

Abstract
Background
Primary spontaneous pneumothorax (PSP) or pulmonary cysts is one of the manifestations of Birt-Hogg-Dube syndrome (BHDS) that is caused by heterozygous mutations in FLCN gene. Most of the mutations are SNVs and small indels, and there are also approximately 10 % large intragenic deletions and duplications of the mutations. These molecular findings are generally obtained by disparate methods including Sanger sequencing and Multiple Ligation-dependent Probe Amplification in the clinical laboratory. In addition, as a genetically heterogeneous disorder, PSP may be caused by mutations in multiple genes include FBN1, COL3A1, CBS, SERPINA1 and TSC1/TSC2 genes. For differential diagnosis, these genes should also be screened which makes the diagnostic procedure more time-consuming and labor-intensive.
Methods
Forty PSP patients were divided into 2 groups. Nineteen patients with different pathogenic mutations of FLCN previously identified by conventional Sanger sequencing and MLPA were included in test group, 21 random PSP patients without any genetic screening were included in blinded sample group. 7 PSP genes including FLCN, FBN1, COL3A1, CBS, SERPINA1 and TSC1/TSC2 were designed and enriched by Haloplex system, sequenced on a Miseq platform and analyzed in the 40 patients to evaluate the performance of the targeted-NGS method.
Results
We demonstrated that the full spectrum of genes associated with pneumothorax including FLCN gene mutations can be identified simultaneously in multiplexed sequence data. Noteworthy, by our in-house copy number analysis of the sequence data, we could not only detect intragenic deletions, but also determine approximate deletion junctions simultaneously.
Conclusions
NGS based Haloplex target enrichment technology is proved to be a rapid and cost-effective screening strategy for the comprehensive molecular diagnosis of BHDS in PSP patients, as it can replace Sanger sequencing and MLPA by simultaneously detecting exonic and intronic SNVs, small indels, large intragenic deletions and determining deletion junctions in PSP-related genes.
Keywords: Primary spontaneous pneumothorax, BHD syndrome, FLCN, Targeted next generation sequencing, CNV analysis
Background
Primary spontaneous pneumothorax (PSP, OMIM #173600) is defined as the spontaneous occurrence of pneumothorax in a healthy individual without obvious underlying lung disease. A positive family history is found in 11.5 % of PSP patients [1]. While some cases can be attributed to several rare inherited monogenic disorders caused by mutations in FBN1, COL3A1, CBS, SERPINA1 and TSC1/TSC2 genes [2–6], a significant proportion of PSP cases are caused by germline mutations of FLCN gene, a causative gene of Birt-Hogg-Dube syndrome (BHD, OMIM #135150) [7–11].
BHD syndrome is an autosomal dominant disease characterized by skin fibrofolliculomas, renal cancer, pulmonary cysts and spontaneous pneumothorax [12–15]. Since the clinical manifestations may be atypical and variable [16, 17], BHD patients could exhibit a pneumothorax-dominant phenotype with no or reduced penetrance of skin or renal manifestations, the syndrome is considered to be under-diagnosed [18–23]. FLCN mutation carriers have been reported to have an increased lifetime risk of developing renal cell carcinoma (RCC) [24, 25], they should be advised to take some preventive measures, largely aimed at early recognition and treatment of RCC. Under the circumstances, it is an imperative diagnostic approach to perform genetic testing in PSP patients [17].
Thus far, the molecular diagnosis of BHD syndrome is mainly based on Sanger sequencing. However, there are different types of mutations in FLCN gene, comprised of base pair substitutions, small indels, deletions and duplications [26–28], which make the diagnostic procedure complicated and inefficient. Moreover, for differential diagnosis, patients with PSP should also be screened for mutations in FBN1, COL3A1, CBS, SERPINA1 and TSC1/TSC2 genes. Together, this requires about 220 primer reactions in Sanger sequencing, plus at least 7 probe sets in Multiple Ligation-dependent Probe Amplification (MLPA) analysis.
Given the complicity, it is not feasible to conduct a comprehensive mutation screening in clinical diagnostics in PSP patients. In recent years, next generation sequencing (NGS) and target enrichment techniques have been further developed, allowing us to focus specifically on genomic regions of interest for cheaper multiplexed sequencing of more cases. It has been successfully used in diagnosis of genetically heterogeneous diseases, such as breast cancer and congenital muscular dystrophy [29, 30].
Herein we sought to develop a NGS-based method using Haloplex target enrichment and the MiSeq platform to identify not only point mutations and indels but also copy number variations (CNV) as an accurate and easier diagnostic tool for simultaneous mutation screening of all the PSP-related genes. Using little input DNA, this system promises a quicker, more affordable and efficient analysis in diagnostic laboratories. To this end, we used Haloplex to screen 7 genes responsible for PSP in 40 PSP patients and aimed to evaluate its performance in identifying SNPs, indels and copy number variations affecting target genes.
Methods
Patients
Forty unrelated patients with PSP who had given written informed consent participated in the genetic screen and were divided into two groups.
Test group
Nineteen patients with different pathogenic mutations of FLCN previously identified by conventional Sanger sequencing and MLPA were included in test group (patients F01 to F19) [7, 8].
Blinded sample group
Twenty-one random PSP patients without any genetic screening were included in blinded sample group (patients F20 to F40).
Subjects were recruited from two tertiary hospitals, Taizhou Hospital of Zhejiang Province and Nanjing Chest Hospital. All participants in this study completed a medical questionnaire. The presence of lung cysts was evaluated by chest computed tomography. Medical and surgical records were reviewed if available. Peripheral blood samples were collected for DNA extraction and subsequent analysis.
Statement of ethical approval
The study was approved by the Ethical Committees of Taizhou Hospital of Zhejiang Province (TZYY-2011106) and the Ethical Committees of Nanjing Chest Hospital (NCH-2011062). Informed consent was obtained from all individual participants included in the study.
DNA extraction, quantification, and quality control
Genomic DNA was extracted from peripheral blood samples of collected family members and controls using a Qiagen DNA Mini blood Kit (Qiagen, Hilden, Germany) according to manufacturer instruction. DNA purity and concentration were assessed by the NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Florida, USA), A260/A280 ratios of 1.8 to 2.0 were accepted. DNA fragmentation was assessed by agarose gel electrophoresis.
HaloPlex design, target enrichment, and next generation sequencing
A custom Haloplex panel was designed using Agilent’s online SureDesign tool (https://earray.chem.agilent.com/suredesign/index.htm). In all, 78 target regions including the whole gene sequences of FLCN, TSC1, TSC2, CBS, COL3A1 and all coding exons, intron-exon boundaries including 50 intronic nucleotides and 5′ UTR (Untranslated Regions), 3′ UTR of FBN1, SERPINA1 were captured for enrichment (Table 1). According to previous reports, intragenic large deletions have been identified in FLCN, TSC1, TSC2, CBS and COL3A1 genes [8, 31–33], most breakpoints of the deletions locate in intronic sequences. In order to assess the performance of breakpoint analysis using NGS system, we captured both exonic and intronic sequences. However, large deletions in SERPINA1 gene were rarely reported. Blyth et al. (2008) suggested that Marfan patients with large deletions in FBN1 gene have a more severe phenotype, and barely manifest as isolated pneumothorax [34]. Therefore, we only targeted coding exons, intron-exon boundaries, 5′- and 3′- UTRs of SERPINA1 and FBN1genes. The predicted target coverage was 98.72 %. The 214 kb target regions were captured using the Agilent HaloPlex Target Enrichment System Kits for Illumina Sequencing (Custom Panel Tier 1, ILM, 48 reactions; Agilent Technologies, Inc. Santa Clare, CA) following Agilent protocols. In brief, 225 ng of genomic DNA was fragmented using eight different restriction enzymes and denatured. Hybridization was performed with a biotinylated custom probe library and sample indexes, and then the target DNA fragments were captured with magnetic streptavidin beads and circularized by ligation. The captured target libraries were amplified by PCR, quality controlled and quantified using the BioAnalyzer 2100 (Agilent Technologies, Inc. Santa Clare, CA). Equimolar amounts of differentially indexed samples were pooled before pair-ended sequencing 300 bp on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) and the mean read depth within the regions of interest was ~300 reads per base.
Table 1.
Genomic regions targeted for PSP genes
| Disease | Gene name | Chromosome location | Capture region | Size (bp) | Exon | Transcript |
|---|---|---|---|---|---|---|
| Birt–Hogg–Dube syndrome | FLCN | chr17:17115527–17145502 | Whole gene | 29976 | 14 | NM_144997 |
| Lymphangioleiomyomatosis | TSC1 | chr9:135766735–135822020 | Whole gene | 55286 | 23 | NM_000368 |
| TSC2 | chr16:2095990–2138713 | Whole gene | 42724 | 42 | NM_000548 | |
| Homocystinuria | CBS | chr21:44473301–44497053 | Whole gene | 23853 | 17 | NM_000071 |
| Ehlers-Danlos syndrome | COL3A1 | chr2:189839046–189877472 | Whole gene | 38527 | 51 | NM_000090 |
| Marfan syndrome | FBN1 | chr15:48700503–48938046 | Exons | 18559 | 66 | NM_000138 |
| α1- Antitrypsin deficiency | SERPINA1 | chr14:94843084–94857030 | Exons | 4863 | 7 | NM_001002236 |
| Capture size | 213788 | 220 |
Bioinformatics analysis for NGS results
The authors implicated in the bioinformatics analysis had no information on the patient data. Post-run sequencing quality was assessed by FastQC (Babraham Bioinformatics, Cambridge, UK). Sequence reads were aligned to the hg19 human reference genome (February 2009 assembly), and analyzed with Agilent software Surecall v2.1.1.13 using the default Haloplex parameters. The mutation reports (*.vcf files) were then annotated using ANNOVAR [35]. After removing all SNVs/indels occurring within non-coding regions, the variations were filtered against dbSNP137 NonFlagged, 1000 genomes and ESP 6500 databases. Rare, non-synonymous, exonic variants were subjected to bioinformatics tool PolyPhen [36] and SIFT [37] to evaluate their possible impact on protein structure and function. To perform extensive analysis, we further analyzed the intron sequences to identify potential splice and branch-site mutations using splice prediction tool ESEfinder [38]. If any potentially harmful mutation identified, validation was performed by Sanger sequencing.
Copy-number analysis
CNVs were identified using a depth-based method. Coverage statistics were generated using GATK Depth of Coverage tool version 3.1 [39, 40]. We normalized coverage in each sample by dividing the average coverage of each exon by the total number of on-target mapped reads for that sample . To identify copy number variations at individual exons, we compared the normalized coverage of each exon of testing patients with that of controls. Coverage data and bars/plots graph were produced using Prism 6. Exons with a normalized coverage ratio below/above 0.7/1.3 of the mean in controls were classified as heterozygously deleted/duplicated and further analyzed by MLPA [8]. A 20-bp interval was used for precise breakpoint determination.
Sanger sequencing
Sanger sequencing was performed to confirm all detected variants. The primers Prism 3130 were designed using the online software Primer3. The amplification reaction mixture (50 μl) was subjected to denaturation at 95 °C for 2 min followed by 30 cycles at 94 °C for 1 min, annealing temperature 60 °C for 1 min, 72 °C for 1 min and by a final extension at 72 °C for 15 min. Bi-directional sequencing was performed using BigDye Terminator v3.1 Cycle Sequencing Kit, version 3.1 (Applied Biosystems, Foster, CA, USA) and analyzed on an ABI genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Results
A total of 40 samples were included in this study. Nineteen patients with 15 different types of FLCN pathogenic mutations previously identified by conventional Sanger sequencing and MLPA were chosen as positive controls to evaluate the performance of the target enrichment method. In addition, 21 blinded samples from genetically untested PSP patients were also examined by this method to identify the underlying mutation(s) responsible for PSP and to assess this method for its diagnostic potential. All the results were re-confirmed by conventional Sanger sequencing and MLPA. The strategy used in this study were detailed in Fig. 1c and Table 1.
Fig. 1.
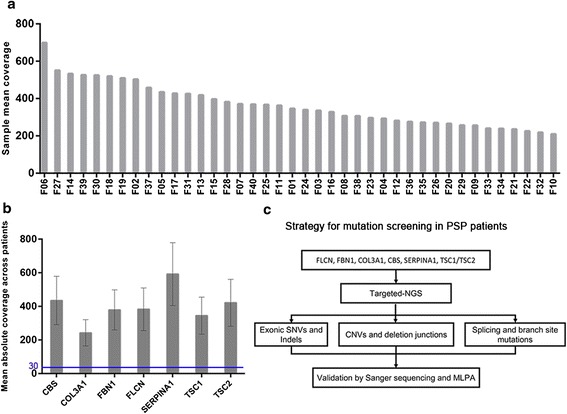
Representative Quality metrics of the targeted-NGS method and strategy for mutation screening in the targeted genes. a Representation of the average depth of each sample. The coverage depth was >200× for each sample. b Mean coverage obtained for the different genes across patients. Bar chart displaying the mean coverage of each gene in all samples. Error bars represent standard deviation. The blue line shows the lower limit of coverage appropriate to perform CNV analysis (30×). c Strategy for comprehensive mutation screening of the targeted genes in PSP cohort
Sequencing quality metrics
The obtained sequencing data was analyzed by FastQC software, which indicated good-quality sequencing metrics, with an average 80.53 % of the sequenced bases above Q30. On average, a mean coverage of 364× was obtained per sample (ranged 210 ×–699×) (Fig. 1a). Different average coverage were obtained for the seven analyzed genes, ranging from 243× (COL3A1) to 592× (SERPINA1) (Fig. 1b). All genes were covered at more than 30× for at least 96 % of the target regions, promising the CNV analysis. The percentage of sequencing on target was 72.35 %.
Identification of SNVs, indels and CNVs in test group: validation of the diagnostic strategy
All 15 different pathogenic sequence changes were correctly identified in the 19 positive control samples (Table 2; patients F01-F19), resulting in a diagnostic rate of 100 % for the detection of pathogenic mutations in our system. These encompassed a variety of mutations, including 2 SNVs (1 stop gain and 1 splice site mutation), 4 insertions/6 deletions ranging from 1 to 23 bases and 3 large intragenic deletions. The false-negative rate was 0 %. All samples were analyzed blind to their mutations.
Table 2.
SNVs, Indels, and exon deletions identified by the assay in Test Group
| Patient ID | Gene | RefSeq | Exon or Intron | Mutation(s) by nucleotide | Mutation(s) by amino acid | Mutation type | Conventional method of detection |
|---|---|---|---|---|---|---|---|
| F01 | FLCN | NM_144997 | exon11 | c.1285dupC | p.H429PfsX27 | Frameshift insertion, protein truncation | Sanger Sequence |
| F02 | FLCN | NM_144997 | exon11 | c.1285dupC | p.H429PfsX27 | Frameshift insertion, protein truncation | Sanger Sequence |
| F03 | FLCN | NM_144997 | exon14 | c.1579_1580insA | p.R527QfsX75 | Frameshift deletion, protein truncation | Sanger Sequence |
| F04 | FLCN | NM_144997 | exon4 | c.182_186dupACAGC | p.P63TfsX69 | Frameshift deletion, protein truncation | Sanger Sequence |
| F05 | FLCN | NM_144997 | exon12 | c.1360dupT | p.C454LfsX2 | Frameshift deletion, protein truncation | Sanger Sequence |
| F06 | FLCN | NM_144997 | exon11 | c.1285delC | p.H429TfsX39 | Frameshift deletion, protein truncation | Sanger Sequence |
| F07 | FLCN | NM_144997 | exon9 | c.946_947delAG | p.S316YfsX73 | Frameshift deletion, protein truncation | Sanger Sequence |
| F08 | FLCN | NM_144997 | exon10 | c.1156_1175del | p.S386DfsX63 | Frameshift deletion, protein truncation | Sanger Sequence |
| F09 | FLCN | NM_144997 | exon14 | c.1648_1658del | p.L550DfsX48 | Frameshift deletion, protein truncation | Sanger Sequence |
| F10 | FLCN | NM_144997 | exon6 | c.469_471delTTC | p.F157del | In-frame deletion | Sanger Sequence |
| F11 | FLCN | NM_144997 | exon13 | c.1522_1524delAAG | p.K508del | In-frame deletion | Sanger Sequence |
| F12 | FLCN | NM_144997 | intron4 | c.250–3_268del | --- | Splice site | Sanger Sequence |
| F13 | FLCN | NM_144997 | exon6 | c.507G > A | p.W169X | Nonsense mutation | Sanger Sequence |
| F14 | FLCN | NM_144997 | exon6 | c.507G > A | p.W169X | Nonsense mutation | Sanger Sequence |
| F15 | FLCN | NM_144997 | intron13 | c.1539–1G > A | --- | Splice site | Sanger Sequence |
| F16 | FLCN | NM_144997 | exon1–3 | c.-504–1355_-25 + 894 del | --- | Large intragenic deletion | MLPA |
| F17 | FLCN | NM_144997 | exon14 | c.1539–476_1740 + 1077 del | --- | Large intragenic deletion | MLPA |
| F18 | FLCN | NM_144997 | exon9–14 | c.872–562_1740 + ?a del | --- | Large intragenic deletion | MLPA |
| F19 | FLCN | NM_144997 | exon9–14 | c.872–562_1740 + ?a del | --- | Large intragenic deletion | MLPA |
aThe breakpoint is within the 3′ flank of the FLCN gene, which was not included in the panel
Detection of exon deletions and breakpoint analysis
We observed that the coverage varied significantly between positions from the same sample, however, the samples captured with an identical protocol had very similar distribution of sequencing depth. In this context, we normalized coverage of each exon for on-target mapped bases for that sample and performed the CNV analysis using a depth-based method. This analysis allowed us to identify 4 patients with heterozygous large intragenic deletions of FLCN gene in our study cohort: 1 deletion in FLCN comprised exons 1–3 (average normalized depth ratio = 0.356). One deletion in FLCN comprised exon 14 (normalized depth ratio = 0.601) and 2 deletions in FLCN comprised exons 9–14 (average normalized depth ratio = 0.533 and 0.539, respectively) (Fig. 2a-d).
Fig. 2.
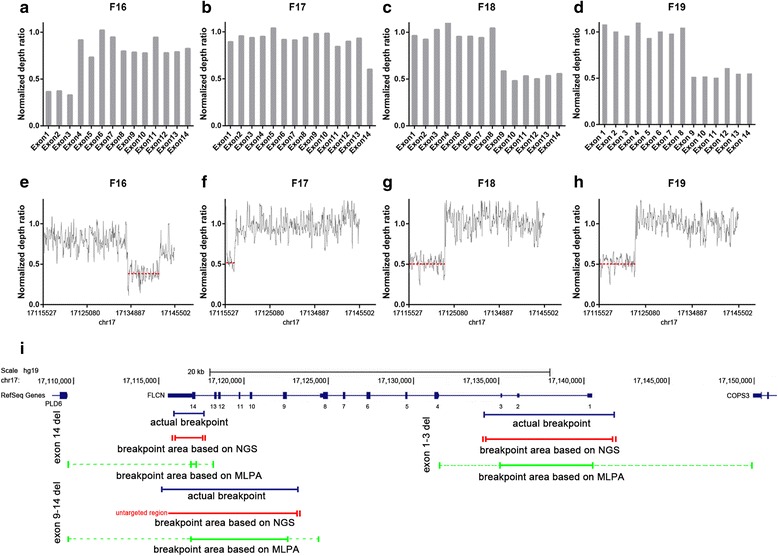
Detection of copy number variants and mapping of deletion junctions. a-d Representation of the normalized depth ratio of each exon in FLCN gene. Exons with a normalized coverage ratio below 0.7 were classified as heterozygously deleted. e-h Representation of the normalized depth ratio of 20-bp intervals in FLCN gene. The red dotted lines represent the average ratio of the deletion area. i The upper panel shows genomic structure of the FLCN gene (data from USCS database). The blue line represents actual deletion size. Deletion size of NGS-based and MLPA-based methods are represented by green line and red line, respectively. The solid line represents the definite deletion size determined by each method. Interval of vertical bars on each side of solid lines represent the range of probable deletion junctions
We subsequently managed to identify exact breakpoints of the deletions by calculating the normalized depth of a set of 20 bp-interval contigs covered the entire FLCN gene (Fig. 2e-h, Table 3). For patient F16, by using MLPA, of which probes are designed in the exons of FLCN gene, we detected a deletion of exons 1–3 [8]. The estimated deletion size is between 5346–18666 bp in length, while with the NGS strategy, we were able to restrict it to about 7650 bp, closed to its actual deletion size (Fig. 2i). Likewise, in patient F17 and F18/F19, we reduced the estimated size of deletion from 1645–8671 bp and 5555–13782 bp down to about 1740 bp and 6117–12017 bp, respectively (Fig. 2i).
Table 3.
Genomic deletions and breakpoints identified by the assay
| Patient ID | Gene | Genomic event | Breakpoint identified by NGS system | Breakpoint identified by amplification of the deletion junction | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Starta | Enda | Size (bp) | Average ratiob | Chromosome | Start | End | Size (bp) | |||
| F16 | FLCN | Deletion exons 1–3 | 17 | 17134237 | 17141880 | 7644 | 0.377 | 17 | 17134286 | 17141828 | 7543 |
| F17 | FLCN | Deletion exons 14 | 17 | 17115904 | 17117646 | 1743 | 0.534 | 17 | 17115898 | 17117706 | 1809 |
| F18 | FLCN | Deletion exons 9–14 | 17 | untargeted regionc | 17123085 | --- | 0.513 | 17 | 17115206 | 17123002 | 7747 |
| F19 | FLCN | Deletion exons 9–14 | 17 | untargeted regionc | 17123085 | --- | 0.516 | 17 | 17115206 | 17123002 | 7747 |
aBreakpoints are defined by the external boundaries of 20 bp-intervals. Breakpoints are flanked by Alu repeats
bAverage of normalized depth ratio of each 20 bp-interval
cThe breakpoint is within the 3′ flank of the FLCN gene, which was not included in the panel
The breakpoints for four large intragenic deletions in FLCN had been identified by genomic amplification of the DNA regions across the deletion junctions, followed by Sanger sequencing. Each large deletion is flanked by Alu repeat sequences that mediate the mutation. Our NGS system accurately identified exon deletions and determined breakpoints on the targeted sequence within about 100 bp, largely narrowed the boundaries of the deletions, which made it much easier to design primers for genomic amplification of the deletion junction.
Mutation analysis in blinded sample group
Our study shown there were an average of 194 variants per sample within the 214 kb targeted region in our blinded PSP cohort of PSP. After removing all SNVs/indels occurring within non-coding regions, the variations were filtered against dbSNP137 NonFlagged, 1000 genomes and ESP 6500 databases, and then submitted to PolyPhen and SIFT to evaluate possible impact of the variations on protein structure and function. Using this filtering criteria, there was 0–1 novel, predicted harmful variant remained for each sample. Of the 21 patients, we detected clearly pathogenic mutations in 3 cases (2 in FLCN, 1 in FBN1) and likely pathogenic mutations in the other 3 cases (Table 4). All of the variants were confirmed by Sanger sequencing.
Table 4.
Mutations identified in the 21 SP patients of the blinded sample group
| Patient ID | Gene | RefSeq | Exon or Intron | Mutation(s) by nucleotide | Mutation(s) by amino acid | Mutation type | Zygosity | Classification | Comments |
|---|---|---|---|---|---|---|---|---|---|
| F29 | FLCN | NM_144997 | exon14 | c.1648_1658del | p.L550DfsX48 | Frameshift deletion | Heterozygous | Definitely Pathogenic | |
| F37 | FLCN | NM_144997 | exon6 | c.473delT | p.I158TfsX19 | Frameshift deletion | Heterozygous | Definitely Pathogenic | |
| F32 | FBN1 | NM_000138 | exon51 | c.6169C > T | p.R2057X | Stopgain SNV | Heterozygous | Definitely Pathogenic | Not reported in any database |
| F27 | FBN1 | NM_000138 | exon19 | c.2269G > C | p.D757H | Missense SNV | Heterozygous | Likely Pathogenic | Situates in the EGF-like calcium-binding domain of FBN1; highly conserved; predicted damaging |
| F33 | TSC1 | NM_000368 | exon15 | c.1631G > A | p.G544E | Missense SNV | Heterozygous | Likely Pathogenic | Highly conserved; predicted damaging |
| F34 | FLCN | NM_144997 | intron10 | c.1177–21G > A | --- | Splicing | Heterozygous | Likely Pathogenic | Branch Site mutation |
Two small frameshift deletions were identified in FLCN gene in patients F29 and F37, both resulting in a truncated protein of FLCN. And one of them, a c.1648_1658 deletion in exon 14 of the FLCN gene had been detected in patient F09. A nonsense mutation were detected in FBN1 gene in patient F32, it was a C to T transition (c.6169 C > T) in exon 51, predicting an early stop codon at position 2057 (p.Arg2057Ter) which causes a premature termination of the protein. This mutation has not been recorded in any database including FBN1 mutation database [41]. In this unscreened cohort of PSP patients, we also identified two missense mutations predicted to be damaging by SIFT and Polyphen2. One of them was a c.2269G > C transversion in exon 19 of the FBN1 gene, which converted the codon GAT for Aspartic at position 757 to CAT, a codon for Histidine. This variant is predicted by ScanProsite to situate in the EGF-like calcium-binding domain of FBN1 [42]. The other was a c.1631G > A transition in exon 15 of the TSC1 gene, which was predicted to substitute a polar uncharged glycine residue for a negatively charged glutamic residue (p.Gly544Glu), presenting a distinct change in amino acid properties. We also detected a G to A point mutation in intron 10 (c.1177–21G > A) of FLCN gene (Fig. 3a). This variant located within the branch-site sequence, by using the ESEfinder, we obtained a matrix score for the c.1177–21G > A substitution (2.2919) of the branch-site sequence, relatively less than a score for the wild type (2.7018) (Fig. 3b), indicating that it may disrupt the branch-site in this gene. However, because of unavailability of the patient’s tissue, we were unable to verify it at transcript level.
Fig. 3.
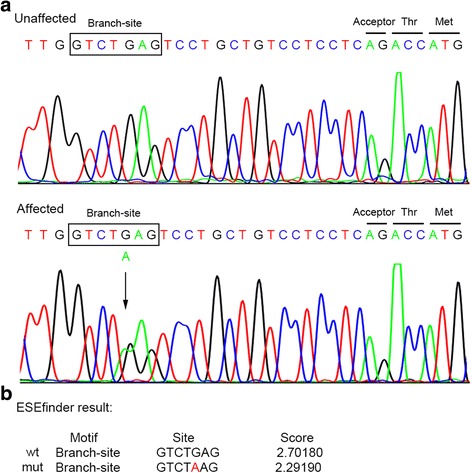
Sanger sequencing and ESEfinder results of the branch-site mutation. a Sequencing chromatograms show a heterozygous mutation c.1177–21G > A in FLCN gene. The arrow indicates the position of the nucleotide mutation and the boxed nucleotides indicate the branch site predicted by ESEfinder. b ESEfinder results for the c.1177–21G > A substitution and for the wild type of the branch-site sequence
In general, NGS method not only successfully identified missense, nonsense, splice site, intronic branch site, small indel, and large deletion changes in positive and blinded samples, but also determined approximate deletion junction within about 100 bp deviation by using a depth-based method.
Discussion
In this study, to establish a genetic diagnostic method for PSP, we evaluated the performance of targeted enrichment next-generation sequencing for the molecular diagnosis of PSP patients. We enriched 7 genes which are known to cause PSP using our self-designed Haloplex panel (Table 1) and sequenced them on Miseq platform. In test group, our target-enrichment NGS method successfully identified all 19 pathogenic mutations including SNVs, Indels and large intragenic mutations previously detected by Sanger sequencing and MLPA. No false negative result in test group was obtained. Moreover, we also detected the breakpoints of the deletions within 100 bp using a depth-based bioinformatics method. In blinded sample group, we detected 3 definitely pathogenic mutations including a novel FBN1 mutation and 3 likely pathogenic mutations including an intronic branch-site mutation. Therefore, we conclude that our system has a more extensive performance than Sanger sequencing and MLPA.
As BHD patients often receive medical attention for their pneumothorax onset in the hospital, genetic screening in PSP patients will help early diagnose of BHD syndrome, thus lead to early recognition and treatment of renal cell carcinoma. However, for screening, Sanger sequencing and MLPA are relatively low throughput, inefficient and expensive. Our targeted next-generation sequencing method successfully and efficiently identifies full spectrum of FLCN gene mutations in a single experiment.
By using a depth-based bioinformatics method, we also narrowed the range of breakpoints to a great extent, but we did not identify the precise breakpoints of the deletions, as it is reported that all FLCN intragenic deletions are Alu repeats-mediated [8], yet the low sensitivity in detecting CNVs in repeat regions is not completely solved [43], and Sanger sequencing is still the gold standard for breakpoint determination. Our NGS approach thus obviates the need for separate testing for genomic deletions/duplications after Sanger sequencing, and can also largely minimize the boundaries of the deletions, which will make it much easier to design primers for long-range PCR amplification. Our limitation is that one of the breakpoint of exon 9–14 deletion was not recovered since our design did not include the 3′ flank of FLCN gene. To optimize our method, the 5′ flank and 3′ flank sequences of tested genes should be included.
In the blinded PSP cohort, two out of twenty-one patients (9.5 %) were identified to carry pathogenic mutations in FLCN gene. This is in concordance with the previous report that up to 10 % of PSP patients are carrying FLCN mutations [7, 8]. A novel nonsense mutation in FBN1 (c.6169C > T; p.Arg2057Ter) was found in a male PSP patient. It was classified as definitely pathogenic mutation and was not recorded in any database. Our finding expand the spectrum of mutations in Marfan syndrome. Interestingly, a novel intronic variant in FLCN (c.1177–21G > A) was also detected by this method, bringing forward the need of raising awareness of intronic variant in pathogenesis, which is often avoided by traditional Sanger sequence.
Among the targeted genes, we also identified three mutations in FBN1 and TSC1 gene in PSP patients in addition to FLCN gene, but no predicted harmful variant was detected in TSC2, CBS, COL3A1 and SERPINA1 gene. Patients with mutations in these genes often express as syndromes and can be easily diagnosed. Isolated spontaneous pneumothorax patients with mutations in these genes are rare [44]. Nevertheless, pulmonologists should consider these syndromes and sequence these genes when treating patients for spontaneous pneumothorax, since early detection improves the prognosis significantly.
Moreover, a cost analysis of NGS approach showed significant savings. The cost with conventional Sanger sequencing is about $1760 per sample for 220 PCR and sequencing reactions, and the cost with 7 MLPA reactions is about $110 per sample. While a total cost of Haloplex target enrichment sequencing is about $400. With our NGS-based assay, a 78 % of cost savings per sample could be achieved in PSP diagnostic procedure.
Conclusions
This is the first study to evaluate the performance of NGS in the detection of exonic and intronic SNVs, Indels and large intragenic deletions, and to develop a cost- and time-effective screening system for the molecular diagnosis of BHDS in PSP cohort. In conclusion, NGS based Haloplex target enrichment technology is a rapid, high-throughput and cost-effective screening strategy for the molecular diagnosis of BHD in PSP patients, as it can replace Sanger sequencing and MLPA by simultaneously detecting exonic and intronic SNVs, small indels, large intragenic deletions and determining deletion junctions in PSP-related genes.
Acknowledgements
We would like to thank all the PSP patients who participated in this study.
Funding
This work was supported by the grant from National Natural Science Foundation of China (No. 81170002), Science and Technology Project of Jiangsu Province (No.BL2014053), Jiangsu Province Health Department of Medicine leading talent and innovation team project (LJ201109), Foundation of Nanjing Municipal Public Health Bureau (ZKX11030), National Natural Science Foundation of China (No. 91543131 and 8151101124), and the grant from the research projects in traditional Chinese medicine industry of China (No.201507004–2).
Abbreviations
- BHD
birt-hogg-dube syndrome
- CNV
copy number variations
- MLPA
multiple ligation-dependent probe amplification
- NGS
next generation sequencing
- PSP
primary spontaneous pneumothorax
- RCC
renal cell carcinoma
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XZ, DM, QG and LY conceived and designed the study, contributed to data analysis and interpretation, manuscript drafting and critical review for intellectual content and final approval of the manuscript. XZ, YD, HM, BZ and WW performed the experiment, contributed to data analysis and manuscript editing. DM, WZ, CZ, BC, MY, MC, YP and LC co-designed experiments, contributed to collection of the clinical data, manuscript editing and discussed analyses, interpretation, and presentation. YW and YJ contributed to study design, data collection and manuscript editing. All authors read and approved the final manuscript.
Contributor Information
Qian Gao, Phone: 8625 83595103, FAX: 862583594755, Email: qian_gao@nju.edu.cn.
Long Yi, Phone: 8625 83595103, FAX: 862583594755, Email: yilong@nju.edu.cn.
References
- 1.Abolnik IZ, Lossos IS, Zlotogora J, Brauer R. On the inheritance of primary spontaneous pneumothorax. Am J Med Genet. 1991;40(2):155–158. doi: 10.1002/ajmg.1320400207. [DOI] [PubMed] [Google Scholar]
- 2.Yellin A, Shiner RJ, Lieberman Y. Familial multiple bilateral pneumothorax associated with Marfan syndrome. Chest. 1991;100(2):577–578. doi: 10.1378/chest.100.2.577. [DOI] [PubMed] [Google Scholar]
- 3.Ishiguro T, Takayanagi N, Kawabata Y, Matsushima H, Yoshii Y, Harasawa K, Yamaguchi S, Yoneda K, Miyahara Y, Kagiyama N, Tokunaga D, Aoki F, Saito H, Kurashima K, Ubukata M, Yanagisawa T, Sugita Y, Okita H, Hatamochi A. Ehlers-Danlos syndrome with recurrent spontaneous pneumothoraces and cavitary lesion on chest X-ray as the initial complications. Intern Med. 2009;48(9):717–722. doi: 10.2169/internalmedicine.48.1818. [DOI] [PubMed] [Google Scholar]
- 4.Bass HN, LaGrave D, Mardach R, Cederbaum SD, Fuster CD, Chetty M. Spontaneous pneumothorax in association with pyridoxine-responsive homocystinuria. J Inherit Metab Dis. 1997;20(6):831–832. doi: 10.1023/A:1005384121649. [DOI] [PubMed] [Google Scholar]
- 5.Serapinas D, Obrikyte V, Vaicius D, Balciuviene R, Valavicius A, Sakalauskas R. Alpha-1 antitrypsin deficiency and spontaneous pneumothorax: possible causal relationship. Pneumologia. 2014;63(1):32–35. [PubMed] [Google Scholar]
- 6.Valentin-Mendoza S, Nieves-Nieves J, Fernandez-Medero R, Fernandez-Gonzales R, Adorno-Fontanez J, Adorno-Fontanez E. Pulmonary lymphangioleiomyomatosis: literature update. Bol Asoc Med P R. 2013;105(3):64–69. [PubMed] [Google Scholar]
- 7.Ren HZ, Zhu CC, Yang C, Chen SL, Xie J, Hou YY, Xu ZF, Wang DJ, Mu DK, Ma DH, Wang Y, Ye MH, Ye ZR, Chen BF, Wang CG, Lin J, Qiao D, Yi L. Mutation analysis of the FLCN gene in Chinese patients with sporadic and familial isolated primary spontaneous pneumothorax. Clin Genet. 2008;74(2):178–183. doi: 10.1111/j.1399-0004.2008.01030.x. [DOI] [PubMed] [Google Scholar]
- 8.Ding Y, Zhu C, Zou W, Ma D, Min H, Chen B, Ye M, Pan Y, Cao L, Wan Y, Zhang W, Meng L, Mei Y, Yang C, Chen S, Gao Q, Yi L. FLCN intragenic deletions in Chinese familial primary spontaneous pneumothorax. Am J Med Genet A. 2015;167(5):1125–1133. doi: 10.1002/ajmg.a.36979. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S, Pavlovich CP, Glenn G, Greenberg CR, Linehan WM, Zbar B. Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69(4):876–882. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, Sharma N, Walther M, Munroe D, Hill R, Maher E, Greenberg C, Lerman MI, Linehan WM, Zbar B, Schmidt LS. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2(2):157–164. doi: 10.1016/S1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 11.Johannesma PC, Reinhard R, Kon Y, Sriram JD, Smit HJ, van Moorselaar RJ, Menko FH, Postmus PE. Prevalence of Birt-Hogg-Dube syndrome in patients with apparently primary spontaneous pneumothorax. Eur Respir J. 2015;45(4):1191–1194. doi: 10.1183/09031936.00196914. [DOI] [PubMed] [Google Scholar]
- 12.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113(12):1674–1677. doi: 10.1001/archderm.1977.01640120042005. [DOI] [PubMed] [Google Scholar]
- 13.Roth JS, Rabinowitz AD, Benson M, Grossman ME. Bilateral renal cell carcinoma in the Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 1993;29(6):1055–1056. doi: 10.1016/S0190-9622(08)82049-X. [DOI] [PubMed] [Google Scholar]
- 14.Toro JR, Glenn G, Duray P, Darling T, Weirich G, Zbar B, Linehan M, Turner ML. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135(10):1195–1202. doi: 10.1001/archderm.135.10.1195. [DOI] [PubMed] [Google Scholar]
- 15.Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, Toure O, Wei MH, Schmidt LS, Davis L, Zbar B, Choyke P, Steinberg SM, Nguyen DM, Linehan WM. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube syndrome. Am J Respir Crit Care Med. 2007;175(10):1044–1053. doi: 10.1164/rccm.200610-1483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maffe A, Toschi B, Circo G, Giachino D, Giglio S, Rizzo A, Carloni A, Poletti V, Tomassetti S, Ginardi C, Ungari S, Genuardi M. Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dube syndrome ascertained for non-cutaneous manifestations. Clin Genet. 2011;79(4):345–354. doi: 10.1111/j.1399-0004.2010.01480.x. [DOI] [PubMed] [Google Scholar]
- 17.Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, Nordenskjold M, Hansen TV, Solly J, Maher ER. Birt-Hogg-Dube syndrome: diagnosis and management. Lancet Oncol. 2009;10(12):1199–1206. doi: 10.1016/S1470-2045(09)70188-3. [DOI] [PubMed] [Google Scholar]
- 18.Frohlich BA, Zeitz C, Matyas G, Alkadhi H, Tuor C, Berger W, Russi EW. Novel mutations in the folliculin gene associated with spontaneous pneumothorax. Eur Respir J. 2008;32(5):1316–1320. doi: 10.1183/09031936.00132707. [DOI] [PubMed] [Google Scholar]
- 19.Graham RB, Nolasco M, Peterlin B, Garcia CK. Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med. 2005;172(1):39–44. doi: 10.1164/rccm.200501-143OC. [DOI] [PubMed] [Google Scholar]
- 20.Gunji Y, Akiyoshi T, Sato T, Kurihara M, Tominaga S, Takahashi K, Seyama K. Mutations of the Birt Hogg Dube gene in patients with multiple lung cysts and recurrent pneumothorax. J Med Genet. 2007;44(9):588–593. doi: 10.1136/jmg.2007.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunogi M, Kurihara M, Ikegami TS, Kobayashi T, Shindo N, Kumasaka T, Gunji Y, Kikkawa M, Iwakami S, Hino O, Takahashi K, Seyama K. Clinical and genetic spectrum of Birt-Hogg-Dube syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet. 2010;47(4):281–287. doi: 10.1136/jmg.2009.070565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Painter JN, Tapanainen H, Somer M, Tukiainen P, Aittomaki K. A 4-bp deletion in the Birt-Hogg-Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet. 2005;76(3):522–527. doi: 10.1086/428455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang CY, Wang HC, Chen JS, Yu CJ. Isolated familial pneumothorax in a Taiwanese family with Birt-Hogg-Dube syndrome. J Postgrad Med. 2013;59(4):321–323. doi: 10.4103/0022-3859.123169. [DOI] [PubMed] [Google Scholar]
- 24.Houweling AC, Gijezen LM, Jonker MA, van Doorn MB, Oldenburg RA, van Spaendonck-Zwarts KY, Leter EM, van Os TA, van Grieken NC, Jaspars EH, de Jong MM, Bongers EM, Johannesma PC, Postmus PE, van Moorselaar RJ, van Waesberghe JH, Starink TM, van Steensel MA, Gille JJ, Menko FH. Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105(12):1912–1919. doi: 10.1038/bjc.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L, Walther M, Choyke P, Weirich G, Hewitt SM, Duray P, Gabril F, Greenberg C, Merino MJ, Toro J, Linehan WM. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11(4):393–400. [PubMed] [Google Scholar]
- 26.Wei MH, Blake PW, Shevchenko J, Toro JR. The folliculin mutation database: an online database of mutations associated with Birt-Hogg-Dube syndrome. Hum Mutat. 2009;30(9):E880–890. doi: 10.1002/humu.21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim DH, Rehal PK, Nahorski MS, Macdonald F, Claessens T, Van Geel M, Gijezen L, Gille JJ, Giraud S, Richard S, van Steensel M, Menko FH, Maher ER. A new locus-specific database (LSDB) for mutations in the folliculin (FLCN) gene. Hum Mutat. 2010;31(1):E1043–1051. doi: 10.1002/humu.21130. [DOI] [PubMed] [Google Scholar]
- 28.Benhammou JN, Vocke CD, Santani A, Schmidt LS, Baba M, Seyama K, Wu X, Korolevich S, Nathanson KL, Stolle CA, Linehan WM. Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dube syndrome. Genes Chromosomes Cancer. 2011;50(6):466–477. doi: 10.1002/gcc.20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A. 2010;107(28):12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valencia CA, Rhodenizer D, Bhide S, Chin E, Littlejohn MR, Keong LM, Rutkowski A, Bonnemann C, Hegde M. Assessment of target enrichment platforms using massively parallel sequencing for the mutation detection for congenital muscular dystrophy. J Mol Diagn. 2012;14(3):233–246. doi: 10.1016/j.jmoldx.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperandeo MP, Panico M, Pepe A, Candito M, de Franchis R, Kraus JP, Andria G, Sebastio G. Molecular analysis of patients affected by homocystinuria due to cystathionine beta-synthase deficiency: report of a new mutation in exon 8 and a deletion in intron 11. J Inherit Metab Dis. 1995;18(2):211–214. doi: 10.1007/BF00711769. [DOI] [PubMed] [Google Scholar]
- 32.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263(13):6226–6232. [PubMed] [Google Scholar]
- 33.Jones AC, Daniells CE, Snell RG, Tachataki M, Idziaszczyk SA, Krawczak M, Sampson JR, Cheadle JP. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum Mol Genet. 1997;6(12):2155–2161. doi: 10.1093/hmg/6.12.2155. [DOI] [PubMed] [Google Scholar]
- 34.Blyth M, Foulds N, Turner C, Bunyan D. Severe Marfan syndrome due to FBN1 exon deletions. Am J Med Genet. 2008;10:1320–1324. doi: 10.1002/ajmg.a.32229. [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 38.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31(13):3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collod-Beroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A, Hyland JC, Holman K, Kaitila I, Loeys B, Matyas G, Nuytinck L, Peltonen L, Rantamaki T, Robinson P, Steinmann B, Junien C, Beroud C, Boileau C. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22(3):199–208. doi: 10.1002/humu.10249. [DOI] [PubMed] [Google Scholar]
- 42.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34(Web Server issue):365–5. doi:10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed]
- 43.Teo SM, Pawitan Y, Ku CS, Chia KS, Salim A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics. 2012;28(21):2711–2718. doi: 10.1093/bioinformatics/bts535. [DOI] [PubMed] [Google Scholar]
- 44.Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med. 2006;12(4):268–272. doi: 10.1097/01.mcp.0000230630.73139.f0. [DOI] [PubMed] [Google Scholar]