

Abstract
Epoxide hydrolase is involved in metabolism of vasoactive and anti-inflammatory epoxyeicosatrienoic acids to their corresponding diols. Consequently, epoxide hydrolase 2 (EPHX2) is a candidate cardiovascular disease (CVD) gene. We investigated EPHX2 for association with subclinical CVD in European American (EA) and African American (AA) families from the Diabetes Heart Study. The R287Q polymorphism was associated with carotid artery calcified plaque (CarCP) in EAs. Other EPHX2 polymorphisms were associated with coronary artery calcified plaque (CorCP), CarCP or carotid artery intima-media thickness (IMT). Polymorphism rs7837347 was associated with all traits in the AAs (p=0.003, 0.001 and 0.017, respectively). Polymorphism rs7003694 displayed association with IMT (p=0.017) and, along with rs747276, a trend towards association with CorCP in diabetic EAs (p=0.057 and 0.080, respectively). These results provide additional evidence that EPHX2 contributes to the risk of subclinical CVD, although the true trait defining polymorphisms may not be identified and the effect size could be small.
Keywords: calcified plaque, cardiovascular disease, gene polymorphisms, type 2 diabetes
Introduction
Cardiovascular disease (CVD) is a complex disorder well known to have both environmental and genetic susceptibility components. Many studies have investigated the contribution of candidate genes to the risk of CVD. Recently, a polymorphism in epoxide hydrolase 2 (EPHX2), the gene encoding soluble epoxide hydrolase (sEH), was found to be associated with subclinical CVD as measured by coronary artery calcified plaque (CorCP).1,2 Epoxide hydrolases catalyze degradation of vasoactive epoxyeicosatrienoic acids (EETs), produced from arachidonic acid by cytochrome p450 2J2 (CYP2J2),3 into their corresponding dihydroxyeicosatrienoic acids (DHETs).4 EETs and DHETs have been shown to promote vasorelaxation of small arteries,5,6 and display anti-inflammatory properties through the inhibition of nuclear factor kappa B (NF-κB) activation.7 These data have led to speculation that variations in the abundance or function of sEH may contribute to the development of pathogenic states in the vasculature through the regulation of these vasoactive and anti-inflammatory compounds. Soluble EH has also been shown to modulate vascular smooth muscle (VSM) cell proliferation,8 providing another potential avenue for the contribution of this enzyme to CVD. Nevertheless, the precise role of sEH in the pathogenesis of calcified atherosclerotic plaque is unclear.
In order to assess further the contribution of this gene to the risk of CVD, we have assessed several variants of the EPHX2 gene in a sample of type 2 diabetes mellitus (T2DM) patients from the Diabetes Heart Study (DHS) and their available unaffected siblings. T2DM increases significantly the risk of CVD. Mortality from CVD is 2–5-fold greater in diabetic versus non-diabetic subjects, with almost 60% of diabetic individuals having CVD complications.9–12 The majority of participants in this study have detectable CorCP. This enrichment of the study population with subjects having extensive subclinical CVD should provide increased power to detect association of EPHX2 with CVD.
Methods
Patients and phenotyping
Recruitment and phenotyping of DHS participants have been previously described.13,14 Briefly, siblings concordant for T2DM were recruited from internal medicine clinics, endocrinology clinics and community advertising. T2DM was defined as a clinical diagnosis of diabetes after the age of 34 years, in the absence of historical evidence of diabetic ketoacidosis, and active treatment at the time of examination. Unaffected siblings, similar in age to the siblings with T2DM, were also invited to participate, as were any additional diabetes-affected siblings. The sample includes European-American (EA) and African-American (AA; approximately 15% of the total) participants.
All protocols were approved by the institutional review board of Wake Forest University School of Medicine and all participants gave their informed consent. Participant examinations were conducted in the General Clinical Research Center of the Wake Forest University Baptist Medical Center. They included interviews for medical history and health behaviours, anthropometric measures, resting blood pressure, fasting blood sampling and spot urine collection. Laboratory assays included urine albumin and creatinine, total cholesterol, high-density lipoprotein cholesterol (HDL), triglycerides, glycosylated haemoglobin (HbA1C), fasting glucose and blood chemistries.
Intima-media thickness (IMT) of the common carotid artery was measured by high-resolution B-mode ultrasonography with a 7.5-MHz transducer and a Biosound Esaote (AU5) ultrasound machine, as previously described.13 Coronary artery (CorCP) and carotid artery (CarCP) calcified plaque were measured using fast-gated helical computerised tomography (CT) scanners; calcium scores were calculated as described previously.14,15
Genetic analysis
Total genomic DNA was purified from whole blood samples obtained from subjects using the PUREGENE® DNA isolation kit (Gentra, Inc., Minneapolis, MN). DNA was quantitated using standardised fluorometric readings on a Hoefer DyNA® Quant 200 fluorometer (Hoefer Pharmacia Biotech Inc., San Francisco, CA). Each sample was diluted to a final concentration of 5 ng/μL.
Six single nucleotide polymorphisms (SNPs) were chosen from the HapMap database (http://www.hapmap.org)16 to cover the 63.84 kb genomic region containing EPHX2 comprehensively, including 5 kb upstream and downstream of the EPHX2 transcript. Four of the SNPs evaluated in this study had a minor allele frequency (MAF) more than or equal to 0.05 in both the Centre d’Etude du Polymorphisme Humain (CEPH) Utah residents with ancestry from northern and western Europe (CEU population) and the Yoruba population from Ibadan, Nigeria (YRI population). Of the remaining two SNPs, one was polymorphic in the YRI population but monomorphic in the CEU population (rs7837347); the other was polymorphic in the CEU population and had no data available for the YRI population (rs7003694). The R287Q polymorphism (rs751141) was selected based on an a priori hypothesis of association with vascular calcified plaque.1 The remaining five SNPs were selected in an effort to maintain consistent SNP spacing (average spacing of 1 SNP / 9 kb). SNP rs1042064 in the 3′ untranslated region of the gene failed to genotype and was removed from the analysis.
SNP genotypes were determined using a MassARRAY® SNP genotyping system (Sequenom, Inc., San Diego, CA), as previously described.17 This genotyping system uses single base extension reactions to create allele-specific products that differ in mass and can be separated and automatically scored in a matrix-assisted laser desorption ionisation-time-of-flight (MALDI-TOF) mass spectrometer. Primers for PCR amplification and extension reactions were designed using the Assay Design software (Sequenom Inc., San Diego, CA).
Statistical analyses
Clinical characteristics were compared between the two ethnic groups using generalised estimating equations (GEE1) as described below. Ethnic-specific allele and genotype frequencies were calculated from unrelated probands and tested for departure from Hardy–Weinberg Equilibrium (HWE) using a chi-squared test. Linkage disequilibrium (LD) was assessed through calculation of D′ and r2 values. To test for an association between each SNP and each phenotype, a series of GEE118 models was evaluated. The correlation between subjects within a family was adjusted for in the analyses by assuming exchangeable correlation among siblings within a pedigree and computing the sandwich estimator of the variance.19 The sandwich estimator is also denoted the robust or empirical estimator of the variance as it is robust to misspecification of the correlation matrix because it estimates the within-pedigree correlation matrix from the first two moments of the data.19
A two degrees of freedom overall test of genotypic association was performed for all SNPs with IMT, CorCP and CarCP. For phenotypes that demonstrated a significant association, the three individual contrasts defined by the a priori genetic models (i.e. dominant, additive and recessive) were computed. This is consistent with the Fisher’s protected least significant difference multiple comparison procedure. The test of the dominant model compares the phenotypic means of the combined “1/2” and “2/2” genotypic class versus the “1/1” genotypic class (i.e. a difference in mean based upon presence versus absence of allele “2”). The additive model tests for a cumulative effect of allele 2. The test of the recessive model compares the phenotypic mean of the “2/2” genotypic class versus the mean of the combined “1/1” and “1/2” genotypic class (i.e. two copies of allele “2”). Within each trait, a sequential Bonferroni multiple comparison adjustment was computed.20 This conservative multiple testing adjustment rank-orders the observed p values, divides the a priori threshold for statistical significance by the p value rank and declares significance if the observed p value is less than the rank-adjusted threshold for significance. Because this investigation contains a strong a priori hypothesis, we report the unadjusted p value and indicate which SNPs retain statistical significance after adjustment by the conservative sequential Bonferroni.
All analyses were conducted incorporating effects of known risk factors (age, gender, diabetes status, smoking status and use of lipid-lowering medication) in the models. IMT, CorCP+1 and CarCP+1 values were natural log-transformed to best approximate the distributional assumptions (e.g. approximate conditional normality, homogeneity of variance) of these tests.
Results
DNA was collected from 982 EA individuals from 367 families and 176 AA individuals from 73 families. Clinical characteristics of participants are summarised in table 1. Obesity, measured through body mass index (BMI), was significantly different between the two ethnic groups (31.8±6.6 kg/m2 in EA versus 33.8±7.2 kg/m2 in AA, p=0.0003). Subclinical CVD, defined as the presence of CorCP and/or CarCP, was prevalent in both EA and AA subjects. EA subjects had greater levels of CorCP (1,254± 2,445 versus 727±1,604, p=0.0091) and CarCP (341±695 versus 192±614, p=0.0096) than AA subjects, and had more prevalent CVD. In general, the participants are representative of a diabetes-enriched collection of older, obese subjects with significant CVD burden.
Table 1.
Clinical characteristics of all participants by ethnicity. Mean ± standard deviation or % (n)
| European-American | African-American | |
|---|---|---|
| N | 982 | 176 |
| Age (years) | 61.6.±9.4 | 58.8+9.2 |
| Female (%) | 53.5 (525) | 67.1 (118) |
| Diabetes diagnosis (%) | 83.6 (821) | 89.8 (158) |
| Diabetes duration (years) | 10.3±7.0 | 10.7±7.9 |
| BMI (kg/m2) | 31.8±6.6 | 33.8±7.2 |
| Lipids | ||
| Cholesterol (mg/dL)* | 188±42 | 191±37 |
| HDL (mg/dL)* | 43±13 | 50±15 |
| LDL (mg/dL)* | 106±32 | 114±31 |
| Smoking (%) current or past | 59.1 (578) | 61.5 (107) |
| Lipid-lowering medication (%) | 42.7 (415) | 33.1 (59) |
| Prevalent CVD (%) | ||
| Myocardial infarction | 19.7 (191) | 8.6 (15) |
| Angina | 18.2 (166) | 16.3 (26) |
| Stroke | 9.5 (92) | 6.4 (11) |
| Coronary Artery Bypass Graft | 14.1 (138) | 5.7 (10) |
| Angioplasty | 15.2 (148) | 7.4 (13) |
| Endarterectomy | 2.2 (21) | 0.6 (1) |
| Vascular imaging | ||
| CorCP>0 | 92.9 (737) | 91.9 (147) |
| Untransformed CorCP score | 1,254±2,445 (793) | 727±1604 (160) |
| Log CorCP1 | 5.2±2.6 (793) | 4.4±2.5 (160) |
| Untransformed CarCP score | 341±695 (897) | 192±614 (168) |
| Log CarCP2 | 3.6±2.7 (897) | 2.6±2.5 (168) |
| Untransformed IMT (mm) | 0.67±0.13 (952) | 0.69±0.13 (163) |
| Log IMT3 | 0.51±0.08 (952) | 0.52±0.08 (163) |
Key: BMI = body mass index; HDL = high-density lipoprotein cholesterol; LDL = low-density lipoprotein cholesterol; CVD = cardiovascular disease; CorCP = coronary artery calcified plaque; CarCP = carotid artery calcified plaque; IMT = intima-media thickness;
to convert mg/dL to mmol/L. multiply by 0.0259;
EA skewness=−0.290; EA kurtosis=−0.106; AA skewness=−0.032; AA kurtosis=−0.351;
EA skewness=−0.216; EA kurtosis=−0.692; AA skewness=0.232; AA kurtosis=−0.787;
EA skewness=1.038; EA kurtosis=2.346; AA skewness=0.961; AA kurtosis=1.410
DNA from these participants was genotyped for six SNPs selected to cover the 73.84 kb genomic region containing EPHX2 completely (table 2). One SNP (rs1042064) failed to genotype using the Sequenom platform, and was therefore excluded from subsequent analysis. The ability of the five remaining SNPs to capture genotypic variation was assessed using the greedy pair-wise tagging algorithm implemented in the Tagger program21 of Haploview.16 In the CEU population, the four polymorphic SNPs (rs7003694, rs751141, rs721619 and rs747276) captured a very high proportion of the genetic variation within this region (mean r2=0.82). In the YRI population, the four polymorphic SNPs (rs7837347, rs751141, rs721619 and rs747276) captured less genetic variation (mean r2=0.37). No data were available on rs7003694 in the YRI population. In each ethnic group, allele and genotype frequencies were consistent with those expected under HWE. SNP rs7837347 was not variable in the EA subjects and had a minor allele frequency of only 0.06 in the AA group. The extent of LD between SNPs was assessed with the D′ statistic implemented in Haploview.22 The four polymorphic SNPs in EAs were contained in a single block of high LD (figure 1a) whereas the four polymorphic SNPs in AAs did not conform to any block structure (figure 1b).
Table 2.
SNPs genotyped in EPHX2. The frequency of allele 2 and the number of observations of each genotype in unrelated probands by ethnicity is shown
| SNP | Position in gene | Alleles 1/2 | European-Americans
|
African-Americans
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | 1/1 | 1/2 | 2/2 | Frequency | 1/1 | 1/2 | 2/2 | |||
| rs7003694 | Promoter | T/C | 0.092 | 277 | 58 | 2 | 0.174 | 44 | 22 | 0 |
| rs7837347 | Intron 2 | C/T | 0.000 | – | – | – | 0.063 | 63 | 8 | 1 |
| rs751141 | R287Q | G/A | 0.101 | 282 | 61 | 6 | 0.146 | 49 | 22 | 1 |
| rs721619 | Intron 11 | C/G | 0.320 | 165 | 145 | 41 | 0.102 | 53 | 16 | 1 |
| rs747276 | Intron 12 | G/C | 0.139 | 259 | 81 | 9 | 0.667 | 14 | 29 | 26 |
Key: SNP = single nucleotide polymorphism
Figure 1.
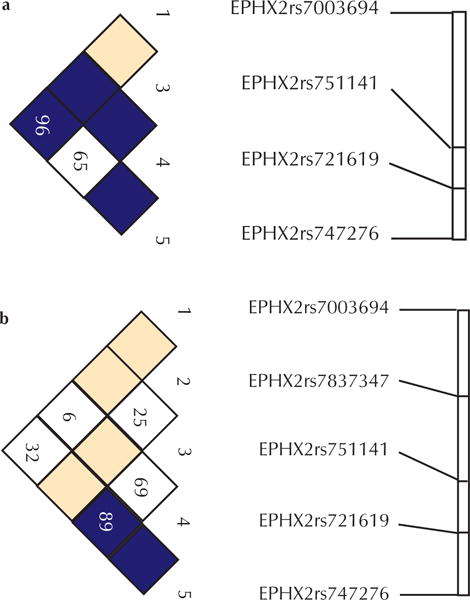
LD between the EPHX2 polymorphic SNPs genotyped in (a) European-Americans and (b) African-Americans
Each SNP in EPHX2 was assessed for association with IMT, CorCP and CarCP in each ethnic group (table 3). Additionally, EAs with diabetes were analysed as a separate subgroup. Due to the small number of AAs available for study, only those with diabetes were included in the analysis in order to maximise the homogeneity of the sample.
Table 3.
P values* for association of EPHX2 SNPs with measures of subclinical atherosclerosis
| SNP | IMT | CorCP | CarCP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EA | dEA | AA | EA | dEA | AA | EA | dEA | AA | |
| rs7003694 | 0.058 | 0.017 | 0.560 | 0.266 | 0.057 | 0.374 | 0.791 | 0.700 | 0.569 |
| rs7837347 | – | – | 0.017 | – | – | 0.003 | – | – | 0.001 |
| rs751141 | 0.446 | 0.448 | 0.579 | 0.188 | 0.757 | 0.194 | 0.048 | 0.077 | 0.184 |
| rs721619 | 0.331 | 0.193 | 0.850 | 0.284 | 0.348 | 0.648 | 0.932 | 0.610 | 0.498 |
| rs747276 | 0.312 | 0.261 | 0.559 | 0.244 | 0.080 | 0.722 | 0.209 | 0.342 | 0.246 |
Key: EA = European-American; dEA = diabetic European-American; AA = African-American; SNP = single nucleotide polymorphism; IMT = intima-media thickness; CorCP = coronary artery calcified plaque; CarCP = carotid artery calcified plaque;
based on 2 degrees-of-freedom test
SNP rs751141 (coding for R287Q), previously reported to be associated with CorCP in AAs but not EAs,1 was not associated with CorCP in either ethnic group. There was evidence of association between rs751141 and CarCP (p=0.048) in the EA sample. Although not statistically significant after adjusting for all polymorphisms using the sequential Bonferroni adjustment, this association merits consideration given the biological similarity to CorCP. The geometric mean for CarCP by genotype is shown in table 4, along with the p values for association under the three a priori genetic models. The strongest evidence of association was observed with the additive model, consistent with the stepwise increase in mean CarCP with the addition of each copy of allele 2.
Table 4.
Untransformed mean trait values by genotype and model-specific p values for association
| SNP (Sample) | Trait | Mean ± Standard Deviation (N)* | P value | ||||
|---|---|---|---|---|---|---|---|
| 1/1 | 1/2 | 2/2 | Dominant | Additive | Recessive | ||
| rs7003694 (dEA) | IMT | 0.68±0.13 (608) | 0.68±0.15 (126) | 0.60±0.04 (5) | 0.874 | 0.654 | 0.004 |
| rs7837347 (AA) | IMT | 0.70±0.13 (139) | 0.63±0.11 (17) | 0.75±0.15 (3) | 0.050 | 0.277 | 0.683 |
| CorCP | 659±1612 (137) | 698±1235 (15) | 1021±622 (4) | 0.008 | 0.002 | 0.002 | |
| CarCP | 159±501 (143) | 188±483 (17) | 313±337 (4) | 0.819 | 0.419 | 0.0002 | |
| rs751141 (EA) | CarCP | 324±648 (688) | 426±882 (156) | 486±749 (14) | 0.083 | 0.042 | 0.046 |
Numbers reported reflect the data from individuals with both genotype and phenotype information available.
Key: dEA = diabetic European-Americans; AA = African-Americans; IMT = intima-media thickness; CorCP = coronary artery calcified plaque; CarCP = carotid artery calcified plaque; SNP = single nucleotide polymorphism
The strongest evidence for association was found with SNP rs7837347. This SNP was significantly associated with all three traits in AAs (table 3) and remained statistically significant even after applying the conservative sequential Bonferroni multiple comparison correction. As previously noted, this polymorphism is uncommon among the current AA sample and is not present in the current EA sample. Thus, these results are interesting but should be viewed as exploratory in nature. The association with IMT appears to be driven by a reduction in IMT among the heterozygous individuals (table 4). The dominant genetic model provides the evidence of statistical association (p=0.05) but the two individuals homozygous for the 2 (T) allele have the largest standard deviation. The association between rs7837347 and CorCP is most consistent with either a dominant (p=0.008) or additive (p=0.002) genetic model as demonstrated by an increasing mean as a function of the number of copies of allele 2. In contrast, the association between rs7837347 and CarCP appear to be most consistent with a recessive genetic model. However, this result is suspect given that only three individuals were homozygous for the 2 allele.
SNP rs7003694 is also associated with IMT, particularly in the EA diabetic subset, although the strongest evidence comes from a recessive model and is highly influenced by only five homozygotes (table 4). This SNP, as well as rs747276, displays suggestive evidence of association with CorCP in the diabetic EA group (p=0.057 and 0.080, respectively).
Discussion
While the evidence for association of the EPHX2 gene with any of the three subclinical CVD measures is limited, the results of this study are consistent with a role for the EPHX2 gene in CVD risk, when interpreted in context with the previous reports of association.1,2,23,24
Fornage et al.1 and Wei et al.2 investigated the contribution of the EPHX2 R287Q variant and haplotypes containing this variant to CorCP in the CARDIA cohort of young, largely asymptomatic adults. They were able to include a larger sample than the current study but only 11% of EAs and 7% of AAs had detectable CorCP, which translates to approximately 172 and 89 individuals, respectively. The risk factor profile of the current study is quite different from that of the CARDIA cohort in that the DHS population consists of older, obese, diabetic subjects with a significant CVD burden. Approximately 76% of our sample had detectable CorCP, namely 737 EA and 147 AA individuals, providing substantially increased power to detect an association with this trait, particularly in the EA samples.
Fornage et al.1 were unable to detect association in the Caucasian component of the CARDIA study using single SNP analysis, although a variant in the gene was associated with CorCP in AAs. The DHS15 and other studies25,26 have indicated ethnic differences in arterial calcification. Fornage et al.1 recognised that the EPHX2 gene may account for some of this difference, but also pointed out that other factors whose prevalence differs between races may impact their observed association and contribute to the observed ethnic differences. Our study of EPHX2 suggests that variation within the gene does not account for ethnic differences in arterial calcification. A small effect was detected in the EA individuals with associations of both IMT and calcified plaque in the current study. This difference may be due to the improved power of the current study, or to the specific SNPs selected. As with the CARDIA study, we failed to detect any association of the R287Q polymorphism (rs751141) in EAs. However, we also failed to detect an effect of this SNP in AAs, suggesting that any effect this gene is having in the CARDIA cohort is not necessarily mediated through this variant.
Since the initial report of Fornage et al.,1 more detailed investigations of the association of sequence variation in EPHX2 with cardiovascular risk have been conducted. In the CARDIA cohort, a haplotype tagged by the R287Q polymorphism and a different haplotype tagged by an intron 11 SNP (rs721619) were associated with increased risk for CorCP in AAs and Caucasians, respectively.2 Each of these polymorphisms was also significantly associated with CorCP in the single SNP analyses. The Atherosclerosis Risk in Communities (ARIC) study evaluated EPHX2 polymorphisms for association with ischaemic stroke24 and coronary heart disease (CHD) risk.23 Haplotypes associated with risk for ischaemic stroke were identified in both AAs and Caucasians.24 Interestingly, the haplotype spanning the intron 11 region that was associated with increased CorCP in the Caucasians of the CARDIA cohort2 was also associated with lower ischaemic stroke risk in Caucasians from the ARIC study.24 The ARIC study also identified the K55R variant, and haplotypes containing this variant, as being associated with increased risk for CHD in Caucasians; the R287Q polymorphism was not significantly associated with CHD in either the AAs or the Caucasians.23 In the present study, we have evaluated both the R287Q polymorphism and the intron 11 polymorphism (rs721619) for association with subclinical measures of CVD; however, neither was associated with CorCP, CarCP or carotid IMT in either the EA or AA ethnic groups.
Firm conclusions about association are not possible due to the low allele frequency for most of the SNPs. This increases the likelihood of observing a false positive association, particularly under the recessive model. If there is an association between these polymorphisms and CVD measures, the effect size is likely to be small. Thus, much larger sample sizes will be required to delineate the putative relationships fully. It should be noted that the statistical power in the AA sample of the current study (n=176 individuals) is modest and therefore, results in the AA sample should be interpreted with caution. Specifically, under an additive genetic model and for the minor allele frequencies in table 2, the AA sample has 0.70 and 0.80 power to detect a third to a half of a standard deviation of the respective vascular calcium measures reported in table 1. Additional studies may also benefit from complete resequencing of the gene, including intronic regions, in order to identify additional genetic variation, allowing selection of more informative SNPs and a more thorough description of the LD pattern.
This study consists of an evaluation of several EPHX2 SNPs with three measures of subclinical atherosclerosis and, therefore, includes multiple statistical comparisons. This study was undertaken with an a priori hypothesis for the association of EPHX2 SNPs with the reported phenotypes, based on previous reports from the CARDIA1,2 and ARIC23,24 studies, and should therefore be considered as a confirmation study for the R287Q polymorphism. It should be acknowledged that there is no generally accepted method of application of corrections in studies of this kind where the SNPs are in LD with each other and the traits are also correlated. We do not believe the genomic structure of the region or the interdependence among the traits supports a highly conservative approach such as a Bonferroni correction for multiple comparisons. However, even if we make a conservative sequential Bonferroni adjustment across all traits and polymorphisms, the intron 2 SNP rs7837347 remains significantly associated with CarCP and CorCP.
In summary, the Diabetes Heart Study adds to the evidence that the EPHX2 gene is involved in CVD risk. However, studies in larger at-risk cohorts incorporating complete resequencing of the gene are required to confirm the association of both single SNPs and haplotypes.
Acknowledgments
This study was supported in part by the General Clinical Research Center of the Wake Forest University School of Medicine grant M01 RR07122, and by NHLBI R01 grant HL67348 awarded to DWB and R01 AR48797 awarded to JJC.
KPB was supported by an American Diabetes Association Mentor-based Fellowship.
Footnotes
Conflicts of interest statement
None declared.
Contributor Information
KATHRYN P BURDON, Postdoctoral Research Fellow, Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, NC, US; Postdoctoral Research Fellow, Center for Human Genomics, Wake Forest University School of Medicine, Winston-Salem, NC, US.
ALLISON B LEHTINEN, Postdoctoral Research Fellow, Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, NC, US; Postdoctoral Research Fellow, Center for Human Genomics, Wake Forest University School of Medicine, Winston-Salem, NC, US.
CARL D LANGEFELD, Section Head, Statistical Genetics and Bioinformatics, Department of Public Health Sciences, Wake Forest University School of Medicine, Winston-Salem, NC, US.
J JEFFREY CARR, Professor, Vice Chair for Clinical Research, Department of Radiology, Wake Forest University School of Medicine, Winston-Salem, NC, US.
STEPHEN S RICH, Professor, Department of Public Health Sciences, Wake Forest University School of Medicine, Winston-Salem, NC, US.
BARRY I FREEDMAN, Section Head, John H Felts III, MD, Professor of Internal Medicine/Nephrology, Department of Internal Medicine, Wake Forest University School of Medicine, Winston-Salem, NC, US.
DAVID HERRINGTON, Professor, Department of Internal Medicine, Wake Forest University School of Medicine, Winston-Salem, NC, US.
DONALD W BOWDEN, Professor, Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, NC, US; Associate Director, Center for Human Genomics, Wake Forest University School of Medicine, Winston-Salem, NC, US; Professor, Department of Internal Medicine, Wake Forest University School of Medicine, Winston-Salem, NC, US.
References
- 1.Fornage M, Boerwinkle E, Doris PA, Jacobs D, Liu K, Wong ND. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation. 2004;109:335–9. doi: 10.1161/01.CIR.0000109487.46725.02. [DOI] [PubMed] [Google Scholar]
- 2.Wei Q, Doris PA, Pollizotto MV, et al. Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis. 2007;190:26–34. doi: 10.1016/j.atherosclerosis.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. Faseb J. 1992;6:731–6. doi: 10.1096/fasebj.6.2.1537463. [DOI] [PubMed] [Google Scholar]
- 4.Chacos N, Capdevila J, Falck JR, et al. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;223:639–48. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- 5.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–23. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 6.Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 1998;83:932–9. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- 7.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA. 2002;99:2222–7. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kannel WB, McGee DL. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care. 1979;2:120–6. doi: 10.2337/diacare.2.2.120. [DOI] [PubMed] [Google Scholar]
- 10.Pan WH, Cedres LB, Liu K, et al. Relationship of clinical diabetes and asymptomatic hyperglycemia to risk of coronary heart disease mortality in men and women. Am J Epidemiol. 1986;123:504–16. doi: 10.1093/oxfordjournals.aje.a114266. [DOI] [PubMed] [Google Scholar]
- 11.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 12.Geiss L, Herman W, Smith P. National Diabetes Data Group Diabetes in America. Bethesda, Md: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 1995. [Google Scholar]
- 13.Lange LA, Bowden DW, Langefeld CD, et al. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002;33:1876–81. doi: 10.1161/01.str.0000019909.71547.aa. [DOI] [PubMed] [Google Scholar]
- 14.Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes. 2001;50:861–6. doi: 10.2337/diabetes.50.4.861. [DOI] [PubMed] [Google Scholar]
- 15.Wagenknecht LE, Langefeld CD, Carr JJ, et al. Race-specific relationships between coronary and carotid artery calcification and carotid intimal medial thickness. Stroke. 2004;35:e97–e99. doi: 10.1161/01.STR.0000127081.99767.1d. [DOI] [PubMed] [Google Scholar]
- 16.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bensen JT, Langefeld CD, Hawkins GA, et al. Nucleotide variation, haplotype structure, and association with end-stage renal disease of the human interleukin-1 gene cluster. Genomics. 2003;82:194–217. doi: 10.1016/s0888-7543(03)00123-x. [DOI] [PubMed] [Google Scholar]
- 18.Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42:121–30. [PubMed] [Google Scholar]
- 19.Hardin J, Hilbe J. Generalized estimating equations. New York, New York: Chapman & Hall/CRC; 2003. [Google Scholar]
- 20.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–02. [Google Scholar]
- 21.de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 22.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 23.Lee CR, North KE, Bray MS, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15:1640–9. doi: 10.1093/hmg/ddl085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fornage M, Lee CR, Doris PA, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–37. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman AB, Naydeck BL, Whittle J, Sutton-Tyrrell K, Edmundowicz D, Kuller LH. Racial differences in coronary artery calcification in older adults. Arterioscler Thromb Vasc Biol. 2002;22:424–30. doi: 10.1161/hq0302.105357. [DOI] [PubMed] [Google Scholar]
- 26.Lee TC, O’Malley PG, Feuerstein I, Taylor AJ. The prevalence and severity of coronary artery calcification on coronary artery computed tomography in black and white subjects. J Am Coll Cardiol. 2003;41:39–44. doi: 10.1016/s0735-1097(02)02618-9. [DOI] [PubMed] [Google Scholar]