

Abstract
Objectives:
We describe the largest series of patients with TARDBP mutations presenting with frontotemporal dementia (FTD) and review the cases in the literature to precisely characterize FTD diseases associated with this genotype.
Methods:
The phenotypic characteristics of 29 TARDBP patients, including 10 new French and Dutch cases and 19 reviewed from the literature, were evaluated.
Results:
The most frequent phenotype was a behavioral variant frontotemporal dementia (bvFTD), but a significant proportion (40%) of our patients had semantic (svFTD) or nonfluent variants (nfvFTD) at onset; and svFTD was significantly more frequent in TARDBP carriers than in other FTD genotypes (p < 0.001). Remarkably, only a minority (40%) of our patients secondarily developed amyotrophic lateral sclerosis (ALS). Two patients carried a homozygous mutation but strikingly different phenotypes (bvFTD and ALS) indicating that homozygosity does not result in a specific phenotype. Earlier age at onset in children than parent's generations, mimicking an apparent “anticipation” (21.8 ± 9.3 years, p = 0.001), and possible reduced penetrance were present in most families.
Conclusions:
This study enlarges the phenotypic spectrum of TARDBP and will have important clinical implications: (1) FTD can be the only clinical manifestation of TARDBP mutations; (2) Initial language or semantic disorders might be indicative of a specific genotype; (3) Mutations should be searched in all FTD phenotypes after exclusion of major genes, even in the absence of ALS in the proband or in family history; (4) reduced penetrance and clinical variability should be considered to deliver appropriate genetic counseling.
Frontotemporal dementia (FTD) is characterized by behavioral and language disorders. Predominant symptoms at onset define 3 clinical variants: behavioral (bvFTD), agrammatical/nonfluent (nfvFTD), and semantic (svFTD) variants of FTD.1,2 FTD and amyotrophic lateral sclerosis (ALS) are occasionally associated in patients or within a family, and both disorders share common genetic etiologies, the most frequent one being the C9orf72 GGGGCC repeat expansion.3,4
TARDBP mutations explain ∼3% of familial ALS.5 More than 30 pathogenic missense mutations, mostly clustered in exon 6, have been identified so far. The majority produce isolated ALS, but rare patients presenting dementia or atypical parkinsonism have been reported.5 In this article, we describe the largest series of TARDBP carriers presenting with FTD phenotypes, and we reviewed the cases in the literature to precisely characterize the spectrum of FTD diseases associated with this genotype.
METHODS
Patients.
We evaluated 10 patients (8 probands and 2 relatives) from 8 families (figure 1A) carrying TARDBP missense mutations. The 8 families (F575, F242, F332, F389, F016, FAPP007, D012914, and M008015) were identified through genetic screening of 344 FTD cases. The TARDBP mutations are listed in the table 1. All patients carried heterozygous mutations, except patients F016-003 (presenting FTD) and a relative F016-006 (presenting ALS) who carried homozygous TARDBP mutation, without known familial consanguinity. All patients were negative for MAPT, GRN, and C9orf72 mutations. Two cases (F242 and F389) have been briefly reported previously.6
Figure 1. Pedigrees of the 8 families carrying TARDBP mutations (A) and pathologic examination of patient M008015-001 (B).

(A) Pedigrees of the 8 families carrying TARDBP mutations. Probands are indicated with an arrow. Individuals are represented by a diamond to preserve confidentiality. All affected members are represented in black. Individuals with an uncertain phenotype are represented in gray. AO = age at onset; AAD = age at death; CA = current age; the phenotypes are indicated for each affected individual. Genotypes are indicated for the individuals for whom DNA was available. Family F242: The obligate transmitter parent (004) died of suicide at age 68. Family 389: The mutation was not detected in 007, indicating that the transmitter was 006 who presented with isolated amyotrophic lateral sclerosis (ALS) at age 73 and died at age 74. One sibling (011) developed bulbar ALS without dementia at age 57. (B) Pathologic examination of patient M008015-001. Immunohistochemistry with TDP-43 and p62 antibodies showed TDP-43-positive neuronal inclusions in the hippocampus (a, b) and putamen (c); cytoplasmic p62-positive neuronal inclusions in the frontal cortex (d), hippocampus (e), medulla (f), and glial inclusions in the cerebellum (g).
Table 1.
Phenotypic characteristics and genotypes of ours series and the previously reported frontotemporal dementia (FTD) cases carrying TARDBP mutations


Standard protocol approvals, registrations, and patient consents.
This study was approved by the ethic committee of Pitié-Salpetrière hospital, and all individuals signed informed consent forms for clinical and genetic studies.
Phenotypic evaluation.
All the patients, except the 2 Dutch cases, were examined by expert neurologists of the French FTD/ALS clinical and genetic research network, using standard evaluation procedures, in referral memory or ALS clinics. The basic neurologic examination included evaluation of movement disorders, apraxia, language impairment, oculomotor abnormalities, and symptoms of ALS. Standardized neuropsychological battery was performed when possible, according to the severity of the disease (table e-1 at Neurology.org/ng), additional extended language evaluation was done in patients with predominant aphasic disorders at onset only. Clinical data at onset were also taken from patients' medical records and interviews of relatives. FTD subtypes and ALS were diagnosed according to international criteria.1,2,7 Ages at onset between successive generations within families were compared using an independent sample t test (significant p value <0.05).
Pathologic procedures.
Brain autopsy was performed in case M008015-001 by the Netherlands Brain Bank within 4 hours of death according to the Legal and Ethical Code of Conduct of the Netherlands Brain Bank. The frontal cortex, hippocampus, substantia nigra, caudate, cerebellum, and putamen were studied. Immunohistochemistry was performed with antibodies directed against TDP-43 (ProteinTech Group; 1:100); hyperphosphorylated tau (AT-8, Innogenetics; 1:400); amyloid-β protein (Dako; 1:100); α-synuclein (Zymed Laboratories), polyubiquitin-binding protein p62 (BD Biosciences Pharmingen; 1:200).
Comparisons of the phenotypes in TARDBP, GRN, C9orf72, and MAPT carriers.
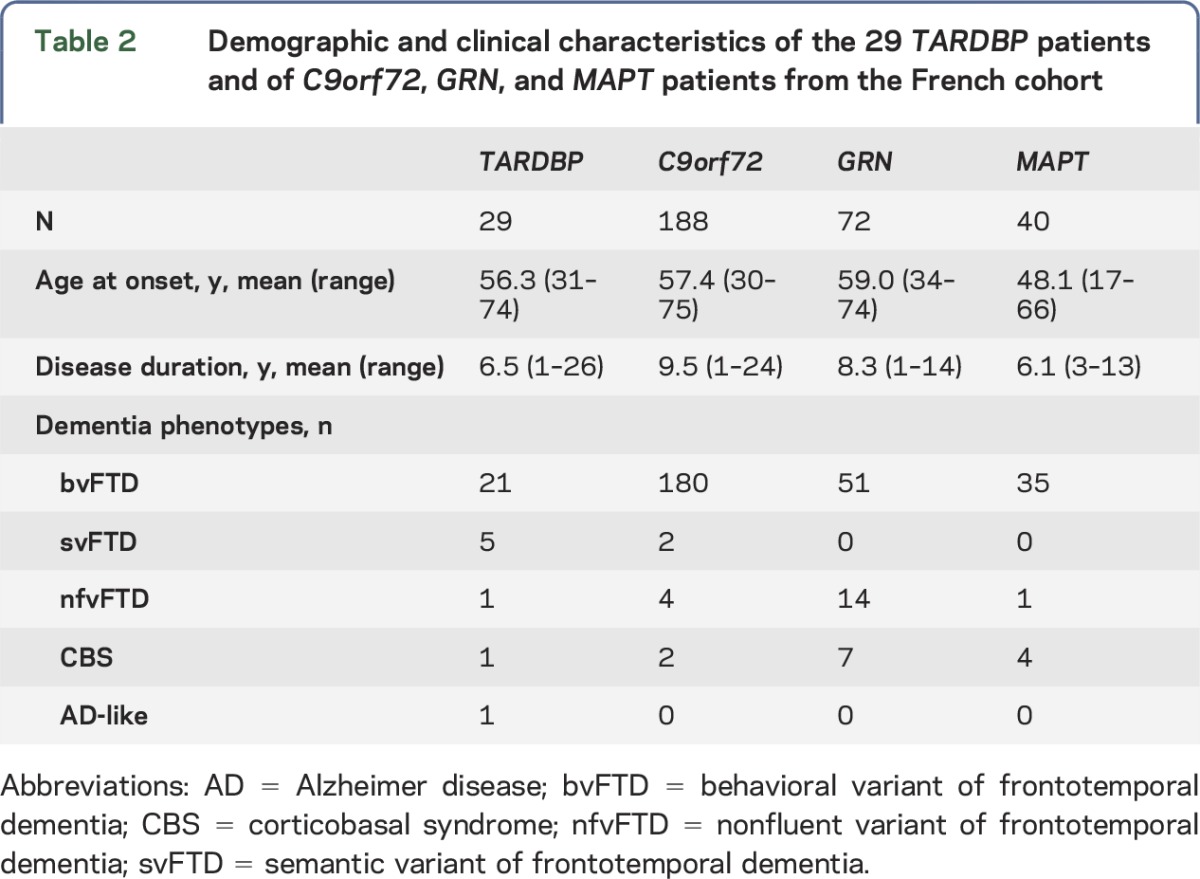
To compare the FTD phenotypes in TARDBP carriers with other FTD genotypes, we used an overall population of 29 TARDBP carriers. This group included our 10 patients and 19 already published cases presenting dementia without ALS at onset, mostly Italians, identified through a comprehensive review of the literature. Most published cases were isolated case reports or small series that were diagnosed according to clinical FTD criteria. Major FTD or ALS genes were excluded in most of the published cases (table 1); 4 of them also carried a C9orf72 expansion (table 1).
TARDBP carriers were compared with 72 GRN, 188 C9orf72, and 40 MAPT carriers issued from the French FTD genetic cohort.8 The populations are described in table 2. The numbers of patients with bvFTD, nfvFTD, svFTD, and corticobasal syndrome (CBS) phenotypes were compared in each group using the Fisher exact test (significant p value <0.05, z-score >2.5).
Table 2.
Demographic and clinical characteristics of the 29 TARDBP patients and of C9orf72, GRN, and MAPT patients from the French cohort
RESULTS
Phenotype of the patients.
Patients carrying TARDBP mutations (figure 1A) are described below; their clinical characteristics are summarized in tables 1 and 2. The ages at onset were significantly earlier in affected children compared with their affected parents (21.8 ± 9.3, p = 0.001).
Family F389.
Proband 014 presented with semantic deficit at age 59. At age 61, language and neuropsychological evaluations evidenced predominant verbal and visual semantic deficits. The semantic score of the Boston Diagnostic Aphasia Examination (BDAE) test was 63/72 (table e-2); recognition of famous faces (3.5/30) and knowledge of public events (0/10) were also impaired. He later developed apathy, social avoidance, indifference to others, bulimia, rituals, cleansing activities, and mild executive dysfunction (Wisconsin card sorting test: 6 categories, 7 errors, 5 perseverations). The Frontal Behavioral Inventory (FBI) score was 26/72. Brain SPECT study revealed bilateral temporopolar hypoperfusion (figure 2). Bulbar symptoms developed at age 61. At age 64, he had severe lingual atrophy and diffuse motor deficit scored 4/5. The ALS-FRS bulbar score was 2/12. Death occurred at age 66. One sibling (011) developed bulbar ALS without dementia at age 57. His father had isolated ALS at age 73 and died at age 74.
Figure 2. Neuroimaging characteristics of TARDBP carriers.

Brain 99mTc-ethylcysteinate dimer-SPECT, FDG-PET, and MRI of 6 patients: F242-001 (A), F575-001 (B), F242-002 (C and E), F389-014 (D), FAPP007-001 (F and H), and M008015-001 (G). Note the hypoperfusion and hypometabolism of the temporal pole in the 3 patients F575-001, F389-014, and FAPP007-001 presenting with semantic dementia.
Family F242.
Proband F242-001 was apathetic, disinhibited, and developed collectionism, hyperorality, and stereotypies at age 52. Neuropsychological evaluation 2 years later revealed short-term memory and attentional deficits (table e-1). The FBI score was 36/72. Brain MRI and 99mTc-ethylcysteinate dimer (99mTc-ECD)-SPECT showed bilateral predominantly right frontotemporal involvement (figure 2). At that time, he developed dysarthria, dysphagia, and, few months later, left lower limb motor deficit and fasciculations. The diagnosis of ALS was confirmed by electromyograms. At age 57, language was reduced to verbal stereotypies; he had severe swallowing disorders, tetraparesis, and was bedridden. The ALS-FRS bulbar score was 3/12, and the motor score was 12/28. He died at age 58. His obligate transmitter parent (004) died of suicide at age 68.
His cousin (002) developed progressive language disorders characterized by anarthric and reduced speech output with agrammatism consistent with nonfluent aphasia at age 65. The BDAE test showed oral expression difficulties, but preserved verbal comprehension (table e-2). Repetition of words and sentences was impaired, with production of phonemic paraphasias. Reading was normal; spelling words was impaired. Oral sentence comprehension was mildly impaired, mainly for long sentences. The oral confrontation naming score was 67/80. There was no buccofacial apraxia or swallowing disorders. Three years later, he developed right limb apraxia and right parkinsonian rigidity. He had no signs of ALS. Brain MRI and 99mTc-ECD-SPECT showed left frontoinsular and parietal involvement (figure 2). The diagnosis was revised to a CBS. His obligate transmitter parent (003) was neurologically healthy at age 91.
Family F575.
Proband 001 presented with predominant verbal and visual semantic deficits consistent with an svFTD at age 71. Loss of initiative and projects, indifference, joviality, food fads, and imitation behaviors developed at age 73. The FBI was scored 45/72 at age 74. Neuropsychological evaluations revealed initiation and conceptualization deficits, reduced phonetic and semantic fluencies, and impaired performance with picture naming (table e-1). Brain MRI revealed bifrontal and predominant temporopolar atrophy. Brain 99mTc-ECD-SPECT showed marked bilateral temporopolar and frontal hypoperfusion (figure 2). Spinal ALS occurred at age 74 and was confirmed by electromyograms. The patient died at age 78. A proband's child (002) had ALS at age 35 and died 3 years later.
Family F016.
Proband 003 had behavioral disorders at age 65. He was apathetic, disinhibited, had fixed ideas, motor stereotypies, and eating disorders including bulimia with food fads, eating only cheese. Language was reduced with verbal stereotypies. Neuropsychological tests revealed frontal dysfunction (table e-1). The FBI score was 36/72. Brain MRI and 99mTc-ECD-SPECT study showed predominant frontotemporal anterior and mild parietal involvement. A diagnosis of bvFTD was made. At age 67, he developed spinal ALS confirmed by electromyograms, and died of respiratory distress at age 68. One sib, 006, presented with spinal ALS at age 65 and died 1 year later without behavioral or cognitive changes. The transmitting parent 001 had chronic psychiatric disorders and died at age 73, without obvious cognitive or motor disorders.
Family F332.
Proband 001 presented with excessive spending, coarseness, irritability, bulimia, and disinhibition at age 63. Brain CT scan showed bilateral frontotemporal atrophy. Neuropsychological evaluation revealed a frontal cognitive syndrome consistent with a diagnosis of bvFTD. Behavioral disorders worsened slowly; he had no motor symptoms at age 87 years and died at age 89.
One child (002) presented with indifference, disinhibition, social withdrawal, and logorrhea at age 51. Brain MRI revealed predominantly right temporopolar atrophy associated with moderate frontal and hippocampal atrophy. Brain 99mTc-ECD-SPECT showed bitemporal, lateral, and right inferior prefrontal hypoperfusion. Neuropsychological evaluation at age 55 revealed a predominant dysexecutive syndrome with deficits in attention, planning, and conceptualization, associated with semantic deficits (table e-1). A diagnosis of bvFTD was made. At age 61, he developed intermittent dysphagia, but had no other motor symptoms. He died at age 61.
Family FAPP007.
Proband 001 presented with verbal and visual semantic deficits suggesting semantic variant of FTD, at age 51. The BDAE scores evidenced semantic impairment (word discrimination: 64/72) without phonological, repetition, or writing deficits at age 54 (tables e-1 and e-2). The Pyramid and Palm Tree test9 demonstrated semantic verbal (43/50) and visual (39/50) deficits. Recognition of famous faces (41/54) and picture denomination (15/40) were impaired. Brain MRI and fluorodeoxyglucose (FDG)-PET revealed bilateral predominantly left temporopolar involvement (figure 2). Neurologic examination was normal, without motor deficit at age 55. There was no family history of neurologic disorders; his parents died at age 78 and 86 without neurologic diseases.
Dutch family D012914.
Proband 001 presented with behavioral changes, excessive spending, impulsivity, and indifference, was less accurate in housekeeping, and had prosopagnosia at age 58. A diagnosis of bvFTD was established. The patient showed slowly progressive behavioral disturbances and secondarily developed verbal semantic deficit and parkinsonism, but no ALS symptoms. He died at age 81. A parent presented with psychiatric symptoms and suicide attempt at age 57 and died at age 64. One sib had dementia with behavioral changes, without motor symptoms, before age 70.
Dutch family M008015.
Proband 001 presented with behavioral disturbances such as marked behavioral disinhibition, chasing unknown young women, and calling random people in the night, loss of interest, social withdrawal, and loss of motivation at age 68. The Mini-Mental State Examination score was 25/30. Brain MRI showed severe bilateral temporal atrophy (figure 2). He died at age 72 without symptoms of ALS. One parent had Parkinson disease, and the other died at age 50 without dementia or other neurologic disorders.
Pathologic characteristics of patient M008015-001.
Macroscopically, brain examination showed severe atrophy in the temporal neocortex, and moderate involvement of frontal gyri, frontal basal gyri, and insular cortex. TDP-43-positive neuronal inclusions were present in the hippocampus and in the frontal cortex. Abundant TDP-43-positive inclusions were also present in the putamen. Other subcortical regions, including the substantia nigra, caudate, and cerebellum, did not show TDP-43-positive inclusions. p62 staining showed cytoplasmic positive neuronal inclusions in the frontal cortex, hippocampus, medulla (d, e, and f in figure 1B, respectively), and many p62-positive inclusions in glial cells of cerebellar white matter, as well as few neuronal inclusions in olivary nuclei. The cerebellar granular layer did not contain p62-positive TDP-43 inclusions. Amyloid, hyperphosphorylated tau, and α-synuclein stainings were negative (figure 1B).
Comparisons of the phenotypes in TARDBP, GRN, C9orf72, and MAPT carriers.
The frequency of svFTD phenotype was higher in TARDBP carriers than in GRN, C9orf72, and MAPT carriers (p < 0.001, z = 4.2). The frequency of nfvFTD was not different in TARDBP carriers compared with others groups, but was higher in GRN carriers compared with C9orf72 carriers (p < 0.001; z = 5.2). The frequencies of bvFTD and CBS phenotypes were similar in the 4 groups (figure 3).
Figure 3. Frequencies of major FTD phenotypes according to the genotype in TARDBP, GRN, C9orf72, and MAPT carriers.
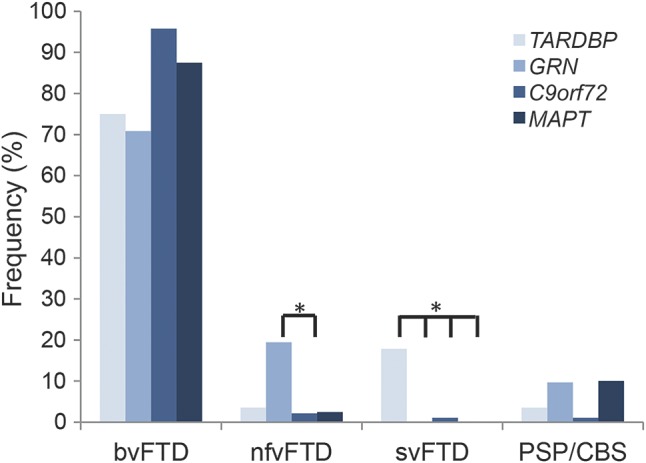
The frequency of svFTD phenotype was higher in TARDBP carriers compared with GRN, C9orf72, and MAPT carriers (p < 0.001). The frequency of nfvFTD was not different in TARDBP carriers compared with others groups, but was higher in GRN carriers compared with C9orf72 carriers (p < 0.001). The frequencies of bvFTD and corticobasal syndrome (CBS) phenotypes were similar in the 4 groups. FTD = frontotemporal dementia; nfvFTD = nonfluent variant of frontotemporal dementia; svFTD = semantic variant of frontotemporal dementia.
DISCUSSION
Mutations in the C9orf72 gene is the most frequent genetic cause of FTD and ALS.3,4 TARDBP mutations are much rarer, and the majority produce isolated ALS.5 Isolated FTD symptoms at onset are much less frequent. In this article, we describe 10 new TARDBP patients initially presenting with isolated FTD symptoms, which is the largest series with this less common phenotype described so far, and 19 other cases described separately in the literature. Notably, 4 cases among the latter10,11 also carried a pathogenic C9orf72 expansion that could contribute by itself to the determinism of the clinical presentation.
A remarkably wide range of ages at onset (31–74; mean 56.3 ± 12.6) characterizes TARDBP carriers (tables 1 and 2). In our French-Dutch series, age at onset also varied greatly within families, with a mean difference of 21.8 ± 9.3 years in successive generations (p = 0.001). Age at onset was earlier in children than in parents' generations, thus mimicking an apparent clinical “anticipation” phenomenon in most families, but this observation should be validated in larger cohorts. This was particularly remarkable in family F575, in which an affected offspring developed disease symptoms ∼35 years earlier than his transmitting parent. This observation and the apparently “sporadic” case FAPP007-001, whose parents died after age 75 without neurologic disorders, suggest reduced penetrance. This is further supported by the obligate transmitter F242-003 who was neurologically healthy at age 91. A precise evaluation of age-dependent penetrance in larger cohorts of TARDBP families is crucial, therefore, to deliver appropriate counseling to the families.
The behavioral variant of FTD was the most frequent presentation in all FTD cases of the literature (15/19, table 1) and in our series (6/10). Of note, nearly half of our patients (4/10) had linguistic or semantic disorders at onset. One of them had the diagnosis of nfvFTD, well documented by detailed clinical and imaging characteristics, and was secondarily diagnosed with CBS based on the occurrence of parietal signs and parkinsonism. Three patients developed svFTD, which was reported previously in only 2 TARDBP carriers in the literature.12,13 Notably, 1 published case also had tau-pathology at autopsy, possibly implicating a pathologic comorbidity.12 The 3 new cases of our series now convincingly demonstrate the direct, not coincidental, association between this rare phenotype and TARDBP mutations. Finally, the frequency of svFTD and nfvFTD phenotypes is relatively high, reaching 40% in our cases and 21% in the overall series.
Intriguingly, 3 relatives (including 2 obligate transmitters) had a medical history of psychiatric disorders, a feature frequently associated in C9orf72 disease. This observation suggests that psychiatric disturbances could be prodromal symptoms of TARDBP disease, and should be further investigated in this genotype also.
Less than half of our patients secondarily developed ALS, 2 years after onset of behavioral disorders. Of note, 6 patients of our series had no ALS symptoms. Notably, 3 of them (F332-001, F332-002, and D012914-001) never developed motor symptoms even after long disease duration (10–26 years), demonstrating that FTD can be the only clinical manifestation of TARDBP mutations during a long time, and within a family. This indicates that the TARDBP gene should be more systematically analyzed after exclusion of the most frequent FTD genes, even in families with isolated FTD or in patients with long-lasting FTD.
Notably, 5 families carried the p.Ile383Val mutation, possibly inherited in some of them from an ancestral founder. This mutation has been identified previously in 5 other families, all presenting ALS except one,14,15,12 showing no obvious phenotype–genotype correlations. Two patients of family F016 carried homozygous p.Ile383Val mutation possibly resulting from unknown consanguinity. Of interest, both patients had onset at age 65 but strikingly different phenotypes (bvFTD in one, ALS in the other), indicating that homozygosity does not result in a specific phenotype. The symptoms do not appear earlier (age at onset: 65.0 ± 0.0 vs 59.8 ± 7.5 years) or the disease seems more severe than in heterozygous carriers, which suggests that a gain of function could be the predominant pathogenic mechanism.
So far, there are only 4 other neuropathologic reports of FTD cases carrying TARDBP mutations that showed variable association of TDP-43 inclusions12,16,17 and other proteinopathies such as tau,12 amyloid,16 and α-synuclein.17 In our case, a TDP-43 pathology was associated with p62-positive inclusions without any tau, amyloid, or α-synuclein accumulation. The association of TDP-43 and p62 inclusions can also be observed in patients carrying C9orf72 expansions. However, the absence of p62-positive TDP-43-negative inclusions in the cerebellar granular layer differentiates our case from C9orf72 expansions.
Together, our and other cases showed that TARDBP mutations may be responsible for a large spectrum of phenotypes including bvFTD, svFTD, nfvFTD, and CBS, in addition to ALS phenotypes. A large phenotypic variability is also observed in GRN carriers.18 The clinical presentations are less variable in C9orf72 carriers, the majority presenting with bvFTD, ALS, or both disorders, but rarely nfvFTD or svFTD.8 Our study in a French genetic population revealed that semantic variants are more frequent in TARDBP than in GRN, C9orf72, and MAPT carriers (p < 0.001), whereas nfvFTD is more frequent in GRN than in other genotypes (figure 3). This indicates that initial semantic or language disorders might be indicative of a specific genotype, a finding that could have important implications for diagnostic strategies in clinical practice.
Finally, approximately 890 patients with FTD have been screened for TARDBP mutations so far (table e-3). Compiling these studies, the frequency of mutations is close to 1% in FTD, although variations may exist between countries. It is important to underline that this frequency is in the same range as mutation frequency in isolated patients with ALS (0.6%–3.3%).14,19,5 This study thus clearly suggests that TARDBP analysis should also be proposed in all FTD phenotypes, after exclusion of GRN, C9orf72, and MAPT, even in the absence of a personal or family history of ALS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Lydia Guennec, Isabelle Lagroua, Sylvie Forlani, and Christelle Dussert (DNA and cell bank of CR-ICM, Hôpital de la Salpêtrière, Paris) and Federico D'Agata (University of Turin, Italy) for technical assistance.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- CBS
corticobasal syndrome
- FBI
Frontal Behavioral Inventory
- FTD
frontotemporal dementia
- 99mTc-ECD
99mTc-ethylcysteinate dimer
- nfvFTD
nonfluent variant of frontotemporal dementia
- svFTD
semantic variant of frontotemporal dementia
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
P. Caroppo: drafting/revising the manuscript for content, including medical writing for content; analysis or interpretation of data; obtaining funding; statistical analysis. A. Camuzat, L. Guillot-Noel, T.H. Wong, S. Lattante, F. Clot, B. Dubois, and J.C. van Swieten: drafting/revising the manuscript for content, including medical writing for content; analysis or interpretation of data. C. Thomas-Anterion, P. Couratier, M. Teichmann, V. Golfier, S. Auriacombe, S. Belliard, and B. Laurent: drafting/revising the manuscript for content, including medical writing for content; acquisition of data. S. Millecamps: drafting/revising the manuscript for content, including medical writing for content. A. Brice: drafting/revising the manuscript for content, including medical writing for content; study supervision or coordination; obtaining funding. I. Le Ber: study concept and design; drafting/revising the manuscript for content, including medical writing for content; analysis or interpretation of data; obtaining funding; study supervision or coordination.
STUDY FUNDING
The research leading to these results has received funding from the program “Investissements d'avenir” ANR-10-IAIHU-06. This study was funded by the French Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla, France) (to A.B. and S.M.), ANR FTDGenes (to I.L.B.), PHRC FTLD-exomes (to I.L.B.), Roger de Spoelberch Foundation (to A.B., contract R12123DD). Dr. Paola Caroppo received a PhD Fellowship from IRCCS Foundation Carlo Besta Neurological Institute, Milano, Italy.
DISCLOSURE
Dr. Caroppo, Ms. Camuzat, Ms. Guillot-Noel, and Dr. Thomas-Anterion report no disclosures. Dr. Couratier has served on the editorial board of Revue Neurologique. Dr. Wong reports no disclosures. Dr. Teichmann has received speaker honoraria from Lundbeck and Novartis. Dr. Golfier reports no disclosures. Dr. Auriacombe has served on the scientific advisory boards of Eisai and Novartis; has received travel funding/speaker honoraria from Janssen Cilag, Novartis, and Eisai; and has been an employee of Eisai, Novartis, and Janssen Cilag. Dr. Belliard has served on the editorial board of Revue de Neuropsychologie. Dr. Laurent and Dr. Lattante report no disclosures. Dr. Millecamps receives research support from Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla, France) and Association française contre les myopathies (AFM, France). Dr. Clot reports no disclosures. Dr. Dubois has been a consultant for Eli Lilly. Dr. van Swieten has received research support from AFTD (Association for Frontotemporal Dementias), the Dioraphte Foundation, The Netherlands Organization for Scientific Research, the Netherlands Alzheimer Foundation, Accountants KPMG, and ISAO. Dr. Brice has served on the scientific advisory boards of FWO (Research Foundation Flanders), ERC (European Research Council), and BMBF (Bundesministeriums für Bildung and Forschung-Berlin Germany); has served on the editorial boards of Neurology and Clinical Neuroscience, Parkinsonism and Related Disorders, Brain, Neurodegenerative Diseases, The Cerebellum, and Neurogenetics; and has received research support from the French Research Agency, UE, French program “Investissements d'avenir” (ANR-10-IAIHU), FP (France Parkinson Association), RDS (Roger de Spoelberch Foundation), FDF (Fondation de France), and FRM (Fondation pour la Recherche Médicale). Dr. Le Ber reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioral variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat 2013;34:812–826. [DOI] [PubMed] [Google Scholar]
- 6.Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 2009;65:470–473. [DOI] [PubMed] [Google Scholar]
- 7.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci 1994;124(suppl):96–107. [DOI] [PubMed] [Google Scholar]
- 8.Le Ber I, Camuzat A, Guillot-Noel L, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis 2013;34:485–499. [DOI] [PubMed] [Google Scholar]
- 9.Howard D, Patterson K. The Pyramids and Palm Trees Test: A Test of Semantic Access From Words and Pictures. Bury St. Edmunds: Thames Valley Company; 1992. [Google Scholar]
- 10.Origone P, Accardo J, Verdiani S, et al. Neuroimaging features in C9orf72 and TARDBP double mutation with FTD phenotype. Neurocase 2014;20:1–6. [DOI] [PubMed] [Google Scholar]
- 11.Kaivorinne AL, Moilanen V, Kervinen M, et al. Novel TARDBP sequence variant and C9ORF72 repeat expansion in a family with frontotemporal dementia. Alzheimer Dis Assoc Disord 2014;28:190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gelpi E, van der Zee J, Turon Estrada A, Van Broeckhoven C, Sanchez-Valle R. TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol 2014;40:225–230. [DOI] [PubMed] [Google Scholar]
- 13.Floris G, Borghero G, Cannas A, et al. Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J Neurol 2015;262:375–384. [DOI] [PubMed] [Google Scholar]
- 14.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008;4:e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ticozzi N, LeClerc AL, van Blitterswijk M, et al. Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol Aging 2011;32:2096–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreno F, Rabinovici GD, Karydas A, et al. A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol Commun 2015;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovacs GG, Murrell JR, Horvath S, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord 2009;24:1843–1847. [DOI] [PubMed] [Google Scholar]
- 18.Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131:732–746. [DOI] [PubMed] [Google Scholar]
- 19.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borroni B, Bonvicini C, Alberici A, et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat 2009;30:E974–E983. [DOI] [PubMed] [Google Scholar]
- 21.Borroni B, Archetti S, Del Bo R, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res 2010;13:509–517. [DOI] [PubMed] [Google Scholar]
- 22.Corrado L, Ratti A, Gellera C, et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat 2009;30:688–694. [DOI] [PubMed] [Google Scholar]
- 23.Synofzik M, Born C, Rominger A, et al. Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol Aging 2014;35:1212.e1–1212.e5. [DOI] [PubMed] [Google Scholar]
- 24.Chiang HH, Andersen PM, Tysnes OB, Gredal O, Christensen PB, Graff C. Novel TARDBP mutations in Nordic ALS patients. J Hum Genet 2012;57:316–319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.