

Abstract
Skeleton and liver are preferred organs for cancer dissemination in metastatic melanoma negatively impacting quality of life, therapeutic success and overall survival rates. At the target organ, the local microenvironment and cell-to-cell interactions between invading and resident stromal cells constitute critical components during the establishment and progression of metastasis. Mesenchymal stem cells (MSCs) possess, in addition to their cell progenitor function, a secretory capacity based on cooperativity with other cell types in injury sites including primary tumors (PT). However, their role at the target organ microenvironment during cancer dissemination is not known. We report that local MSCs, acting as pericytes, regulate the extravasation of melanoma cancer cells (MCC) specifically to murine bone marrow (BM) and liver. Intra-arterially injected wild-type MCC fail to invade those selective organs in a genetic model of perturbed pericyte coverage of the vasculature (PDGF-Bret/ret), similar to CD146-deficient MCC injected into wild type mice. Invading MCC interact with resident MSCs/pericytes at the perivascular space through co-expressed CD146 and Sdf-1/CXCL12-CXCR4 signaling. Implanted engineered bone structures with MSCs/pericytes deficient of either Sdf-1/CXCL12 or CD146 become resistant to invasion by circulating MCC. Collectively, the presence of MSCs/pericytes surrounding the target organ vasculature is required for efficient melanoma metastasis to BM and liver.
Keywords: mesenchymal stem cells (MSCs), pericytes, metastatic melanoma, CD146, Sdf-1
Mesenchymal stem cells (MSCs) have long been described as cellular progenitors of mesenchymal lineages including bone, cartilage, fat, muscle and other connective tissues.1 In addition, a secretory capacity has been identified with both immunomodulatory and trophic activities, exerted at sites of injury where they interact with other local cellular components.2 The recent identification of the MSC niche in the perivascular space as pericytes3–5 has opened the possibility that MSC may have additional roles directly controlling tissue homeostasis from their cardinal abluminal location.
Metastasis is a leading prognostic indicator for cancer survival and a major contributor to cancer mortality. The skeleton and liver are preferred organs for cancer dissemination in various malignancies including malignant melanoma. Although the preponderance of distant metastases implies a selective advantage for arriving disseminating cells, recent studies have determined that the rates of tumor growth, invasion and metastasis are in fact independent pathological traits governed by different sets of genes.6 Nevertheless, they share a common feature, namely their dependence on the vasculature that provides access to oxygen and nutrients, as well as a route for cancer cell dissemination. Current cancer therapies are designed to alter not only specific biological functions in cancer cells, but also to target components of the tumor microenvironment (TME)/stroma, especially the vasculature.
Pericytes are a specialized cell type that function abluminally covering and stabilizing blood vessels following their recruitment to forming vessels as progenitor cells via PDGF-B/PDGFRB signaling.7 Pericyte involvement in primary tumors growth constitutes a novel therapeutic target based on compelling evidence showing superior reduction in size when targeted in parallel with endothelial cells (ECs).8 However, pericyte coverage of the vasculature seems to differentially regulate tumor growth and metastatic potential, as intravasation of cancer cells is increased in primary tumors grown in mice with a genetically determined deficient pericyte coverage.9 These findings have led to the appreciation of pericytes in primary tumors as gatekeepers for cancer dissemination.10 In parallel, in response to tumor signals, BM-derived progenitor cells of mesenchymal origin (as MSC) are recruited to the tumor stroma localizing in perivascular sites and helping to assemble a supporting vascular network critical for tumor growth.11 Taken together, BM-derived MSC (BM-MSC) appear to play a critical role during primary tumor formation, growth and subsequent invasive potential.
In contrast, the role of pericytes at the target organ microenvironment during cancer cell extravasation is not known. Recently, data suggest that cellular and molecular stromal elements in the BM are related to the establishment and progression of skeletal metastasis. For example, blocking PDGFB signaling with a multitarget tyrosine kinase inhibitor (Sunitinib) impairs bone invasion of circulating osteotropic lung cancer cell lines due to altered tumor cell-BM stroma interactions.12 In addition, it has been established that invading cancer cells physically associate with mesenchymal-derived cells in the BM stroma affecting various biological activities of engrafted cancer cells, including dormancy/quiescence, resistance to chemotherapy and metastatic growth.13,14 However, the specific identity of the cellular and molecular elements, as well as the precise location where the sequence of events occur during extravasation is still not well understood.
The mechanisms governing skeletal metastasis involve comparable details to those used by hematopoietic stem cells (HSC) entering the BM. This “homing” behavior relies on the existence of a specific physical niche within the BM where other cellular players, including MSC, favor the constant trafficking of such progenitors across the sinusoidal wall. Shiozawa et al. showed that invading osteotropic prostate cancer cells enter the HSC niche competing with resident cells and thus establishing physical anchors for further growth inside the BM.15 With the notion of BMMSC as pericytes, a proposal can be constructed centered around the physical interaction of invading cancer cells and resident BMMSC occurring at the abluminal space of BM sinusoids as a determinant step in the initiation and fate of skeletal metastasis.
Here, we show that altering the physical interaction between vascular components of the target organ microenvironment (ECs and MSC/pericytes) via genetic manipulation of PDGF-B, dramatically impairs the engraftment of intra-arterially delivered MCC, thus reducing the frequency of osteolytic bone metastasis. Through in vitro and in vivo approaches, including a humanized assay in which fully functional extraskeletal bones are engineered with human MSC (hMSC), we establish essential molecular players and mechanisms involved in the extravasation of circulating MCC to the BM, which become disrupted in the absence of sinusoidal MSC/pericytes. In parallel, we made the observation that the situation in the BM is replicated in the liver exclusively, where no invasion by melanoma was noticed in mutant mice. Thus, we propose that the presence of MSC as pericytes surrounding BM and liver sinusoids is required for extravasation of MCC, and that the effects of the EC/pericyte dissociation at the metastatic target organ do not mirror its effects during intravasation at the primary tumor.
Material and Methods
PDGF-Bret/ret mice
PDGF-Bret/ret transgenic mice (PDGF-B mutant) were kindly provided by Dr. Betsholtz and Genové (Karolinska Institute, Stockholm, Sweden). These transgenic mice (C57Bl/6 background) express a mutant PDGF-B that lacks a C-terminal retention motif required to confine this growth factor to the EC compartment, necessary for the recruitment of pericyte progenitors expressing PDGFRB.7 The impaired PDGF-B binding results in defective pericyte recruitment and coverage of microvessels with fewer pericytes and their partial abluminal detachment from the vessel wall.7,10,16 Given that the Pdgfbret allele is hypofunctional and PDGF-Bret/+ mice are indistinguishable from PDGF-B+/+ mice16, adult (10-week-old) Het, WT and PDGF-B mutant littermate mice (n = 5 per group) were used in all experiments.
Bioluminescence imaging
Bioluminescence imaging (BLI) was performed after subcutaneous injection of 200 μl of 12.5 mg/ml of luciferin substrate (Biosynth, Cat# L-8220) using a Xenogen IVIS 200 series system. Fifteen minutes after B16F10 cell infusion, an early BLI was performed to evaluate cell distribution throughout the body. Later, images at Days 3, 7 and 12 were obtained to evaluate cancer cells engraftment and their temporal progression as growing metastases. To quantify tumor invasion to target organs, BLI signal was analyzed (d12) in terms of photon flux (photons/sec/cm2/steradian) and the area covered by signal (cm2/e) taken at specific locations (extremities and spine after adrenal glands removal) using a predefined geometrical shape.
Gene silencing in B16F10 MCC and hMSC cells
CD146 was silenced in B16F10 MCC using a validated shRNA murine sequence cloned in a regular pLKO.1-puro vector, bacterially amplified, sequence verified and delivered as lentiviral transduction particles ready to use (MISSION® RNAi clone ID: NM_023062.1-656s1c1; Sigma Aldrich, St. Louis, MO). CD146 and Sdf-1/CXCL12 were silenced in BM-derived hMSC using validated human shRNA sequences cloned into an inducible pLKO-puro-IPTG-3xLacO vector [Isopropyl β-D-1-thiogalactopyranoside (IPTG)-dependent transcriptional induction], also delivered as viral particles (MISSION® RNAi clone IDs: CD146: NM_006500.1-1322s1c1; Sdf-1: NM_000609.4-157s21c1, Sigma Aldrich). The use of an inducible system is intended to avoid the effects of silencing CD146 and Sfd-1/CXCL12 during the formation of both bone and the sinusoidal network inside the ossicles. For the transendothelial migration (TEM) assay, Sdf-1/CXCL12 gene silencing was induced 5 days before the assay. Details are described in Supporting Information Materials and Methods.
In vitro TEM assay
A modified Boyden chamber cell migration assay was used to quantitate the invasion potential of B16F10 cancer cells in two different conditions, relative to prelabeled hMSCs with the cationic lipophilic dye DiI for their fluorescence detection: (i) When the MSC/pericytes are in close contact with the membrane but silenced for Sdf-1/CXCL12 and (ii) When the distance between the membrane (acting as an endothelium) and the MSC/pericytes is increased, reminiscent of the PDGF-Bret/ret mutant mice “anatomic” phenotype (in vitro counterpart). Details are described in the Supporting Information Materials and Methods.
Humanized heterotopic bone formation assay
A total of 4.5 × 106 nontransduced hMSC and hMSC expressing inducible vectors for nontarget (NT), CD146 (CD146.shRNA) and Sdf-1 (Sdf-1.shRNA) were vacuum-loaded into sterile porous ceramic cube carriers (hydroxyapatite/tricalcium phosphate 40/60; Zimmer, Warsaw, IN) precoated with a 100 μg/ml solution of fibronectin17. The cubes were subcutaneously implanted into immunocompromized mice (CB17-Prkdc SCID) for 8 weeks to form extraskeletal bone structures (ossicles). Every animal (n = 8) received 4 cubes, each representing one of the conditions tested. In order to minimize any potential anatomical effect, their relative positions were changed in each animal. After 8 weeks, gene silencing at the protein level in hMSC was accomplished by administering IPTG (12.5 mM) in the drinking water for 7 days. At this time, 1 × 106 murine B16F10 MCC were intra-arterially injected. BLI images at Days 1, 3, 7 and 12 were taken to evaluate MCC engraftment and growth. Two weeks after cell injections, the animals were sacrificed, imaged and the implanted ossicles analyzed by histology.
Statistical comparisons
BLI data from injected animals (photon flux and area covered by tumors) were pooled individually (extremities and spine of each mouse) and statistical difference between PDGF-B mutant and Het mice calculated using a paired t test. BLI data from engineered cubes (photon flux) were compared and statistical difference calculated following a one-way ANOVA with Tukey’s multiple comparison tests (contrasted to control cubes).
Results
Skeletal and liver melanoma tumor burden is reduced in PDGF-bret/ret mice
In PDGF-B mutant mice no underlying bone and gross vascular phenotypes (Supporting Information Fig. S1A) were observed. Twelve days after B16F10 cell injection, both heterozygous (Het) and wild type (WT) mice exhibited marked clinical deterioration including severe cachexia (32 ± 3% body weight reduction vs. 20 ± 2% in PDGF-B mutant mice, p < 0.05), restricted mobility/ambulation, hunched backs, increased movement-evoked limb lifting and respiratory distress, not observed in PDGF-B mutant mice (Supporting Information Movies S1 and S2; representative mice shown).
BLI assessment shows that WT and Het mice exhibit an increased skeletal tumor burden as compared with PDGF-B mutants, determined by the number of metastatic foci in the extremities, pelvis and spine, and by their signal quantification (Fig. 1). In PDGF-B mutant mice (n = 3) some metastatic foci disappeared in time (Fig. 1a—yellow circles). The extensive compromise of long bones seen in Het controls was linked with functional bone osteolysis, analyzed by two (2D)- and three (3D)-dimensional volumetric micro computed tomography (μCT) reconstruction (Supporting Information Fig. S1B).
Figure 1.
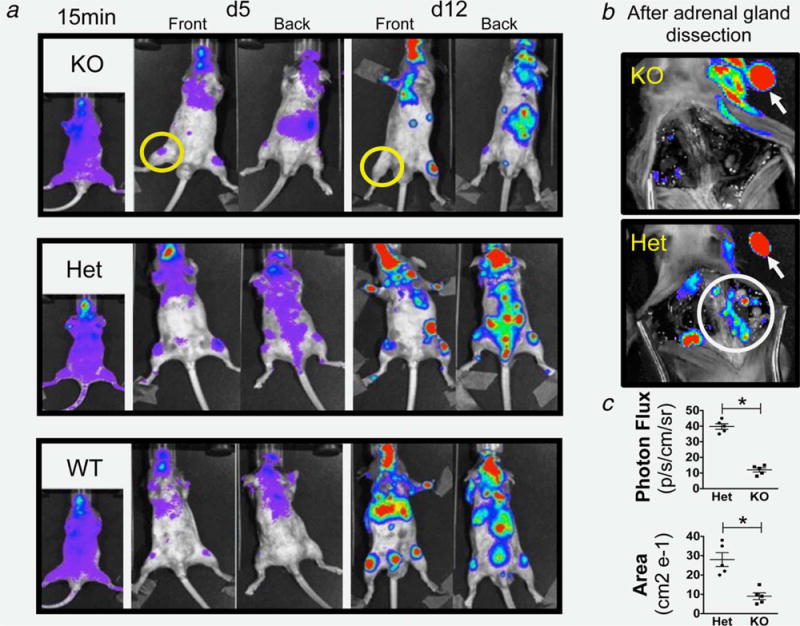
BLI of injected PDGF-B mutant and control mice: (a) Imaging 15 min, 5 and 12 days postinjection, showing increased skeletal invasion (limbs and spine) in WT and Het mice compared to PDGF-B mutant mice and the disappearance of a metastatic signal at 12 days in PDGF-B mutant mice (yellow circles). (b) Absence in PDGF-B mutant and persistence in Het mice of the spine signal (white circle) after adrenal gland removal (white arrows) confirming the nonspinal origin of the signal in PDGF-B mutant spine (yellow arrow in A). (c) Quantification of signal (photon flux and area covered by tumors) showing statistical difference between PDGF-B mutant and Het mice (* = p < 0.01). Data are represented as mean ± SEM. Representative mice of n = 15 (5 per group).
Animal dissections confirmed the overall significant reduction in skeletal invasion in PDGF-B mutant mice compared with Het controls (Fig. 2a), also evident in liver but not in other melanoma target organs including adrenal glands, lungs and brain (Fig. 2b). In addition to the significantly reduced overall macroscopic tumor burden, histological analysis revealed comparable reductions in tumor size in PDGF-B mutant mice (Fig. 3a). Similarly, no metastatic foci were observed histologically in PDGF-B mutant livers compared with the multifocal invasion in WT and Het mice (Fig. 3b).
Figure 2.
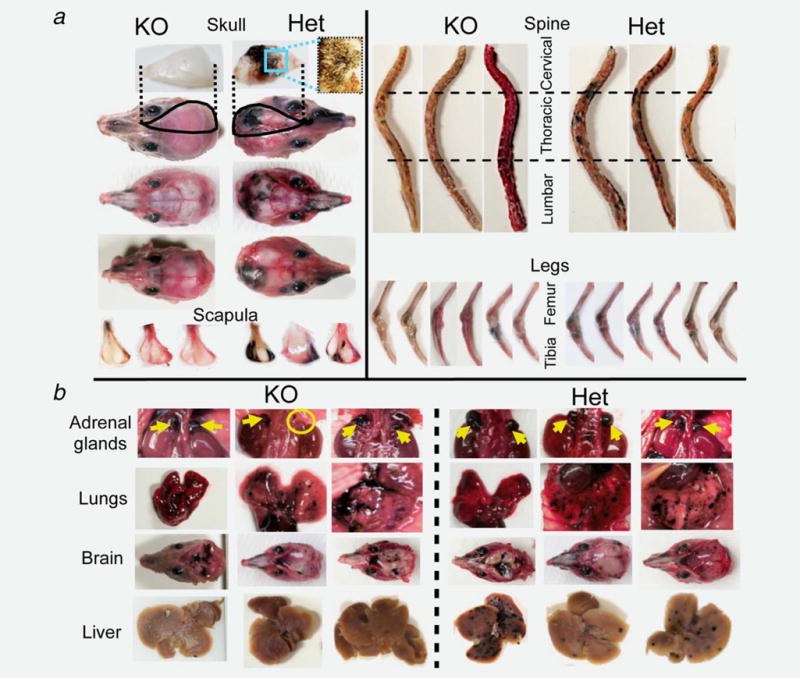
Gross inspection of distant melanoma dissemination: (a) Craniofacial and appendicular invasion by MCC exhibiting a significant reduction in PDGF-B mutant mice. All skulls and scapulae in the mutants were clear of metastases or had only few/small foci, which contrasts sharply with the multiple major/multifocal invasion observed in Het mice. Spines in PDGF-B mutant mice were clear of metastasis in 2/5 mice, or harbored only 1–2 small foci restricted to one vertebral segment in the remaining three animals. WT and Het mice had multiple multisegment metastases in all animals. Both distal femur and proximal tibia were compromised bilaterally in all WT and Het controls, while bilateral invasion was observed in only one of the five PDGF-B mutant mice, with remaining four mice exhibiting only one compromised leg that was restricted to the proximal tibia. (b) Melanoma invasion to other target organs with comparable results in both genotypes except for liver (reduced in PDGF-B mutant mice). Yellow arrows: adrenal glands. Yellow circle: adrenal gland agenesis. Three representative mice shown of n = 15 (5 per group).
Figure 3.
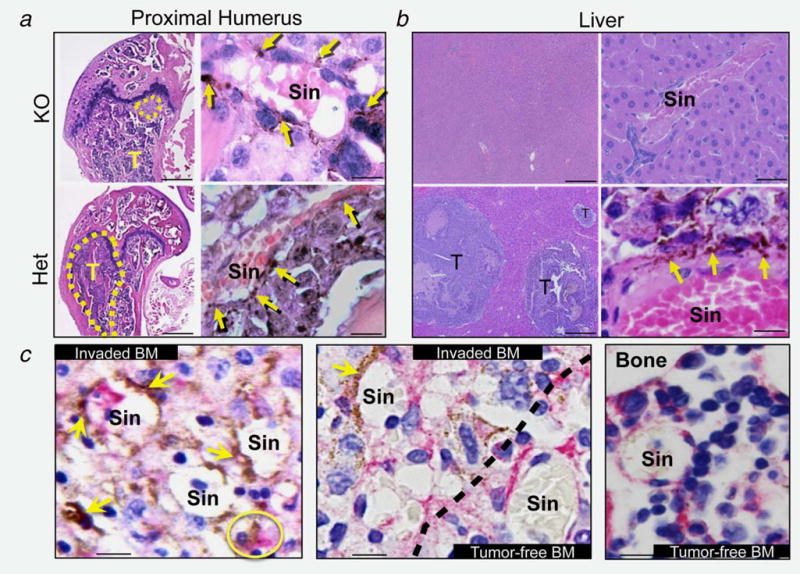
Histology of metastatic bone tumors. (a, b) Metastatic tumors (T) in distal femur (a) and liver (b) are smaller or absent in PDGF-B mutant mice. Bars = 200 μm (low magnification) and 10 μm (high magnification). Melanin-producing B16F10 cells (yellow arrows) localize in the abluminal side of BM sinusoids (Sin), extending to the tissue parenchyma (impaired in PDGF-B mutant mice). (c) CD146 IHC in BM sections from Het mice. Engrafted B16F10 cells (yellow arrows) physically associate with CD146-positive BMMSC/pericytes (pink signal) at the perivascular space and inside the parenchyma (yellow circle). Dotted line = boundary between tumor-invaded and tumor-free BM. Bar = 10 μm.
MCC establish a perivascular niche during engraftment/invasion in the skeleton and liver where they interact with resident murine MSC/pericytes
High power microscopy analysis of all histological specimens across genotypes revealed the absence of micrometastatic foci in regions where invasion was not visible macroscopically. Melanin-producing cancer cells were observed abluminally with respect to BM and liver sinusoids, adopting a perivascular location. In WT and Het mice, the invaded MCC colonized the BM parenchyma where they appear to reside in physical association with CD146-expressing resident MSC/pericytes (Fig. 3c). This is in contrast to PDGF-B mutant-derived samples where no further advancement of invading cells was observed beyond the perivascular space (Fig. 3a, high magnification).
This physical association and the constitutive expression of CD146 by both MCC and MSC/pericytes prompted us to evaluate CD146 as a potential mechanism for an intercellular adhesion between them at the perivascular space generated during extravasation.
CD146 silencing in MCC impairs their ability to extravasate to skeleton and liver
We first assessed the role of CD146 from the invading cell perspective. A high CD146 silencing efficiency (~90%) was obtained after lentiviral transduction of B16F10 cells with the constitutive CD146_shRNA vector as compared with the NT (nontargeting)_shRNA control (Supporting Information Fig. S2A). These engineered cells were then intra-arterially injected into WT mice (n = 6). Animals that received control cells (NT_shRNA; n = 3) exhibited a more rapid and dramatic clinical deterioration compared with animals that received cells with silenced CD146, including a more pronounced weight loss (34 ± 3% vs. 18 ± 2%, p < 0.05), ambulation difficulties, restricted limb movements and hunched backs (Supporting Information Movies S3 and S4). Similar to PDGF-B mutant mice, invasion was compromised selectively to skeleton and liver in mice that received CD146-silenced B16F10 cells compared to control cells (Fig. 4).
Figure 4.
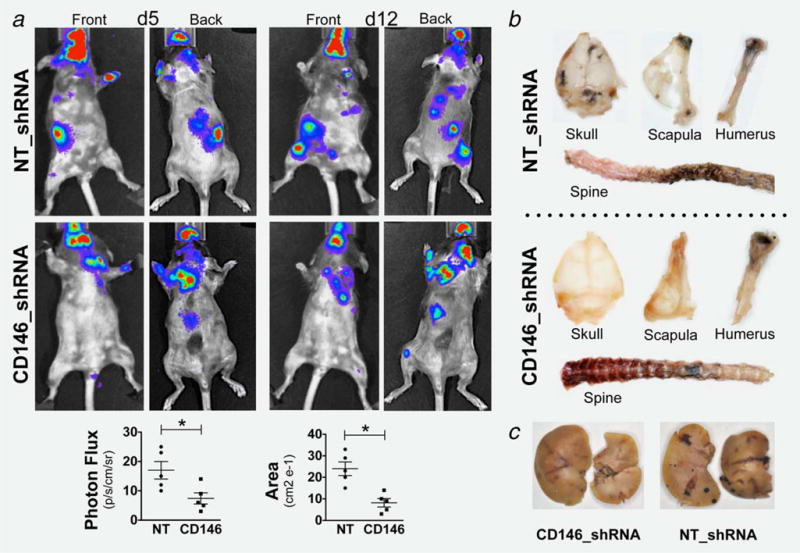
Invasion of engineered B16F10 MCC: Reduced invasion of CD146-silenced MCC to craniofacial, appendicular structures and liver (compared to NT-shRNA), evaluated by BLI (a) and gross examination (b and c). BLI signal reached statistical significance (* = p < 0.01). Data are represented as mean ± SEM. Representative mice of n = 6 (3 per group).
CD146 silencing in BM-derived hMSC/pericytes impairs MCC invasion to humanized extraskeletal bone structures in mice
As CD146 has been shown to exhibit homotypic interactions, we next evaluated the role of CD146 silencing now from the resident MSC/pericyte perspective. The two concentrations of IPTG tested resulted in a comparable silencing efficiency of CD146 in hMSC (~75%) as assessed by immunolocalization of the protein in cultured cells (Supporting Information Fig. S2B). Engineered cells were used to create osteogenic ceramic cubes (depicted in Supporting Information Fig. S4), which after implantation in immunocompromised mice generated humanized extraskeletal bones (ossicles) that fully recapitulate the native bone structure including the formation of blood vessels (sinusoids) and sequentially functional hematopoietic tissue. Within these humanized osseous structures, donor-derived hMSC form the bone tissue and assemble as pericytes during the formation of vascular structures, as shown by Sacchetti et al.,18 and confirmed by our observations with immunolocalization of the hMSC marker CD271 using a specific anti-human antibody (Supporting Information Fig. S5A). This humanized assay allowed us to circumvent the use of a CD146 KO mouse to monitor the effects of CD146 deficiency on the ability of resident perivascular cells to drive melanoma cell extravasation into the BM. As a control, untransduced WT hMSC (control) and a subset of hMSC with pathologically slow dividing activity after being transduced with the NT_shRNA vector and passaged several times (SD) were used. These two controls represent normal (control) and an aberrant (SD) formation of the ossicles and all their structural components of which the SD cells reflected the reduced osteogenic capacity of hMSC after serial passaging. Eight weeks after subcutaneous ossicle implantation, IPTG (12.5 mM for 7 days) was administered through the drinking water to induce the in vivo gene silencing within these bone structures, evidenced by immunolocalization of CD146 in sections (Supporting Information Fig. S5B), and consistent with the in vitro silencing. Intra-arterially injected MCC were found to invade the skeleton and the control hMSC-made ossicles as evidenced by the intense BLI signals that were observed in all structures (5/5) (Fig. 5a). In contrast, ossicles made with hMSC that lacked CD146 generated a dramatic reduction in the invasion of MCC, detected in only one structure (1/5) with a faint signal. As expected, ossicles made with slow dividing hMSC (SD) were not invaded by MCC, as these structures lacked substantial bone formation, vascular structures and hematopoietic tissue observed in a histological analysis (Fig. 5b). Finally, like the situation in the murine BM, invading MCC were found in humanized ossicles in close spatial relationship with resident hMSC/pericytes identified by immunolocalization of CD146 (Fig. 5c).
Figure 5.
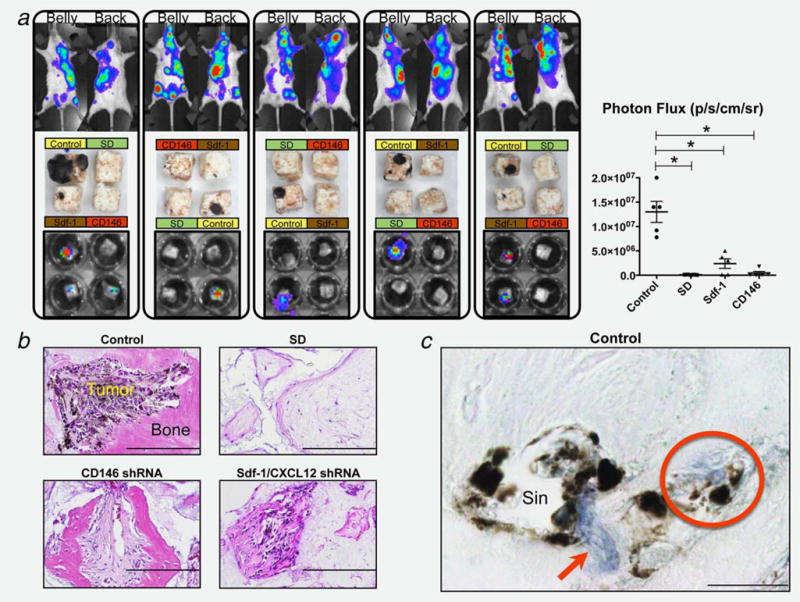
B16F10 MCC invasion to humanized extraskeletal ossicles. (a) MCC invasion of the skeleton (top row BLI) and specific implanted ossicles, evaluated by direct examination after harvesting (middle row) and by BLI (bottom row). The signal from the cubes BLI was quantified (photon flux) giving statistical difference of all groups compared to control (* = p < 0.01). Data are represented as mean ± SEM. Representative mice and cubes of n = 8. (b) Histological analysis (H&E staining) of harvested ossicles shows significant melanoma invasion in structures made with control MSC while significantly reduced or absent with Sdf-1 and CD146-silenced cells. SD cubes exhibit no bone and vasculature formation. Bar = 200 μm. (c) Immunolocalization of CD146 (blue signalred arrow) in sections from Control ossicles showing invading MCC physically associated with MSC/pericytes at the perivascular space surrounding sinusoids (Sin), and advancing towards the tissue parenchyma as a cell complex (red circle). Bar = 10 μm.
Sdf-1/CXCL12 silencing in BM-derived hMSC/pericytes is required for MCC to invade humanized extraskeletal bone structures and to migrate in vitro
Sdf-1/CXCL12 has been described as a potent attractant to CXCR4-expressing cells, including MCC.19,20 Its expression in the BM has been demonstrated to come primarily from perivascular cells (i.e., MSC/pericytes).21,22 Therefore, we assessed its potential contribution during extravasation of MCC into bone, through its silencing in resident MSC/pericytes using the humanized extraskeletal bone formation assay. The two concentrations of IPTG tested to induce Sdf-1/CCXCL12 gene silencing generated a comparable ~55% reduction relative to untreated parental hMSC, or to NT_shRNA-transduced hMSC treated with IPTG (Supporting Information Fig. S2C). Sdf-1/CXCL12 silencing in hMSC/pericytes showed a significant reduction both in the number of ossicles invaded (2/5) and the intensity of the BLI signal obtained (Fig. 5a), as well as on the size of the secondary tumors where present (Fig. 5b).
In an in vitro modified TEM assay (Fig. 6a), fluorescence microscopy revealed DiI prelabeled hMSC that were seeded at the bottom of an 8 μm pore membrane forming colony-like structures (Fig. 6b—top row). Bright-field microscopy showed the interaction of B16F10 cells (after migrating from the upper chamber) with parental hMSC (i.e., NT_shRNA control cells) in that same plane, an event that was noticeably absent in their Sdf-1-deficient counterparts and when skin fibroblasts and no cells were used as controls (Fig. 6b— bottom row). Similarly, an increased distance between MCC and hMSC prevented their migration throughout the membrane regardless of whether the hMSC were silenced not for Sdf-1/CXCL12 (Supporting Information Fig. S3).
Figure 6.
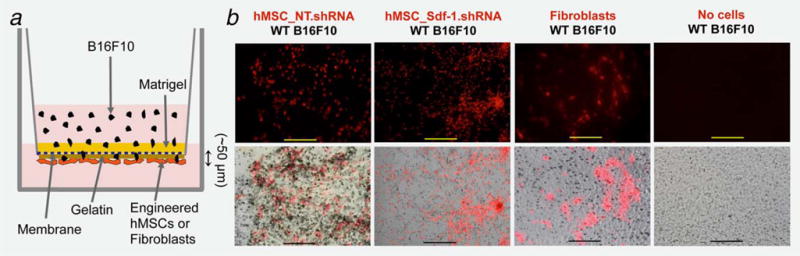
In vitro transendothelial migration assay: (a) Schematic representation of the modified TEM. (b–top row) Fluorescence microscopy of DiI-labeled (red) hMSC expressing either nontarget (NT) or Sdf-1 silencing vectors (Sdf-1_shRNA) and papillary dermal Fibroblasts seeded at the bottom surface of an 8 μm pore diameter insert membrane. (b–bottom row) Merged bright field and fluorescence microscopy showing B16F10 melanoma cell invasion to the membrane and interaction with seeded cells (Bar = 200 μm). Representative pictures from three independent experiments.
Discussion
Through the use of complementary in vitro and in vivo approaches, including a humanized assay of bone metastases in mice, we documented details of the function of MSC/pericytes in mediating the extravasation and the initial metastatic seeding of MCC at the BM and liver microvasculature. Mechanistically, we describe the participation of the cell surface molecule CD146 and the chemokine Sdf-1/CXCL12 as critical determinants of the molecular events occurring during the dissemination process resulting in the physical association between the invading cancer cell and the MSC/pericyte at the target organ microenvironment (i.e., perivascular space). Importantly, genetic ablation of abluminal positioning of pericytes alleviates these untoward events, and as such, we propose that circulating MCC follow an Sdf-1/CXCL12 gradient that facilitates their access to endothelial pores and resident MSC/pericytes. These MSC/pericytes specifically associate with CD146-positive MCC and promote their extravasation into the target organ parenchyma (depicted in Supporting Information Fig. S6).
The concept of a physical association between invading cancer cells and stromal cells in the BM has been previously reported as critical for the progression and fate of metastatic tumors.13 In addition, the participation of MSC during the endothelial transmigration of low metastatic breast cancer cells has been suggested using in vitro models.14 Nevertheless, our study constitutes the first direct in vivo evidence of the participation of MSC as pericytes during the process of melanoma dissemination, as well as the description of the precise location (i.e., perivascular space) and the molecular players involved in the cell-to-cell association that lead to the establishment of distant metastases. We observed a close proximity between invading MCC and resident MSC/pericytes at both the sinusoidal perivascular space and the tissue parenchyma. These observations further suggest the physical interaction between those two cell types during the extravasation of cancer cells at the target organ and establishes a previously underappreciated sentinel role of MSC (as pericytes) during the process of cancer cell invasion.
MSC have been historically seen as precursors of mesenchymal tissues including bone, cartilage, fat and muscle.1 Nevertheless, our data supports the recent proposition that MSC reside in a perivascular niche and that they arise from perivascular cells,3,5 while suggesting that they participate in parallel homeostatic functions exerted at their strategic abluminal location including cancer invasion to target organs. Indeed, the function of pericytes during cancer dissemination is not limited solely to their known effect on vascular stability,7 but instead indicates a more active role during the process of cancer cell extravasation. In fact, pericytes have been described as gatekeepers of tumor metastasis, since their absence promotes cancer cell dissemination to target tissues from primary tumors in mice.9,10 Using the same genetic model of pericyte disturbed coverage reported by Xian et al.(PDGF-Bret/ret mice), we now present evidence that invasion of circulating MCC is significantly and selectively reduced in the skeleton and liver, but remains surprisingly intact at other target organs (e.g., brain, adrenal glands and lungs). These apparent discrepancies may be explained by the following arguments. First, Xian et al. focused on the process of cancer cells intravasation from a primary tumor (insulinoma) where an “abnormal” tumor-forming vasculature is further destabilized in the mutant mice, thereby increasing the number of tumor cells capable of escaping into the vasculature. This increased number of circulating cancer cells might account for the distant colonization observed in a fraction of the animals. In stark contrast, we focused on the process of extravasation of a predetermined, fixed number of intra-arterially injected MCC from a vessel network at the target organ impacted only by the expression of the transgene. Second, Xian et al. studied an insulinoma primary tumor, which makes comparisons of distal invasion difficult to assess because insulinoma cells typically do not invade the BM and liver as proficiently as MCC. Third, it has been proposed that primary tumors secrete factors that “prepare” the target organ for future invasion, a process known as premetastatic niche formation.23 In contrast to the model of Xian et al., our study bypassed the formation of a primary tumor, thus preventing this contributing phenomenon. Finally, Xian et al. described both lymphatic and hematogenous pathways as contributors of distant dissemination. Our model does not involve a lymphatic-mediated mechanism as we inject the cancer cells intra-arterially. In addition, truly lymphatic-dependent bone metastases are still controversial.24
The impaired melanoma invasion to BM and liver observed in the mutant mice suggests the presence of unique mechanistic traits within these two organ sites that are not shared with other target organs. These traits may only become evident in the absence of perisinusoidal cells (MSC/pericytes in the BM and stellate cells in the liver), conferring them “resistance” to metastasis from melanoma. One potential explanation for this phenomenon takes into consideration that BM and liver have hematopoietic capabilities and may share a similar HSC niche, which is known to be targeted by osteotropic cancer cells for the establishment of footholds during skeletal metastasis.15 Recently, hepatic stellate cells were observed to represent pericyte-like, CD146-positive liver resident MSC that associate with sinusoidal ECs in close contact with hematopoietic progenitor cells.25 This situation is reminiscent of the molecular mechanisms we establish herein for MSC/pericytes in the BM. In addition, hepatic stellate cells share a similar pattern of recruitment and alignment around vessel walls as other pericyte cells, and as such, they are highly dependent on the secretion of PDGF-B by ECs.26 Thus, both of these target organs share similar structural patterns surrounding sinusoids (i.e., stem cell niche), which apparently determine the invasive capacity of circulating MCC.
There are a number of reported mechanisms involved in the regulation of metastatic MCC to target organs, including the interaction between MCC and ECs,27 and platelets,28 all of which are mediated through adhesion molecules such as integrins and selectins. CD146 (MCAM, MUC18) is a transmembrane glycoprotein that belongs to the immunoglobulin superfamily. Its aberrant expression regulates the tumorigenicity of MCC,29 and is associated with the aggressiveness, poor prognosis and metastatic potential of human melanoma when compared to normal skin.30,31 It has been reported as part of a “metastases aggressiveness gene expression signature” present in melanoma metastases in human patients,32 establishing an essential role of the tumor cell-target organ microenvironment interactions during metastasis, as we demonstrated here between circulating MCC and abluminal MSC/pericytes. In addition, CD146 has been shown to affect melanoma cell extravasation to lung during dissemination, by interacting with ECs and mediating VEGF-induced vessel permeability.33
Homotypic interactions between CD146 molecules have been proposed as a mechanism for increasing cohesive cell-cell interactions between similar and different cell types including MCC (e.g., B16 MCC and B-1 lymphocytes).34 Our results demonstrate the necessity of CD146 to be expressed in both the invading MCC and resident MSC/pericytes for efficient extravasation. This notion is supported by the significant reduction in invasion observed in both WT mice injected with CD146-deficient MCC and the ossicles made with CD146-silenced hMSC/pericytes. This novel humanized assay approach, to the best of our knowledge, represents the first time functional extraskeletal bone structures are created with engineered human cellular precursors (i.e., MSC) to manipulate the target organ microenvironment and to test the role of specific molecular targets during invasion by MCC. This approach provides information that corroborates the human findings in terms of CD146 relevance directly using humanized tissues in rodents. In addition, it not only permits direct side-by-side comparisons between the different genetic backgrounds obtained with engineered ossicles implanted in the same animal, but also to contrast the invasion rates with the animal skeleton used as an internal control. Another advantage with this approach is that we were able to rule out a potential contribution of ECs-derived CD146 during cancer cell extravasation to bone, as these are unmodified host-derived cells, and it has been reported that BM ECs do not express CD146.18 This CD146 cell source discrimination permits to establish a mechanistic difference in melanoma extravasation between BM and other organs (e.g., lung). Unlike the lung, where EC-derived CD146 is indispensable,33 MSC/pericyte in the BM supply CD146 during extravasation.
We then hypothesize that the absence of CD146 impacts MCC extravasation by at least two parallel mechanisms. First, CD146-deficiency prevents the formation of cellular complexes between invading MCC and resident MSC/pericytes at the perivascular space, thereby limiting further penetration to the parenchyma. And second, loss of CD146 elicits cell detachment and subsequent apoptosis (anoikis), a process initiated by the loss of cell interactions with the extracellular matrix and/or neighboring cells.35 CD146 has been shown to promote cancer cell resistance against anoikis by facilitating cell-cell interactions, which may upregulate anti-apoptotic mechanisms,36 a mechanism disrupted when CD146 is not present in either invading MCC and/or the resident MSC/pericytes. This would explain the impaired extravasation observed in the in vivo model of deficient vascular pericyte coverage where the absence of CD146-expressing MSC/pericytes fails to provide anoikis resistance and the generation of the translocating cellular complex. In addition, this mechanism would also explain the disappearance of the signal obtained with MCC over time in some mutant mice.
Using a similar in vivo approach we established the role of Sdf-1/CXCL12 secreted by resident MSC/pericytes during extravasation, observing a drastic reduction of melanoma cell invasion into humanized ossicles silenced for Sdf-1/CXCL12 in MSC/pericytes. Sdf-1/CXCL12 is a pro-survival chemokine that serves as a potent chemoattractant for circulating hematopoietic precursors,20 as well as various bone-metastasizing cancers, including melanoma, expressing its cognate receptor CXCR4.19,37 In the BM, various cell types including perivascular, endothelial and osteoblastic cells express Sdf-1/CXCL12.21,22,38–40 However, it has been demonstrated that perisinusoidal cells (i.e., MSC/pericytes) referred by Sugiyama et al. as CXCL12-abundant reticular cells (CAR) constitute the main source of this factor, compared with a reduced expression and secretion by ECs.21,22,41 These results with Sdf-1/CXCL12 emphasize the striking similarities in the entry (and potentially exit) to the BM between circulating osteotropic MCC and HSCs, further supporting the findings with prostate cancer cells by Shiozawa et al. (2011). In addition, the host-derived low secretion of ECs in the ossicles vasculature might explain the reduced (not absent) MCC invasion to structures made with silenced Sdf-1/CXCL12.
Taken together, the pivotal role of MSC/pericytes at their perivascular niche regulating the extravasation of circulating MCC to bone and liver further expands our knowledge about novel functions of these adult stem cells. In parallel, the identified molecular mechanisms involving intercellular adhesion molecules (i.e., CD146) and secreted chemokines (i.e., Sdf-1/CXCL12) strengthens the concept of cellular cooperativity reported at sites of injury and primary tumors and now expanded to target organs during cancer dissemination. Finally, these mechanisms may serve as a platform for the development of novel therapies aimed at controlling the establishment and progression of skeletal and liver metastasis from melanoma by targeting MSC/pericytes.
Supplementary Material
What’s new?
While in primary tumors Mesenchymal Stem Cells (MSCs) possess a cell progenitor function and secretory capacity based on intercellular cooperativity, their role in the target organ microenvironment during cancer dissemination remains unknown. This study shows that MSC/pericytes function as sentinels regulating cancer cell dissemination, with a differential effect during intravasation at the primary tumor and extravasation at the target organ. The molecular mechanisms, cellular players, and locations identified during the establishment of melanoma metastasis to bone marrow and liver may help design novel therapeutic strategies for reducing engraftment by closing the gate through which metastatic cells enter the target organ.
Acknowledgments
We thank Drs. Christer Betsholtz and Guillem Genové for the PDGF-Bret/ret mutant mice. Amad Awadallah for the histological processing. Lan Wang for the surgeries involving catheter insertion and removal during intra-arterial melanoma cell injections. Don Lennon and Margie Harris for the assistance with the cubes and hMSC preparation.
Grant sponsor: NIH T32 GM007250 (to P.L.)
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 2.Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11–15. doi: 10.1016/j.stem.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caplan AI. All MSCs are pericytes? Cell Stem Cell. 2008;3:229–230. doi: 10.1016/j.stem.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 5.da Silva Meirelles L, Caplan AI, Nardi NB. In search of the in vivo identity of mesenchymal stem cells. Stem Cells. 2008;26:2287–2299. doi: 10.1634/stemcells.2007-1122. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 7.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 8.Bergers G, Song S, Meyer-Morse N, et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xian X, Hakansson J, Stahlberg A, et al. Pericytes limit tumor cell metastasis. J Clin Invest. 2006;116:642–651. doi: 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerhardt H, Semb H. Pericytes: gatekeepers in tumour cell metastasis? J Mol Med. 2008;86:135–144. doi: 10.1007/s00109-007-0258-2. [DOI] [PubMed] [Google Scholar]
- 11.Bergfeld SA, DeClerck YA. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010;29:249–261. doi: 10.1007/s10555-010-9222-7. [DOI] [PubMed] [Google Scholar]
- 12.Catena R, Luis-Ravelo D, Antón I, et al. PDGFR signaling blockade in marrow stroma impairs lung cancer bone metastasis. Cancer Res. 2011;71:164–174. doi: 10.1158/0008-5472.CAN-10-1708. [DOI] [PubMed] [Google Scholar]
- 13.Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun. 2005;328:679–687. doi: 10.1016/j.bbrc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 14.Corcoran KE, Trzaska KA, Fernandes H, et al. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS One. 2008;3:e2563. doi: 10.1371/journal.pone.0002563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiozawa Y, Pedersen EA, Havens AM, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121:1298–1312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindblom P, Gerhardt H, Liebner S, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis JE, Konstantakos EK, Arm D, et al. In vivo osteogenesis assay: a rapid method for quantitative analysis. Biomaterials. 1998;19:1323–1328. doi: 10.1016/s0142-9612(97)00170-1. [DOI] [PubMed] [Google Scholar]
- 18.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 19.Bartolome RA, Galvez BG, Longo N, et al. Stromal cell-derived factor-1alpha promotes melanoma cell invasion across basement membranes involving stimulation of membrane-type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 2004;64:2534–2543. doi: 10.1158/0008-5472.can-03-3398. [DOI] [PubMed] [Google Scholar]
- 20.Mendez-Ferrer S, Lucas D, Battista M, et al. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 21.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiyama T, Kohara H, Noda M, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards JR, Williams K, Kindblom LG, et al. Lymphatics and bone. Hum Pathol. 2008;39:49–55. doi: 10.1016/j.humpath.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 25.Kordes C, Sawitza I, Götze S, et al. Hepatic stellate cells support hematopoiesis and are liver-resident mesenchymal stem cells. Cell Physiol Biochem: Int J Exp Cell Physiol Biochem pharmacol. 2013;31:290–304. doi: 10.1159/000343368. [DOI] [PubMed] [Google Scholar]
- 26.Lee JS, Semela D, Iredale J, et al. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 27.Fritzsche J, Simonis D, Bendas G. Melanoma cell adhesion can be blocked by heparin in vitro: suggestion of VLA-4 as a novel target for antimetastatic approaches. Thromb Haemost. 2008;100:1166–1175. [PubMed] [Google Scholar]
- 28.Nash GF, Turner LF, Scully MF, et al. Platelets and cancer. Lancet Oncol. 2002;3:425–430. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 29.Schlagbauer-Wadl H, Jansen B, Muller M, et al. Influence of MUC18/MCAM/CD146 expression on human melanoma growth and metastasis in SCID mice. Int J Cancer. 1999;81:951–955. doi: 10.1002/(sici)1097-0215(19990611)81:6<951::aid-ijc18>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 30.Luca M, Hunt B, Bucana CD, et al. Direct correlation between MUC18 expression and metastatic potential of human melanoma cells. Melanoma Res. 1993;3:35–41. doi: 10.1097/00008390-199304000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Talantov D, Mazumder A, Yu JX, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- 32.Xu L, Shen SS, Hoshida Y, et al. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jouve N, Bachelier R, Despoix N, et al. CD146 mediates VEGF-induced melanoma cell extravasation through FAK activation. Int J Cancer. 2015;137:50–60. doi: 10.1002/ijc.29370. [DOI] [PubMed] [Google Scholar]
- 34.Johnson JP, Bar-Eli M, Jansen B, et al. Melanoma progression-associated glycoprotein MUC18/MCAM mediates homotypic cell adhesion through interaction with a heterophilic ligand. Int J Cancer. 1997;73:769–774. doi: 10.1002/(sici)1097-0215(19971127)73:5<769::aid-ijc26>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 35.Kim YN, Koo KH, Sung JY, et al. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012;2012:1–11. doi: 10.1155/2012/306879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int J Cell Biol. 2012;2012:340296. doi: 10.1155/2012/340296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scala S, Ottaiano A, Ascierto PA, et al. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin Cancer Res. 2005;11:1835–1841. doi: 10.1158/1078-0432.CCR-04-1887. [DOI] [PubMed] [Google Scholar]
- 38.Dar A, Goichberg P, Shinder V, et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nature Immunol. 2005;6:1038–1046. doi: 10.1038/ni1251. [DOI] [PubMed] [Google Scholar]
- 39.Netelenbos T, van den Born J, Kessler FL, et al. Proteoglycans on bone marrow endothelial cells bind and present SDF-1 towards hematopoietic progenitor cells. Leukemia. 2003;17:175–184. doi: 10.1038/sj.leu.2402738. [DOI] [PubMed] [Google Scholar]
- 40.Yun H-J, Jo D-Y. Production of stromal cell-derived factor-1 (SDF-1)and expression of CXCR4 in human bone marrow endothelial cells. J Korean Med Sci. 2003;18:679–685. doi: 10.3346/jkms.2003.18.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nature Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.