

Abstract
Quantitative data is presented that shows significant changes in cellular metabolism in a head and neck cancer cell line 30 min after irradiation. A head and neck cancer cell line (UM-SCC-22B) and a comparable normal cell line, normal oral keratinocyte (NOK) were each separately exposed to 10 Gy and treated with a control drug for disrupting metabolism (potassium cyanide; KCN). The metabolic changes were measured live by fluorescence lifetime imaging of the intrinsically fluorescent intermediate metabolite nicotinamide adenosine dinucleotide (NADH) fluorescence; this method is sensitive to the ratio of bound to free NADH. The results indicated a prompt shift in metabolic signature in the cancer cell line, but not in the normal cell line. Control KCN treatment demonstrated expected metabolic fluxes due to mitochondrial disruption. The detected radiation shift in the cancer cells was blunted in the presence of both a radical scavenger and a HIF-1α inhibitor. The HIF-1α abundance as detected by immunohistochemical staining also increased substantially for these cancer cells, but not for the normal cells. This type of live-cell metabolic monitoring could be helpful for future real-time studies and in designing adaptive radiotherapy approaches.
INTRODUCTION
Typical clinical radiotherapy regimens consist of daily scheduled “fractions,” which are delivered over minutes to tens of minutes (1). Tissue oxygenation is a proven key modulator in the effectiveness of radiation therapy (2), and several studies have shown that radiation itself impacts oxygen consumption (3–5). Continued improvements in radiation therapy are important for the large number of cancer patients receiving radiation either as their primary treatment or concomitant with other treatment modalities. Since metabolism and oxygen are intrinsically linked (6, 7), prompt changes in metabolism could cause changes in the tumor microenvironment, such as the availability of oxygen.
Under aerobic conditions, cells usually produce energy via oxidative phosphorylation, since it is considerably more efficient [36 adenosine triphosphate (ATP) per glucose molecule] than glycolytic pathways (2 ATP per glucose molecule); however, many types of cancers utilize glycolysis as their primary source of energy production, even under aerobic conditions. This unique metabolic behavior of tumor cells is known as the “Warburg effect” (8–11). In addition to previously reported metabolic states such as the Warburg effect, it is thought that cancer cells may respond differently metabolically to microenvironmental perturbations (12–14). We hypothesized that, after exposure to radiation, detectable and quantifiable changes in metabolism might occur, very quickly, from the physical effects of the radiation itself. This hypothesis is based on the observation that in vivo, the oxygen level quickly changes after exposure to large doses of radiation, well ahead of any cell death (4).
Nicotinamide adenosine dinucleotide (NADH) is an intrinsically fluorescent intermediate metabolite that has been utilized in a number of studies (15–22). Its fluorescent lifetime (the measured amount of time in the excited state) depends greatly on its chemical state (23, 24). When NADH is bound to a protein complex, its fluorescence lifetime is considerably longer (~2.5 ns) relative to its fluorescence lifetime in the free state (~400 ps) (23, 25). Therefore, bound NADH utilized in the mitochondrial electron transport chain (ETC) exhibits a considerably longer mean fluorescent lifetime, whereas free NADH in the cytosol exhibits a comparatively shorter fluorescent lifetime (21, 26, 27).
This intrinsic fluorescent lifetime contrast for NADH was used here to monitor cellular metabolism, since it allows label-free quantitative assessment of key metabolic changes such as glycolysis (28). Not surprisingly, the ability of time-correlated fluorescence lifetime imaging microscopy (FLIM) (23, 25) to monitor lifetime with a spatial context has been used in a multitude of applications (29–31), including therapeutic research such as characterization of metabolic changes in various breast cancer lines in response to chemotherapeutic drugs (32). FLIM has also been applied in assessing precancerous legions in the head and neck region as well as in the breast (26, 33). A major reason for the growth in FLIM-based metabolic studies has been its unique ability to noninvasively monitor cellular metabolism with high resolution, its ability to quantitatively discriminate between bound and free NADH forms without being influenced by intensity levels and to detect without the mitigating effects of fluorescence variations as seen in traditional fluorescence-based assays (34).
Because a large portion of cancer patients receive radiation therapy as part of their therapeutic regimen, it is important to continue to investigate the biological mechanisms that determine its efficacy. In this study, we demonstrate detectable metabolic fluxes resulting after irradiation of live cells as measured by FLIM. We further assessed the interplay of these metabolic fluxes with changes in oxygen consumption and HIF-1α signaling.
METHODS AND MATERIALS
Cell Culture
Two human head and neck cell lines: UM-SCC-22B cancer cells and normal oral keratinocytes (NOK) were used in this study to compare a cancerous and a noncancerous cell line from human head and neck cancers. UM-SCC-22B cells (35) were obtained from Professor Tom Carey, University of Michigan and cultured in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/dl glucose, 10% fetal bovine serum (FBS), 1% hydrocortisone, penicillin (100 units/ml) and streptomycin (100 mg/ml) (35). NOK cells were a kind gift of Dr. Karl Munger, Tufts University, and cultured in keratinocyte serum-free media supplemented with both EGF and bovine pituitary extract. Cells were maintained as monolayer cultures at 37°C in a humidified atmosphere of 5% CO2.
Irradiation and Treatment with KCN, GSH and Chrysin
Cells were irradiated (10 Gy) with a 320 kVp small animal and cell culture irradiator X-RAD 320 (Precision X-ray Inc., North Branford, CT) with a metal ceramic anode and filtration of 1.5 mm Al + 0.25 mm Cu + 0.75 mm Sn. Cells were plated to approximately 50% confluence in 60 mm glass bottom dishes with 35 mm cover slip wells [cat. no. D60-30-1 5-N; Cellvis (In Vitro Scientific), Mountain View, CA] in 4 ml of cell media. For controls of both the imaging and cell culture conditions and to show the FLIM method was sensitive to metabolic flux, 10 mM potassium cyanide (KCN) was administered to untreated cells during imaging to produce a positive control effect (mitochondrial inhibition), and the negative control cells were simply imaged without any treatment (radiation or drug). A small subset of UM-SCC-22B cells were also irradiated in the presence of 10 mM glutathione for free radical scavenging. Similarly, a separate group of NOK and UM-SCC-22B cells were treated with 50 μM HIF-1α inhibitor chrysin (Santa Cruz Biotechnologyt® Inc., Dallas, TX) before irradiation.
Multiphoton Fluorescence Lifetime Imaging
Time-correlated fluorescence lifetime imaging was performed using a custom multiphoton laser-scanning microscope with SPC-830 Becker & Hickl FLIM hardware (Berlin, Germany) (22, 36) at the Laboratory for Optical and Computational Instrumentation (LOCI; University of Wisconsin-Madison, Madison, WI). Fluorescence of cellular NADH was excited using multiphoton excitation at a wavelength of 740 nm utilizing the Mai Tai® Ti:Sapphire laser (Spectra-Physics®, Santa Clara, CA). FLIM was used to map the NADH-based metabolism of cell lines in vitro subjected to different treatment conditions. Five regions of interest per dish were imaged 30 min after irradiation, and during imaging cells were maintained at 37°C and 5% CO2 in a LiveCell™ stage top incubator. A 450/70 nm emission bandpass filter [Semrock (IDEX Corp.), Lake Forest, IL] was utilized to isolate the NADH autofluorescence. For image analysis, we used Becker and Hickl’s SPCImage program to calculate fluorescence lifetimes using fits to a two-component exponential decay model as follows:
| (1) |
The terms in this model are defined as follows: I(t) is the signal intensity as it decays with time, τ1 is the fluorescence lifetime for free NADH and τ2 is the fluorescence lifetime of protein-bound NADH. The α1, α2 terms fit the intensity of each compartment (free and bound NADH) and can be used to create the mean lifetime for the image τm through the relationship τm=α1τ1+ α2τ2. Shorter lifetimes correspond to a higher concentration of free NADH and therefore are associated with glycolysis. Conversely, longer lifetimes correspond to a higher concentration of protein-bound NADH and therefore are associated with more oxidative phosphorylation. In essence, the τm parameter is simply the weighted average of fluorescent lifetime of both the short and long components.
Seahorse XF Analyzer
Metabolism was also monitored using the Seahorse XFe96 Analyzer (Seahorse Bioscience, North Billerica, MA) to measure oxygen consumption rate (OCR) for assessment of respiratory activity in cells. Cells were plated on two separate 96-well plates (8 rows, 12 columns): one nonirradiated and one 10 Gy irradiated. On each plate, six columns were plated with NOK cells and the other six were plated with UM-SCC-22B cells. Each cell group had three control columns and three columns where the Mito stress test kit was administered. For the Mito stress test kits each cell group had 24 replicates, whereas the controls had 22 replicates. This difference is due to the fact that each corner well was left blank to give baseline oxygen consumption measurements. The Mito stress test kit consists of a series of three injections into the wells of oligomycin, FCCP and rotenone/antimycin. Basal respiration was measured before injections under each treatment condition to assess normal oxygen consumption under each condition. Error bars shown in Fig. 3 are the standard deviations of the measurements at each respective time point. More information on the function of each injection can be found in the XF cell Mito stress test kit user guide. Relative well-to-well cell counts were measured using crystal violet staining followed by resolubilizing with acetic acid according to the protocol set forth by Kueng et al. (37).
FIG. 3.
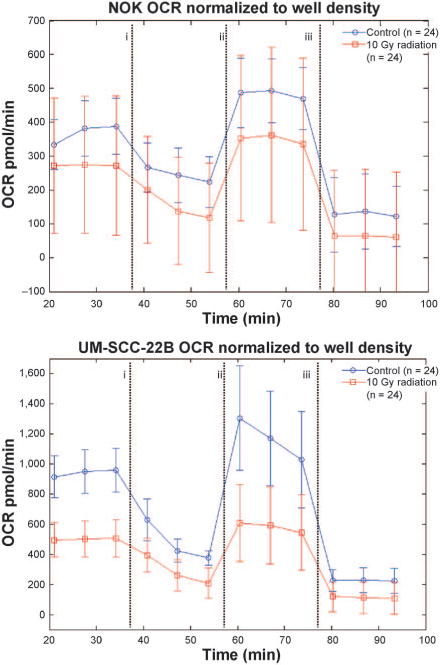
Oxygen consumption rates for each cell group with control (blue line, circles) and irradiated (red line, squares). The vertical dashed lines indicate injection of Mito stress test kit reagents: (i) oligomycin, (ii) FCCP and (iii) rotenone/antimycin A, respectively. Note the difference in OCR scale between NOK and UM-SCC-22B. This suggests that UM-SCC-22B cells are much more energetic as one might expect with more proliferative cells. Irradiation took place at t = 0, the intervening time was due to transport of the samples and the mixing of solutions for each injection port.
Immunohistochemical Staining
To measure HIF-1α expression, cells were plated in two separate 8-well chamber slides, a negative control slide and a 10 Gy irradiated slide. Thirty minutes after irradiation, the slides were washed three times with cold PBS and subsequently fixed in a 10 min cold acetone bath. Cells were then stained with rabbit anti-HIF-1α (cat. no. ab2185; Abcam, Cambridge, UK), dilution 1:500 and Cy3 conjugated goat anti-rabbit IgG (H+L; cat. no. 111-167-003 Jackson Immuno Research Laboratories Inc., West Grove, PA) 1:400 was the secondary antibody (red). Hoechst 33342 (3.75 mg/ml; Sigma-Aldrich®, St. Louis, MO) dilution 1:3,000 was used for nuclear staining (blue). Images were quantified using the image-procesing program FIJI (ImageJ) (38) to integrate HIF-1α intensity normalized to the number of nuclei in each field of view. The color channels in RGB images were split to allow for the application of intensity thresholds, and an intensity threshold also removed artifacts from the images. In all images, there were no counts observed in the green channel. Objects on the edges of images were excluded from quantification, and only objects 25 pixels in size or larger were considered to eliminate extracellular speckle.
Clonogenic Cell Survival Assay
Clonogenic cell survival assay was performed as previously described (39) after irradiation using a 137Cs irradiator (JL Shepherd, San Fernando, CA) at a dose rate of approximately 400 cGy/min. Briefly, cells were plated at low density, irradiated and after 10–15 days colonies containing more than 50 cells were counted and the surviving fraction calculated. Resulting data points were fit to a linear-quadratic model. Each point represents the mean surviving fraction calculated from 6 replicates for each condition. Error bars represent the standard deviation (Fig. 6).
FIG. 6.
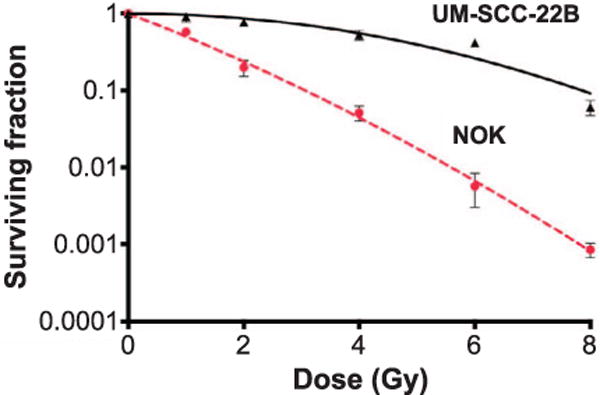
Clonogenic survival over a range of radiation doses for immortalized keratinocyte (NOK, dashed line, circles) and head and neck squamous cell carcinoma (UM-SCC-22B, solid line, triangles) cell lines (n = 6 per condition).
Statistical Analysis
Changes in τm from fluorescence lifetime imaging were assessed for statistical significance using an unpaired, two-sample t test in MATLAB® software (The Mathworks® Inc., Natick, MA). Differences in HIF-1α expression among irradiated and nonirradiated cells were normalized to cell count in each field of view and were assessed for significance using an unpaired two-sample two-tailed t test (MATLAB). For assessment of clonogenic survival after irradiation, the curves were compared using the extra sum-of-squares F test in GraphPad Prism v. 6.0f (La Jolla, CA).
RESULTS
To characterize and demonstrate the metabolic effects of radiation on these representative cancerous and noncancerous cells we divided the two cell lines, the highly proliferative stable UM-SCC-22B and highly stable NOK, into three experimental groups: 1. untreated cells; 2. control cells treated with the mitochondrial inhibitor potassium cyanide (KCN); and 3. irradiatied cells. All cells were imaged using FLIM acquisition integrated over 120 s of imaging time. We used mean fluorescence lifetime (τm) as an indicator of metabolic shifts both before and after treatments and also assessed metabolism by measuring oxygen consumption rates utilizing the Seahorse assay. We also examined HIF-1α expression dynamics in response to radiation.
As expected, τm values dropped significantly in cells subjected to known mitochondrial inhibitors (P < 0.001). Figure 1 shows three sample images of UM-SCC-22B cells that were untreated (left panel), KCN treated (center panel) and irradiated (right panel). When averaged over 35 different fields of view, UM-SCC-22B cells had their τm drop from 711.50 ± 52.65 ps (n = 45 for control group) to 638.08 ± 15.22 ps (n = 40, P < 0.001) when treated with the control drug KCN. This drop occurs because cyanide inhibits mitochondrial complex IV, and this inhibition results in a significant metabolic shift. When NOK cells were subjected to the same treatment with KCN, they showed a similar drop in τm from 789.57 ± 40.66 ps (n = 45 for control group) to 638.96 ± 31.92 ps (n = 40, P < 0.001).
FIG. 1.
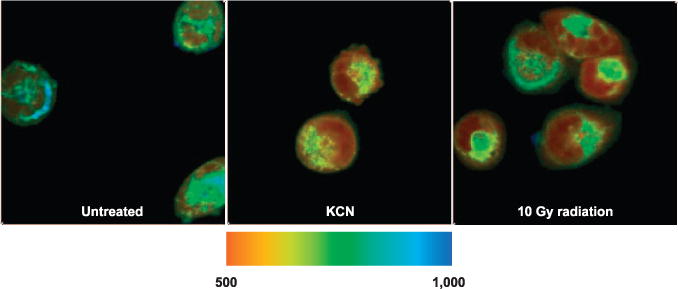
Sample FLIM images of UM-SCC-22B with τm in ps overlaid on each pixel in the field of view. Blue pixels represent longer τm values, whereas red pixels represent shorter τm values. Mean lifetime decreases after both treatment with KCN or 10 Gy dose for the cancer cells.
We then measured the metabolic shift due to a large dose (compared to standard clinical fraction dose) of radiation (10 Gy). The UM-SCC-22B cells responded to radiation with a drop in τm from 711.50 ± 52.65 ps to 617.76 ± 13.68 ps similar to the mitochondrial inhibition therapy (see Table 1 and Fig. 2). Conversely, the τm in 10 Gy irradiated NOK cells changed from 789.57 ± 40.66 ps to 823.02 ± 38.26 ps (n = 40, P < 0.001). The noncancerous NOK cells exhibited a very small but statistically significant change in metabolic signature as measured by FLIM resultant after irradiation, whereas the cancerous UM-SCC-22B cells showed a much larger and directionally opposite change in metabolic signature.
TABLE 1.
The Values for τm Tabulated for UM-SCC-22B and NOK Cells after Treatment with KCN and Radiation along with Each Distribution’s Standard Deviation
| Cell line | Untreated | KCN | 10 Gy radiation |
|---|---|---|---|
| UM-SCC-22B | 711.50 ± 52.65 | 636.94 ± 30.40 | 610.95 ± 21.33 |
| NOK | 789.19 ± 40.57 | 638.96 ± 31.92 | 823.32 ± 38.26 |
Notes. There is a clear contrast in the celluar respective response to radiation. The intra-cell line trends are the key result as opposed to the different baseline τm values.
FIG. 2.
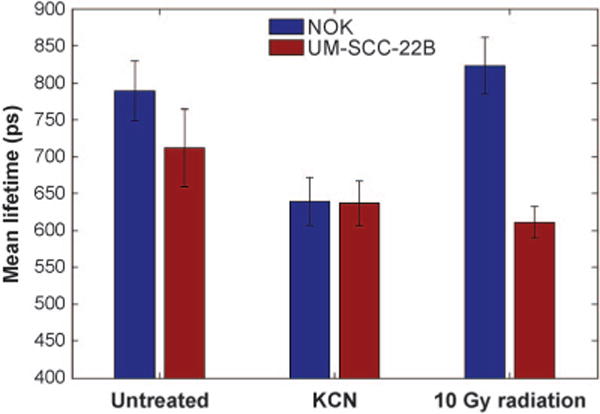
A graph comparing the mean lifetimes of NADH in NOK (blue) and UM-SCC-22B (maroon) cells before and after various treatments. Error bars are the standard deviations for each treatment group. Note the significant drop in τm for UM-SCC-22B cells after irradiation. Again, of note are the trends within cell lines as opposed to baseline measurements.
We also measured the OCR of both irradiated and untreated cells, using the Seahorse XFe96 analyzer (see Fig. 3 for data). The OCR dropped for both cell lines as expected after irradiation (3). From the OCRs, it is clear that the UM-SCC-22B cells are much more energetic than the comparatively less proliferative NOK cells. Furthermore, the resulting changes in oxygen consumption after irradiation in UM-SCC-22B are much more dramatic. The first three points in the OCR curves are the measurements of basal respiration. Approximately 35 min after irradiation, mitochondrial complex V inhibitor oligomycin is added to halt oxygen consumption due to respiration. The rise in OCR at approximately 55 min is due to the administration of FCCP, which permeabilizes the mitochondrial membranes to stimulate maximal respiration. Finally, at approximately 80 min, rotenone (mitochondrial complex I inhibitor) is administered to shut down all mitochondrial activity. This gives a measurement of oxygen consumption due to nonmitochondrial respiration.
It is particularly interesting that the OCR curves for UM-SCC-22B when treated with oligomycin alone (ATP synthase inhibitor), oxygen consumption dropped 573 pmol/min from 949 pmol/min to 376 pmol/min (above 225 pmol/min of nonmitochondrial respiration). When irradiated, oxygen consumption dropped 294 pmol/min from 502 pmol/min to 208 pmol/min (above 112 pmol/min of nonmitochondrial respiration). For unirradiated NOK, the drop in oxygen consumption was 145 pmol/min from 368 pmol/min to 223 pmol/min. After irradiation the drop was 154 pmol/min from 273 pmol/min to 119 pmol/min. This suggests that, for the cancer cells, there is a larger decrease in ATP production from respiration resulting after irradiation compared to the untreated cells.
Exposure to ionizing radiation induces biological effects by the formation of reactive oxygen species (ROS) and free radicals through the radiolysis of water molecules within the cells. These radicals and ROS are responsible for single-and double-strand breaks that damage DNA. Since previously reported studies (40, 41) have shown that free radicals stabilize HIF-1α, and that HIF-1 plays a key role in regulating metabolism, we measured the expression of HIF-1α in both NOK and UM-SCC-22B cells using immunohistochemical staining for HIF-1α antibody stain. Moreover, we observed metabolic signatures in response to radiation by FLIM of both cell lines when treated with radical scavenger glutathione (GSH) and HIF-1α inhibitor chrysin.
For the immunohistochemical staining, HIF-1α fluorescence are indicated with red staining and the nuclei are indicated with blue staining in the images. From the sample images below, after irradiation, it appears that HIF-1α expression increases in the UM-SCC-22B cells; however, no such increase is apparent in the NOK cells. It should be noted that the images shown in Fig. 4 account for considerably less than 0.1% of the total area analyzed.
FIG. 4.
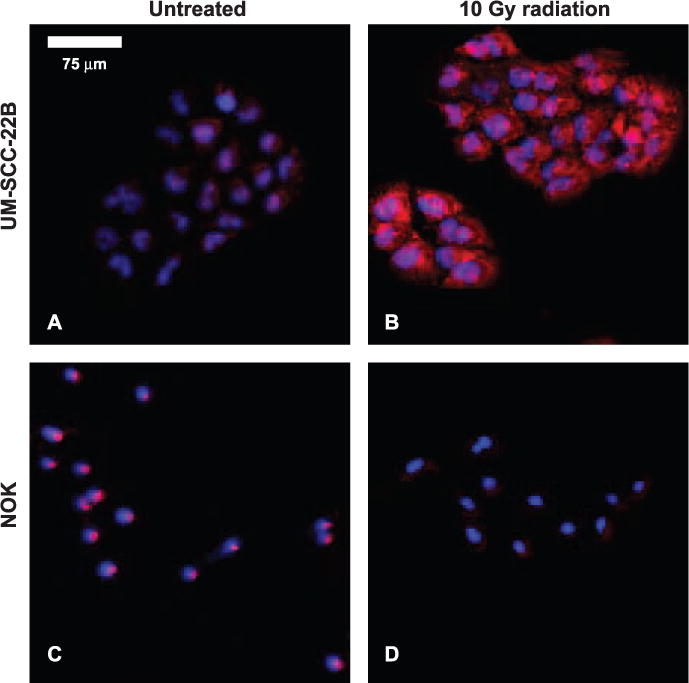
A mosaic of slides, stained for HIF-1α (red) and nuclei (blue) in vitro, of NOK and UM-SCC-22B, both treated with 10 Gy doses and untreated. These images do not represent the same field of view. A minimum size threshold for objects of interest removed the speckle from image quantification.
The raw values for image quantification are given in Supplementary Table S1. After averaging over four fields of view for each case, (NOK: untreated and 10 Gy dose; UM-SCC-22B: untreated and 10 Gy dose) the difference in HIF-1α expression after irradiation was found to be statistically significant between the cell lines (P < 0.05). NOK cells showed a decrease in HIF-1α expression by approximately 40% after irradiation; conversely UM-SCC-22B cells showed an increase in HIF-1α expression per nucleus by 200% (see Supplementary Table S1). It was important to normalize raw HIF-1α pixel counts to the number of nuclei present in the field of view to account for variations in cell count in each field of view. For irradiated UM-SCC-22B malignant cells, metabolic signatures with decreased τm corresponded to an increase in HIF-1α expression, but in the NOK cells, a comparatively slight decrease in HIF-1α corresponded to little change in metabolic signature.
In the parallel FLIM study shown in Fig. 5, NOK cells treated with GSH alone had an observed τm of 830.64 ± 23.13 ps, and NOK cells treated with GSH and 10 Gy dose had an observed τm of 821.67 ± 37.26 ps. This represents no statistically significant change (n = 30, P > 0.05). When treated with chrysin alone, NOK cells had an observed τm of 722.38 ± 21.77 ps, and after irradiation τm was observed to be 746.15 ± 30.42 ps (see Fig. 5A). The UM-SCC-22B cells treated with GSH alone had a τm of 754.73 ± 28.63 ps; after irradiation, τm was observed to be 753.43 ± 23.66 ps. When treated with chrysin alone, UM-SCC-22B cells had an observed τm of 732.24 ± 48.97 ps; after irradiation, τm was observed to be 719.64 ± 45.45 ps. It should be noted that for each UM-SCC-22B cell paired treatment group (drug alone vs. drug with radiation), the observed variations were not found to be statistically significant (n = 30, P > 0.05). However, the slight increase in τm for NOK after irradiation was preserved in the case of control chrysin compared to chrysin with concomitant irradiation (n = 30, P < 0.001). Table 2 gives a concise summary of all these values.
FIG. 5.
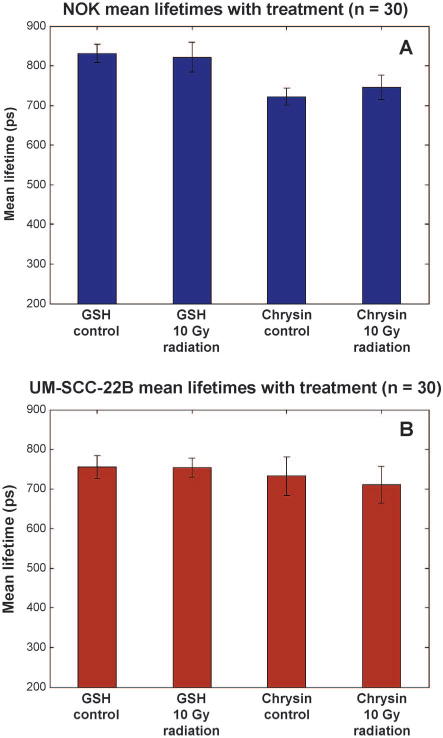
Collected τm values for NOK cells (panel A, blue) and UM-SCC-22B cells (panel B, maroon) with control treatments and treatments plus radiation (n = 30). Error bars shown are standard deviations of each treatment group. Note the consistency of the values between each treatment group.
TABLE 2.
Mean Lifetime Value of Each Cell Group with Standard Deviation for Each Population
| NOK
|
UM-SCC-22B
|
|||
|---|---|---|---|---|
| Control | 10 Gy | Control | 10 Gy | |
| GSH | 830.64 ± 37.26 | 821.67 ± 37.26 | 754.64 ± 28.63 | 753.43 ± 23.66 |
| Chrysin | 722.38 ± 21.77 | 746.15 ± 30.42 | 732.24 ± 48.97 | 719.64 ± 45.45 |
Note. The only change that is statistically significant is for the 10 Gy + chrysin NOK cell group from its chrysin control (P > 0.05).
Clonogenic cell survival was also evaluated for irradiated NOK and UM-SCC-22B cells to quantify radiosensitivity. These survival curves are shown in Fig. 6. The UM-SCC-22B cells are relatively radioresistant (approximately two decades of survival percentage) at high dose levels compared to NOK cells (P < 0.0001).
DISCUSSION AND CONCLUSION
The current study demonstrates that the NOK and UM-SCC-22B cell lines each exhibit distinct and prompt responses to radiation treatment. The UM-SCC-22B cells showed a large change in metabolic signature, whereas the NOK cells showed little metabolic response as measured by FLIM, each within 30 min of treatment. The UM-SCC-22B cellular response was considerably muted when treated with a radical scavenger GSH. Moreover, when a HIF-1α inhibitor was administered, change in metabolic signature was similarly muted. Based on the evidence presented here, we propose that an increase in HIF-1α expression in the UM-SCC-22B cells may be at least partially responsible for the observed changes in metabolic signatures.
Previously published studies have shown that radiation-induced free radical formation caused abnormal interactions with cellular-signaling pathways (42–44). Note that radiation induced a short-lived but spatially-global spike in charges and free radicals to the tumor or cellular microenvironment. One of these interactions takes place with the protein hypoxia inducible factor 1 (HIF-1) (40). HIF-1 is a heterodimer consisting of HIF-1α and HIF-1β subunits; the HIF-1β subunit is independent of oxygen concentration. Under normoxia, the HIF-1α subunit is hydroxylated and subsequently degraded by the von Hippel-Lindau tumor suppressor protein (45). Consequently, the HIF-1 heterodimer does not form due to an insufficient concentration of HIF-1α.
Chandel et al. (40) observed that ROS and free radicals can stabilize HIF-1α, allowing the HIF-1 complex to form. Under these conditions, HIF-1 subsequently regulates a multitude of processes downstream. These processes include stimulation of angiogenesis and erythropoietin production, as well as the regulation of glucose transport enzymes (40, 43, 46–51). The nature of HIF-1α gives it a key role in sensing cellular oxygen levels (40, 52). The free radicals produced by radiation can affect the cells in a multitude of ways, including confounding cellular oxygen sensing and changing oxygen consumption rates.
Since HIF-1 regulates glycolytic enzymes and glucose transport enzymes, it is reasonable to suggest that an increase in HIF-1α and thus HIF-1 might result in metabolic switching. Indeed, the cells with stronger HIF-1 response to radiation (UM-SCC-22B) showed a larger decrease in oxygen consumption compared to NOK cells. This suggests that there may be an increase in aerobic glycolysis after irradiation for UM-SCC-22B cells that is not present in the NOK cells, resulting from upregulated HIF-1 complex-driven signaling pathways. While the mechanism for this is outside the scope of this study, we show a clear correlation between metabolic switching and HIF-1α signaling. This correlation is particularly significant because tumors with upregulated HIF-1α have been associated with more aggressive phenotypes; these tumors are sometimes more metastatic (50, 53–55) and at times more radioresistant (48).
As shown in Fig. 6, the UM-SCC-22B cell line is relatively radioresistant compared to the NOK cell line in vitro (56, 57). Interestingly, the more radioresistant cells were found to have changes in metabolic signature, as measured by FLIM, and were also found to have a great increase in HIF-1α expression after irradiation. It is possible that this increase in HIF-1α expression is a driving factor in the contrast in radiosensitivity between these two cell lines.
The correlation between HIF-1α expression and these FLIM-measured metabolic signatures showed the potential of FLIM as a tool for assessing the overall radiosensitivity and/or aggression of a tumor. Moreover, these measurements show metabolic fluctuations that occur 30 min after irradiation. These metabolic fluctuations may be indicative of modulations in radiosensitivity during treatment or may potentially serve as predictors of radiosensitivity for subsequent treatments. These types of measurements could be influential, particularly for hypofractionated radiotherapy regimens that are more sensitive to intrafraction dynamics (58). Future studies are needed to further elucidate the mechanistic underpinning of these changes in metabolism, such as incorporating flavin adenine dinucleotide (FAD) fluorescence intensity imaging coupled with NADH intensity to measure potential fluxes in cellular redox ratio. Also, while in vitro models provide a tractable system in which it is possible to establish these changes, future studies should use in vivo animal models to ensure the same trends are observed, since other factors may influence metabolism during and after irradiation. Studies of this nature could lead to a deeper understanding of the prompt biological effects of radiation on the tissue microenvironment, and could be valuable in improving therapeutic regimens.
With the reported data and future studies to examine radiation-induced changes in metabolism from a mechanistic and in vivo perspective, there is a great opportunity to consider future clinical ramifications. Recently, there have been efforts to consider the role of dose both spatially and temporally and in the context of the cellular response (51, 59). This study complements previously reported studies (60–62) by asserting that clinical radiotherapy could be guided and improved by monitoring key cellular processes such as metabolism. Since metabolism plays an important role in cancer invasion and progression (63–65), metabolic profiling may have potential for guiding radiation treatments in the future.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health (NIH grant nos. T32 CA009206 and R00 CA160639). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR14093.1) contains supplementary information that is available to all authorized users.
SUPPLEMENTARY MATERIALS
Supplementary materials are available online at: http://dx.doi/10.1667/RR4093.1.S1.
References
- 1.Shaikh M, Burmeister J, Joiner M, Pandya S, Zhao B, Liu Q. Biological effect of different IMRT delivery techniques: SMLC, DMLC, and helical tomotherapy. Med Phys. 2010;37:762–70. doi: 10.1118/1.3284369. [DOI] [PubMed] [Google Scholar]
- 2.Hall EJ, Giaccia AJ. Radiobiology for the radiologist. 7th. Philadelphia: Wolters Kluwer Health; 2012. [Google Scholar]
- 3.Olive PL. Radiation-induced reoxygenation in the SCCVII murine tumor- evidence for a decrease in oxygen-consumption and an increase in tumor perfusion. Radiother Oncol. 1994;32:37–46. doi: 10.1016/0167-8140(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 4.Bussink J, Kaanders J, Rijken P, Raleigh JA, Van der Kogel AJ. Changes in blood perfusion and hypoxia after irradiation of a human squamous cell carcinoma xenograft tumor line. Radiat Res. 2000;153:398–404. doi: 10.1667/0033-7587(2000)153[0398:cibpah]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 5.Ljungkvist ASE, Bussink J, Kaanders J, Wiedenmann NE, Vlasman R, van der Kogel AJ. Dynamics of hypoxia, proliferation and apoptosis after irradiation in a murine tumor model. Radiat Res. 2006;165:326–36. doi: 10.1667/rr3515.1. [DOI] [PubMed] [Google Scholar]
- 6.Carlson DJ, Stewart RD, Semenenko VA. Effects of oxygen on intrinsic radiation sensitivity: a test of the relationship between aerobic and hypoxic linear-quadratic (LQ) model parameters. Med Phys. 2006;33:3105–15. doi: 10.1118/1.2229427. [DOI] [PubMed] [Google Scholar]
- 7.Palcic B, Skarsgard LD. Reduced oxygen enhancement ratio at low-doses of ionizing-radiation. Radiat Res. 1984;100:328–39. [PubMed] [Google Scholar]
- 8.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–30. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Lazaro M. The Warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem. 2008;8:305–12. doi: 10.2174/187152008783961932. [DOI] [PubMed] [Google Scholar]
- 10.Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg effect: Insights from the past decade. Pharmacol Ther. 2013;137:318–30. doi: 10.1016/j.pharmthera.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 12.Guaragnella N, Giannattasio S, Moro L. Mitochondrial dysfunction in cancer chemoresistance. Biochem Pharmacol. 2014;92:62–72. doi: 10.1016/j.bcp.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 13.Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol. 2014;92:43–54. doi: 10.1016/j.bcp.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Warmoes MO, Locasale JW. Heterogeneity of glycolysis in cancers and therapeutic opportunities. Biochem Pharmacol. 2014;92:12–21. doi: 10.1016/j.bcp.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, et al. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun. 2014;5:9. doi: 10.1038/ncomms4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Digman MA, Caiolfa VR, Zamai M, Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94:L14–6. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneckenburger H, Wagner M, Weber P, Strauss WSL, Sailer R. Autofluorescence lifetime imaging of cultivated cells using a UV picosecond laser diode. J Fluoresc. 2004;14:649–54. doi: 10.1023/b:jofl.0000039351.09916.cc. [DOI] [PubMed] [Google Scholar]
- 18.Skala MC, Fontanella A, Lan L, Izatt JA, Dewhirst MW. Longitudinal optical imaging of tumor metabolism and hemodynamics. J Biomed Opt. 2010;15 doi: 10.1117/1.3285584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stringari C, Geyfman M, Wang H, Crosignani V, Kumar V, Takahashi JS, et al. Circadian metabolic oscillations in the epidermis stem cells by fluorescence lifetime microscopy of NADH in vivo. Biophys J. 2014;106:24A. [Google Scholar]
- 20.Stringari C, Pate KT, Edwards RA, Waterman ML, Gratton E. Metabolic imaging of colon cancer tumors in vivo by phasor fluorescence lifetime microscopy of NADH. Biophys J. 2013;104:342A–3A. [Google Scholar]
- 21.Conklin MW, Provenzano PP, Eliceiri KW, Sullivan R, Keely PJ. Fluorescence lifetime imaging of endogenous fluorophores in histopathology sections reveals differences between normal and tumor epithelium in carcinoma in situ of the breast. Cell Biochem Biophys. 2009;53:145–57. doi: 10.1007/s12013-009-9046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Provenzano PP, Eliceiri KW, Keely PJ. Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis. 2009;26:357–70. doi: 10.1007/s10585-008-9204-0. [DOI] [PubMed] [Google Scholar]
- 23.Becker W. Fluorescence lifetime imaging - techniques and applications. J Microsc. 2012;247:119–36. doi: 10.1111/j.1365-2818.2012.03618.x. [DOI] [PubMed] [Google Scholar]
- 24.Chance B. Metabolic heterogeneities in rapidly metabolizing tissues. J Appl Cardiol. 1989;4:207–21. [Google Scholar]
- 25.Lakowicz JR, Szmacinski H, Nowaczyk K, Johnson ML. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci U S A. 1992;89:1271–5. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skala MC, Riching KM, Bird DK, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, et al. In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia. J Biomed Opt. 2007;12 doi: 10.1117/1.2717503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci U S A. 2007;104:19494–9. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, et al. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res. 2005;65:8766–73. doi: 10.1158/0008-5472.CAN-04-3922. [DOI] [PubMed] [Google Scholar]
- 29.Wakita M, Nishimura G, Tamura M. Some Characteristics of the fluorescence lifetime of reduced pyridine-nucleotides in isolated mitochondria, isolated hepatocytes, and perfused rat liver in-situ. J Biochem. 1995;118:1151–60. doi: 10.1093/oxfordjournals.jbchem.a125001. [DOI] [PubMed] [Google Scholar]
- 30.Yaseen MA, Sakadzic S, Wu W, Becker W, Kasischke KA, Boas DA. In vivo imaging of cerebral energy metabolism with two-photon fluorescence lifetime microscopy of NADH. Biomed Opt Express. 2013;4:307–21. doi: 10.1364/BOE.4.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leppert J, Krajewski J, Kantelhardt SR, Schlaffer S, Petkus N, Reusche E, et al. Multiphoton excitation of autofluorescence for microscopy of glioma tissue. Neurosurgery. 2006;58:759–67. doi: 10.1227/01.NEU.0000204885.45644.22. [DOI] [PubMed] [Google Scholar]
- 32.Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL, et al. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013;73:6164–74. doi: 10.1158/0008-5472.CAN-13-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Phipps JE, Meier J, Hatami N, Poirier B, Elson DS, et al. Endoscopic fluorescence lifetime imaging for in vivo intraoperative diagnosis of oral carcinoma. Microsc Microanal. 2013;19:791–8. doi: 10.1017/S1431927613001530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaue D, Ratikan JA, Iwamoto KS. Cellular autofluorescence following ionizing radiation. PLoS ONE. 2012;7:7. doi: 10.1371/journal.pone.0032062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–26. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wokosin DL, Squirrell JM, Eliceiri KW, White JG. Optical workstation with concurrent, independent multiphoton imaging and experimental laser microbeam capabilities. Rev Sci Instrum. 2003;74:193–201. doi: 10.1063/1.1524716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kueng W, Silber E, Eppenberger U. Quantification of cells cultured on 96-well plates. Anal Biochem. 1989;182:16–9. doi: 10.1016/0003-2697(89)90710-0. [DOI] [PubMed] [Google Scholar]
- 38.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimple RJ, Vaseva AV, Cox AD, Baerman KM, Calvo BF, Tepper JE, et al. Radiosensitization of epidermal growth factor receptor/her2-positive pancreatic cancer is mediated by inhibition of Akt independent of Ras mutational status. Clin Cancer Res. 2010;16:912–23. doi: 10.1158/1078-0432.CCR-09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia - A mechanism of O-2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 41.Guzy RD, Hoyos B, Robin E, Chen H, Liu LP, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Hirayama R, Ito A, Noguchi M, Matsumoto Y, Uzawa A, Kobashi G, et al. OH Radicals from the indirect actions of X-rays induce cell lethality and mediate the majority of the oxygen enhancement effect. Radiat Res. 2013;180:514–23. doi: 10.1667/RR13368.1. [DOI] [PubMed] [Google Scholar]
- 43.Dewhirst MW. Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat Res. 2009;172:653–65. doi: 10.1667/RR1926.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hernansanz-Agustin P, Izquierdo-Alvarez A, Sanchez-Gomez FJ, Ramos E, Villa-Pina T, Lamas S, et al. Acute hypoxia produces a superoxide burst in cells. Free Radic Biol Med. 2014;71:146–56. doi: 10.1016/j.freeradbiomed.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O-2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 46.Semenza GL. Expression of hypoxia-inducible factor 1: Mechanisms and consequences. Biochem Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- 47.Han Z-B, Ren H, Zhao H, Chi Y, Chen K, Zhou B, et al. Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF) Carcinogenesis. 2008;29:1853–61. doi: 10.1093/carcin/bgn066. [DOI] [PubMed] [Google Scholar]
- 48.Moeller BJ, Cao YT, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–41. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 49.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8:S62–7. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 50.Vleugel MM, Greijer AE, Shvarts A, van der Groep P, van Berkel M, Aarbodem Y, et al. Differential prognostic impact of hypoxia induced and diffuse HIF-1 alpha expression in invasive breast cancer. J Clin Pathol. 2005;58:172–7. doi: 10.1136/jcp.2004.019885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meijer TWH, Kaanders JHAM, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18:5585–94. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- 52.Semenza GL. Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatr Res. 2001;49:614–7. doi: 10.1203/00006450-200105000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1 alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5. [PubMed] [Google Scholar]
- 54.Halle C, Aarnes E-K, Holm R, Kristensen GB, Lyng H. HIF1 alpha signaling contributes to an aggressive hypoxic phenotype in cervical cancer. Cancer Res. 2013;73 [Google Scholar]
- 55.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16:5928–35. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73:4791–800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park JW, Nickel KP, Torres AD, Lee D, Lambert PF, Kimple RJ. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol. 2014;113:337–44. doi: 10.1016/j.radonc.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kirkpatrick JP, Cardenas-Navia LI, Dewhirst MW. Predicting the effect of temporal variations in Po-2 on tumor radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;59:822–33. doi: 10.1016/j.ijrobp.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 59.McCall KC, Barbee DL, Kissick MW, Jeraj R. PET imaging for the quantification of biologically heterogeneous tumours: measuring the effect of relative position on image-based quantification of dose-painting targets. Phys Med Biol. 2010;55:2789–806. doi: 10.1088/0031-9155/55/10/001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kissick MW, Campos D, Van Der Kogel A, Kimple RJ. On the importance of prompt oxygen changes for hypofractionated radiation treatments. Phys Med Biol. 2013;58:N279–85. doi: 10.1088/0031-9155/58/20/N279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crokart N, Jordan BF, Baudelet C, Ansiaux R, Sonveaux P, Gregoire V, et al. Early reoxygenation in tumors after irradiation: determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys. 2005;63:901–10. doi: 10.1016/j.ijrobp.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 62.Schoenfeld JD, Harris JR. Abbreviated course of radiotherapy (RT) for breast cancer. Breast. 2011;20:S116–27. doi: 10.1016/S0960-9776(11)70308-3. [DOI] [PubMed] [Google Scholar]
- 63.Bredfeldt JS, Liu Y, Conklin MW, Keely PJ, Mackie TR, Eliceiri KW. Automated quantification of aligned collagen for human breast carcinoma prognosis. J Pathol Inform. 2014;5:28. doi: 10.4103/2153-3539.139707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen Is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glunde K. Imaging of collagen I fibers in primary breast cancer to predict metastasis. Amino Acids. 2013;45:589. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.