzymes and to study their activity in the context of the physiological processes they partake in, both in vitro and in situ. In the first instance developed for serine hydrolases and cysteine proteases,28,29 ABPs can on paper be designed for any enzyme, provided that a covalent enzyme–substrate adduct emerges during enzyme action. Retaining glycosidases that employ a Koshland two-step double displacement mechanism fulfill this requirement, and we have reported previously on the versatility of the natural product, cyclophellitol, as a scaffold for activity-based glycosidase probe design. In this work, we capitalize on this generic design principle by the development of a set of α-configured cyclophellitol aziridines as in-class GH31 retaining α-glucosidase ABPs. Our probes label GH31 retaining α-glucosidases in a tissue-dependent fashion and allow detection and identification of these by in-gel fluorescence, by cell imaging, and by chemical proteomics. They are highly selective toward GH31 retaining α-glucosidases when applied in the appropriate concentration and even at higher concentrations show little to no cross-reactivity, apart from labeling the retaining β-glucosidase, GBA. The ABPs label subcellular α-glucosidases optimally at the pH at which the α-glucosidases display maximal enzymatic activity. Thus, whereas GAA in fibroblasts is maximally labeled at pH 4.0–5.0, GANAB, or ER-α-glucosidase II, is optimally labeled at neutral pH. This finding underscores that our probes report on functional enzymes, rather than protein levels, an observation that is corroborated by the X-ray structure we obtained of CjAgd31B rGAA, in which ABP precursor 2 has reacted with the active site nucleophile, Asp412.
The tissue-specific and pH-dependent mode of action of our ABPs allows for probing each of the targeted α-glucosidases independently and within their physiological context, as well as in health and disease. The diagnostic value of our probes is demonstrated in the assessment of the lack of GAA activity in infantile and adult Pompe disease tissue (Figure 6 and Figure S8). Our in-class broad-spectrum GH31 α-glucosidase probes may also find use in the discovery of inhibitors specific for either GAA or GANAB in a competitive ABPP assay. Selective inhibitors for GAA or GANAB would be of interest: GAA inhibitors for pharmacological chaperone discovery in the context of Pompe disease and GANAB inhibitors for antiviral or anticancer drug discovery (GANAB activity being a crucial factor in the quality control of nascent N-linked glycoproteins). We performed an initial competitive ABPP in which fibroblasts expressing GAA and GANAB were first treated with varying concentrations of the iminosugars 18–22 and maltose 23 followed by incubation at both pH 4.0 and pH 7.0 with ABP 5. As can be seen in Figure S9, these compounds efficiently inhibit both enzymes. Indeed no selective inhibitors for either enzyme are known, and our ABPs may be of help in identifying such compounds. Our ABPs finally also efficiently target and identify intestinal α-glucosidases, which are key players in glucose assimilation and the targets of the antidiabetic drugs, miglitol and acarbose. The ability to also study these intestinal enzymes in detail in their physiological surroundings holds promise for the future identification of more effective (and selective with respect to GAA/GANAB inhibition) inhibitors, which may come from natural sources and become part of nutraceutical regimes.
Acknowledgments
We thank the China Scholarship Council (CSC, PhD Grant to J.J.), The Netherlands Organization for Scientific Research (NWO, ChemThem Grant to J.M.F.G.A. and H.S.O.), and the European Research Council (ErC-2011-AdG-290836, to H.S.O., and ERC-2012-AdG-322942, to G.J.D.) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00057.
Author Contributions
J.J. and C.-L.K. contributed equally to this work. J.J. and C.F. synthesized the inhibitors and probes under the guidance of J.D.C.C. and G.A.v.d.M. J.J., C.-L.K., E.v.M., and W.W.K. conducted the enzyme inhibition and biochemistry assays under the guidance of R.G.B. L.W. and G.J.D. designed and performed the X-ray diffraction experiments. J.J., C.-L.K., and B.I.F. performed the chemical proteomics experiments. H.S.O. and J.M.F.G.A. conceived of the idea, supervised the project, and with J.J. and G.J.D. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hoefsloot L. H.; Hoogeveen-Westerveld M.; Reuser A. J.; Oostra B. A. Characterization of the human lysosomal alpha-glucosidase gene. Biochem. J. 1990, 272, 493–497. 10.1042/bj2720493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S.; Hiromi K.; Minamiura N.; Ohnishi M.; Shimomura T.; Suga K.; Suganuma T.; Tanaka A.; Tomioka S.-i.; Yamamoto T. Quantitative Study on Anomeric Forms of Glucose Produced by α-Glucosidases. J. Biochem. 1979, 85, 1135–1141. [PubMed] [Google Scholar]
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. J.; Williams S. J. Carbohydrate-active enzymes: sequences, shapes, contortions and cells. Biochem. Soc. Trans. 2016, 44, 79–87. 10.1042/BST20150186. [DOI] [PubMed] [Google Scholar]
- Moreland R. J.; Jin X.; Zhang X. K.; Decker R. W.; Albee K. L.; Lee K. L.; Cauthron R. D.; Brewer K.; Edmunds T.; Canfield W. M. Lysosomal Acid α-Glucosidase Consists of Four Different Peptides Processed from a Single Chain Precursor. J. Biol. Chem. 2005, 280, 6780–6791. 10.1074/jbc.M404008200. [DOI] [PubMed] [Google Scholar]
- Wisselaar H. A.; Kroos M. A.; Hermans M. M.; van Beeumen J.; Reuser A. J. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J. Biol. Chem. 1993, 268, 2223–2231. [PubMed] [Google Scholar]
- Lee S. S.; He S.; Withers S. G. Identification of the catalytic nucleophile of the Family 31 alpha-glucosidase from Aspergillus niger via trapping of a 5-fluoroglycosyl-enzyme intermediate. Biochem. J. 2001, 359, 381–386. 10.1042/bj3590381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. Stereochemistry and mechanism of enzymetic reactions. Biol. Rev. 1953, 28, 416–436. 10.1111/j.1469-185X.1953.tb01386.x. [DOI] [Google Scholar]
- Hers H. G. alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuser A. J.; Kroos M. A.; Hermans M. M.; Bijvoet A. G.; Verbeet M. P.; Van Diggelen O. P.; Kleijer W. J.; Van der Ploeg A. T. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995, 18 (Suppl. 14), S61–S69. 10.1002/mus.880181414. [DOI] [PubMed] [Google Scholar]
- Kishnani P. S.; Hwu W. L.; Mandel H.; Nicolino M.; Yong F.; Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006, 148, 671–676. 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- Winkel L. P.; Hagemans M. L.; van Doorn P. A.; Loonen M. C.; Hop W. J.; Reuser A. J.; van der Ploeg A. T. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J. Neurol. 2005, 252, 875–884. 10.1007/s00415-005-0922-9. [DOI] [PubMed] [Google Scholar]
- Kishnani P. S.; Corzo D.; Nicolino M.; Byrne B.; Mandel H.; Hwu W. L.; Leslie N.; Levine J.; Spencer C.; McDonald M.; Li J.; Dumontier J.; Halberthal M.; Chien Y. H.; Hopkin R.; Vijayaraghavan S.; Gruskin D.; Bartholomew D.; van der Ploeg A.; Clancy J. P.; Parini R.; Morin G.; Beck M.; De la Gastine G. S.; Jokic M.; Thurberg B.; Richards S.; Bali D.; Davison M.; Worden M. A.; Chen Y. T.; Wraith J. E. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007, 68, 99–109. 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- van der Ploeg A. T.; Clemens P. R.; Corzo D.; Escolar D. M.; Florence J.; Groeneveld G. J.; Herson S.; Kishnani P. S.; Laforet P.; Lake S. L.; Lange D. J.; Leshner R. T.; Mayhew J. E.; Morgan C.; Nozaki K.; Park D. J.; Pestronk A.; Rosenbloom B.; Skrinar A.; van Capelle C. I.; van der Beek N. A.; Wasserstein M.; Zivkovic S. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- Witte M. D.; Kallemeijn W. W.; Aten J.; Li K.-Y.; Strijland A.; Donker-Koopman W. E.; van den Nieuwendijk A. M. C. H.; Bleijlevens B.; Kramer G.; Florea B. I.; Hooibrink B.; Hollak C. E. M.; Ottenhoff R.; Boot R. G.; van der Marel G. A.; Overkleeft H. S.; Aerts J. M. F. G. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat. Chem. Biol. 2010, 6, 907–913. 10.1038/nchembio.466. [DOI] [PubMed] [Google Scholar]
- Kallemeijn W. W.; Li K. Y.; Witte M. D.; Marques A. R.; Aten J.; Scheij S.; Jiang J.; Willems L. I.; Voorn-Brouwer T. M.; van Roomen C. P.; Ottenhoff R.; Boot R. G.; van den Elst H.; Walvoort M. T.; Florea B. I.; Codee J. D.; van der Marel G. A.; Aerts J. M.; Overkleeft H. S. Novel activity-based probes for broad-spectrum profiling of retaining beta-exoglucosidases in situ and in vivo. Angew. Chem., Int. Ed. 2012, 51, 12529–12533. 10.1002/anie.201207771. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Beenakker T. J. M.; Kallemeijn W. W.; van der Marel G. A.; van den Elst H.; Codee J. D. C.; Aerts J. M. F. G.; Overkleeft H. S. Comparing Cyclophellitol N-Alkyl and N-Acyl Cyclophellitol Aziridines as Activity-Based Glycosidase Probes. Chem. - Eur. J. 2015, 21, 10861–10869. 10.1002/chem.201501313. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Beenakker T. J. M.; Murray B.; Scheij S.; Kallemeijn W. W.; Boot R. G.; Verhoek M.; Donker-Koopman W. E.; Ferraz M. J.; van Rijssel E. R.; Florea B. I.; Codee J. D. C.; van der Marel G. A.; Aerts J. M. F. G.; Overkleeft H. S. Potent and selective activity-based probes for GH27 human retaining alpha-galactosidases. J. Am. Chem. Soc. 2014, 136, 11622–11625. 10.1021/ja507040n. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Kallemeijn W. W.; Wright D. W.; van den Nieuwendijk A. M. C. H.; Rohde V. C.; Folch E. C.; van den Elst H.; Florea B. I.; Scheij S.; Donker-Koopman W. E.; Verhoek M.; Li N.; Schurmann M.; Mink D.; Boot R. G.; Codee J. D. C.; van der Marel G. A.; Davies G. J.; Aerts J. M. F. G.; Overkleeft H. S. In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 alpha-L-fucosidases. Chem. Sci. 2015, 6, 2782–2789. 10.1039/C4SC03739A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams B. T.; Niccoli S.; Chowdhury M. A.; Esarik A. N.; Lees S. J.; Rempel B. P.; Phenix C. P. N-Alkylated aziridines are easily-prepared, potent, specific and cell-permeable covalent inhibitors of human beta-glucocerebrosidase. Chem. Commun. 2015, 51, 11390–11393. 10.1039/C5CC03828F. [DOI] [PubMed] [Google Scholar]
- Alcaide A.; Trapero A.; Perez Y.; Llebaria A. Galacto configured N-aminoaziridines: a new type of irreversible inhibitor of beta-galactosidases. Org. Biomol. Chem. 2015, 13, 5690–5697. 10.1039/C5OB00532A. [DOI] [PubMed] [Google Scholar]
- Hansen F. G.; Bundgaard E.; Madsen R. A Short Synthesis of (+)-Cyclophellitol. J. Org. Chem. 2005, 70, 10139–10142. 10.1021/jo051645q. [DOI] [PubMed] [Google Scholar]
- Ghisaidoobe A.; Bikker P.; de Bruijn A. C.; Godschalk F. D.; Rogaar E.; Guijt M. C.; Hagens P.; Halma J. M.; Van’t Hart S. M.; Luitjens S. B.; van Rixel V. H.; Wijzenbroek M.; Zweegers T.; Donker-Koopman W. E.; Strijland A.; Boot R.; van der Marel G.; Overkleeft H. S.; Aerts J. M.; van den Berg R. J. Identification of potent and selective glucosylceramide synthase inhibitors from a library of N-alkylated iminosugars. ACS Med. Chem. Lett. 2011, 2, 119–123. 10.1021/ml100192b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J.; Izumi A.; Hemsworth G. R.; Davies G. J.; Brumer H. Structural enzymology of Cellvibrio japonicus Agd31B protein reveals alpha-transglucosylase activity in glycoside hydrolase family 31. J. Biol. Chem. 2012, 287, 43288–43299. 10.1074/jbc.M112.416511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. J.; Planas A.; Rovira C. Conformational Analyses of the Reaction Coordinate of Glycosidases. Acc. Chem. Res. 2012, 45, 308–316. 10.1021/ar2001765. [DOI] [PubMed] [Google Scholar]
- Speciale G.; Thompson A. J.; Davies G. J.; Williams S. J. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole B.; Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 1981, 90, 665–669. 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Patricelli M. P.; Cravatt B. F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14694–14699. 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum D.; Medzihradszky K. F.; Burlingame A.; Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 2000, 7, 569–581. 10.1016/S1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.