

Abstract
Colchicine is a microtubule disruptor that reduces the occurrence of atrial fibrillation (AF) after an operation or ablation. However, knowledge of the effects of colchicine on atrial myocytes is limited. The aim of this study was to determine if colchicine can regulate calcium (Ca2+) homeostasis and attenuate the electrical effects of the extracellular matrix on atrial myocytes. Whole‐cell clamp, confocal microscopy with fluorescence, and western blotting were used to evaluate the action potential and ionic currents of HL‐1 cells treated with and without (control) colchicine (3 nM) for 24 hrs. Compared with control cells, colchicine‐treated HL‐1 cells had a longer action potential duration with smaller intracellular Ca2+ transients and sarcoplasmic reticulum (SR) Ca2+ content by 10% and 47%, respectively. Colchicine‐treated HL‐1 cells showed a smaller L‐type Ca2+ current, reverse mode sodium–calcium exchanger (NCX) current and transient outward potassium current than control cells, but had a similar ultra‐rapid activating outward potassium current and apamin‐sensitive small‐conductance Ca2+‐activated potassium current compared with control cells. Colchicine‐treated HL‐1 cells expressed less SERCA2a, total, Thr17‐phosphorylated phospholamban, Cav1.2, CaMKII, NCX, Kv1.4 and Kv1.5, but they expressed similar levels of the ryanodine receptor, Ser16‐phosphorylated phospholamban and Kv4.2. Colchicine attenuated the shortening of the collagen‐induced action potential duration in HL‐1 cells. These findings suggest that colchicine modulates the atrial electrical activity and Ca2+ regulation and attenuates the electrical effects of collagen, which may contribute to its anti‐AF activity.
Keywords: colchicine, calcium handling, electrophysiology
Introduction
Atrial fibrillation (AF) is the commonest form of sustained arrhythmia and increases the risk of stroke, heart failure and mortality 1, 2. However, AF rhythm control agents are not very useful due to a high recurrence of AF, low adherence and common adverse effects 3, 4. Colchicine, a drug widely used to treat gouty arthritis, was found to reduce the occurrence of post‐operative AF or early AF recurrence after pulmonary vein isolation in the absence of antiarrhythmic drug treatment 5, 6. Colchicine has a potent anti‐inflammatory effect and decreases the occurrence of pericarditis 7, 8, 9, both critical in the pathogenesis of AF. Moreover, colchicine can directly modulate calcium (Ca2+) homeostasis in cardiomyocytes 10. However, it is not clear if colchicine can regulate atrial electrical activity to reduce AF genesis 11.
Colchicine is a microtubule disruptor that inhibits microtubule assembly in cells and inhibits neutrophil and endothelial cell adhesion molecules 5, 12, which play roles in the pathophysiology of AF 13. Microtubules, a major cytoskeletal component of cardiomyocytes, are linked to various pathological conditions and may play a role in modulating both the electrical and mechanical activities of the heart. Moreover, microtubule assembly increases the deposition of extracellular matrix (ECM) proteins, especially type I collagen 14, 15. Higher concentrations of ECM proteins enhance the occurrence of AF through electrical and structural remodelling 16, 17. Colchicine decreases interstitial myocardial fibrosis by interfering with collagen accumulation and reverses the contractile function in failing hearts 18, 19. Therefore, colchicine may attenuate myocardial remodelling through decreasing ECM accumulation and prevent AF occurrence. The purpose of this study was to investigate the effects of colchicine on Ca2+ homeostasis and electrophysiological characteristics in atrial myocytes and find out if colchicine can modulate collagen‐induced action potential (AP) changes.
Materials and methods
Cell culture
HL‐1 cells derived from mouse atrial cardiac muscle cells (kindly provided by Dr. Claycomb, Louisiana State University Health Sciences Center, New Orleans, LA, USA) were cultured in a humidified atmosphere of 5% CO2 at 37°C in Claycomb medium (JRH Biosciences, Lenexa, KS, USA). HL‐1 cells were seeded in fibronectin/gelatin‐precoated dishes as described previously. Cells were plated at a density of 5 × 105 cells/well in 6‐well culture plates. At 24 hrs after cell seeding, we added colchicine (3 nM, which is within the therapeutic concentration) 20 or a vehicle solution to cells for a further 24 hrs as colchicine‐treated or control cells, respectively. We also studied the effect of colchicine on collagen‐treated HL‐1 cells. We incubated HL‐1 cells with collagen (10 mg/ml) and colchicine (3 nM) for a further 24 hrs after cell seeding.
Measurement of intracellular Ca2+
As described previously 21, HL‐1 cells were loaded with fluorescent Ca2+ (10 μmol/l) fluo‐3/acetoxymethyl (AM) ester for 30 min. at room temperature. Excess extracellular dye was removed by changing the bath solution, and intracellular hydrolysis of fluo‐3/AM occurred at 35 ± 1°C after 30 min. Fluo‐3 fluorescence was excited with a 488‐nm line of an argon ion laser. The emission was recorded at >515 nm. Cells were repetitively scanned at 3‐ms intervals for a total duration of 6 sec. Fluorescence imaging was performed with a laser scanning confocal microscope (Zeiss LSM 510, Carl Zeiss, Jena, Germany) and an inverted microscope (Axiovert 100, Carl Zeiss, Jena, Germany). The fluorescent signals were corrected for variations in the dye concentration by normalizing the fluorescence (F) against the baseline fluorescence (F0) to obtain reliable information about intracellular (cytoplasm) Ca2+ ([Ca2+]i) transient changes from baseline values (F/F0) and to exclude variations in the fluorescence intensity with different volumes of injected dye. [Ca2+]i transients and peak systolic and diastolic [Ca2+]i values were measured during 2‐Hz field stimulation with 10‐ms twice‐threshold strength square‐wave pulses.
After achieving steady‐state Ca2+ transients with repeated pulses from −40 to 0 mV (1 Hz for 5 sec.), the total amount of charge crossing the membrane sarcoplasmic reticulum (SR) Ca2+ content (Ccaff) was estimated by integrating the sodium (Na+)–Ca2+ exchanger (NCX) current rapid application of 20 mM caffeine during rest with the membrane potential clamped to −40 mV to cause SR Ca2+ release 22. The total SR Ca2+ content (expressed as mM of cytosol) was determined by use of the equation: SR Ca2+ content = [(1 + 0.12)(Ccaff/F × 1000)]/(Cm × 8.31 × 8.44), where Cm = membrane capacitance; F = Faraday's number; cell surface to volume ratio = 8.44 pF/pL 23, 24, 25.
APs and ionic currents
A whole‐cell patch clamp was used for single isolated cardiomyocytes with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA, USA) at 35 ± 1°C 26. Borosilicate glass electrodes (o.d., 1.8 mm) with tip resistances of 3~5 MΩ were used. Before the formation of the membrane‐pipette seal, the tip potentials were zeroed in Tyrode's solution. Junction potentials between the bath and pipette solution (9 mV) were corrected for AP recordings. APs were recorded in the current‐clamp mode, and ionic currents were measured in the voltage‐clamp mode. The ionic currents and AP were recorded at approximately similar period (3–5 min.) after membrane rupture or perforation by amphotericin B (for ICa‐L) to avoid decay of ion channel activity over time. A small hyperpolarizing step from a holding potential of −50 mV to a test potential of −55 mV for 80 ms was delivered at the beginning of each experiment. The area under the capacitative current was divided by the applied voltage step to obtain the whole‐cell capacitance. The series resistance (Rs) was compensated by 60~80%. APs were elicited from isolated cardiomyocytes without spontaneous activity at a driven rate of 1 Hz for 20 beats. The resting membrane potential (RMP) was measured during the period between the last repolarization and the onset of the subsequent AP. The AP amplitude (APA) was obtained from the RMP to the peak of AP depolarization. AP durations (APDs) at 90%, 50% and 20% repolarization were, respectively, measured as the APD90, APD50 and APD20. Micropipettes were filled with a solution containing (in mM) KCl 20, K aspartate 110, MgCl2 1, MgATP 5, HEPES 10, EGTA 0.5, LiGTP 0.1 and Na2 phosphocreatine 5 (pH 7.2 with KOH) for the AP and potassium currents; containing (in mM) CsCl 130, MgCl2 1, MgATP 5, HEPES 10, EGTA 10, NaGTP 0.1 and Na2 phosphocreatine 5 (pH 7.2 with CsOH) for the L‐type Ca2+ current (ICa‐L); containing (in mM) NaCl 20, CsCl 110, MgCl2 0.4, CaCl2 1.75, tetraethylammonium 20, 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (BAPTA) 5, glucose 5, MgATP 5 and HEPES 10 (pH of 7.25) for the NCX current; and containing (in mM) potassium gluconate 144, MgCl2 1.15, EGTA 5, HEPES 10 and CaCl2 4.2 (pH 7.25 with KOH) for the apamin‐sensitive small‐conductance Ca2+‐activated K+ current (IKAS).
The ICa‐L was recorded using 300‐ms pulses from a holding potential of −50 mV to inactivate the T‐type Ca2+ current 27, to test potentials that varied between −40 and +60 mV in 10‐mV increments at a frequency of 0.1 Hz using a perforated patch clamp with amphotericin B. In the external solution, NaCl and KCl of the normal Tyrode's solution were respectively replaced with TEACl and CsCl.
The NCX current was obtained as the nickel‐sensitive current by subtracting the current in the presence of 10 mM NiCl2 from that of the control. The recording protocol consisted of 300‐ms pulses ranging from −100 to +100 mV from a holding potential of −40 mV at a frequency of 0.1 Hz. The external solution for measuring the NCX contained (in mM): NaCl 140, CaCl2 2, MgCl2 1, HEPES 5 and glucose 10 (pH of 7.4). It was supplemented with strophanthidin (10 μM), nitrendipine (10 μM) and niflumic acid (100 μM).
The transient outward potassium current (Ito) was studied using a protocol consisting of a 30‐ms pre‐pulse from a holding potential of −80 to −40 mV to inactivate sodium channels followed by a 300‐ms test pulse to +60 mV in 10‐mV increments at a frequency of 0.1 Hz in the presence of 200 μM CdCl2 in Ca2+‐free normal Tyrode's solution as described previously 28. The Ito was measured as the difference between the peak outward current and the steady‐state current 29, and the sustained outward potassium current (IKsus) was evaluated as the difference between the holding current and the end of the steady‐state current.
The ultra‐rapid delayed rectifier potassium current (IKur) was measured as 4‐aminopyridine‐sensitive currents. To record IKur without contamination by Ito, IKur was dissected by 1 mM 4‐aminopyridine with a double‐pulse protocol, consisting of a 100‐ms depolarizing pre‐pulse to +40 mV from a holding potential of −50 mV to inactivate Ito, followed by 150‐ms voltage steps from −40 to +60 mV in 10‐mV increments at room temperature to provide adequate temporal resolution 30, 31.
The IKAS was recorded using the whole‐cell mode of the patch‐clamp technique. Stability of the total potassium current (IK) was monitored with a step‐pulse protocol (with a holding potential of −50 mV and test potentials from −120 mV to +70 mV for 300 ms at a frequency of 0.1 Hz). Once the IK became stable (usually for 5 min.), apamin (100 nM) was applied, and a step‐pulse protocol was performed when the IK reached a steady state. The IKAS was calculated as the difference between the absence and presence of apamin. All experiments were performed at 36°C. The external solution contained (in mM): N‐methylglucamine, 140; KCl, 4; MgCl2, 1; glucose, 5; and HEPES, 10 (pH 7.4 with HCl) 32.
Western blot analysis
Control and colchicine‐treated HL‐1 cells were centrifuged and lysed using radioimmunoprecipitation (RIPA) buffer. The protein concentration was determined with a Bio‐Rad protein assay reagent (Hercules, CA, USA). For the analysis, equal amounts of protein from each sample were separated using 4~16% Tris‐acetate polyacrylamide gradient gel electrophoresis. After electrophoresis, protein samples were transferred onto equilibrated polyvinylidene difluoride membranes (Amersham Biosciences, Buckinghamshire, UK). Blots were probed with primary antibodies against ryanodine receptor type 2 (RyR; Affinity BioReagent, Golden, CO, USA), RyR phosphorylation at S2808 and S2814 (RyR‐2808 and RyR‐2814; Badrilla Leeds, UK), SR Ca2+ ATPase (SERCA2a, Santa Cruz Biotechnology, Santa Cruz, CA, USA), NCX (Swant, Bellinzona, Switzerland), Cav1.2 (ICa‐L subunit; Alomone Labs, Jerusalem, Israel), phospholamban (PLB; Thermo, Rockford, IL, USA), Thr17‐phosphorylated PLB (PLB‐Thr17; Badrilla, Leeds, UK), PLB‐Ser16 (Badrilla), Ca2+/calmodulin‐dependent protein kinase II (CaMKII, Abcam, Cambridge, UK), Kv1.4 (Alomone Labs), Kv1.5 (Alomone Labs), Kv4.2 (Abcam) and α‐actin (Sigma‐Aldrich, St. Louis, MO, USA). Blots were reacted with primary antibodies followed by horseradish peroxidase‐conjugated secondary antibodies. Bound antibodies were detected with an enhanced chemiluminescence (ECL) detection system of an ECL Plus Kit (Millipore, Billerica, MA, USA) and analysed with AlphaEaseFC software (Alpha Innotech, San Leandro, CA, USA). Targeted bands were normalized to α‐actin.
Immunofluorescence microscopy of cardiomyocyte microtubules
HL‐1 cells with and without colchicine (3 nM) or collagen (10 μg/ml), cultured in Lab‐Tek™ 4‐well chamber slides (Thermo Scientific, Rochester, NY, USA) for 24 hrs, were fixed in PBS containing 4% paraformaldehyde for 30 min., blocked with 3% bovine serum albumin and incubated in primary antibody used with anti α‐tubulin antibody (Sigma, St Louis, MO, USA) at a dilution of 1:200 for 2 hrs, and washed three times with PBS for 5 min. Cells were then incubated in secondary antibody used with FITC‐conjugated antibody (Life technologies, Paisley, UK) at a dilution of 1:200 for 1 hr. Finally, counterstaining of nuclei was performed with 4′, 6‐diamidino‐2‐phenylindole (DAPI; Sigma, St Louis, MO, USA). The cells were observed and photographed with a Leica TCS SP5 Confocal Spectral Microscope Imaging System (×200, Leica Microsystems, Wetzlar, Germany).
Statistical analysis
All quantitative data are expressed as the mean ± standard error of the mean (S.E.M.). A paired t‐test was used to compare differences between control and colchicine‐treated HL‐1 cells and collagen‐treated HL‐1 cells with or without colchicine. Nominal variables were compared by a Pearson's chi‐square test or Fisher's exact test. A P value of <0.05 was considered statistically significant.
Results
Effect of colchicine on Ca2+ homeostasis and ionic currents in HL‐1 cells
As shown in Figure 1, colchicine (3 nM)‐treated HL‐1 cells had 10% smaller [Ca2+]i transients than control HL‐1 cells. Moreover, colchicine (3 nM)‐treated HL‐1 cells had 47% smaller caffeine‐induced [Ca2+]i transients than the control group, which suggests less SR Ca2+ stores in colchicine (3 nM)‐treated HL‐1 cells. Moreover, colchicine (3 nM)‐treated HL‐1 cells had smaller ICa‐L and outward (reverse mode) NCX current densities than control HL‐1 cells (Fig. 2). Colchicine (3 nM)‐treated HL‐1 cells had a smaller Ito and IKsus compared with control HL‐1 cells (Fig. 3A). Moreover, colchicine (3 nM)‐treated and control HL‐1 cells had similar values of the IKur and IKAS (Fig. 3B and C).
Figure 1.
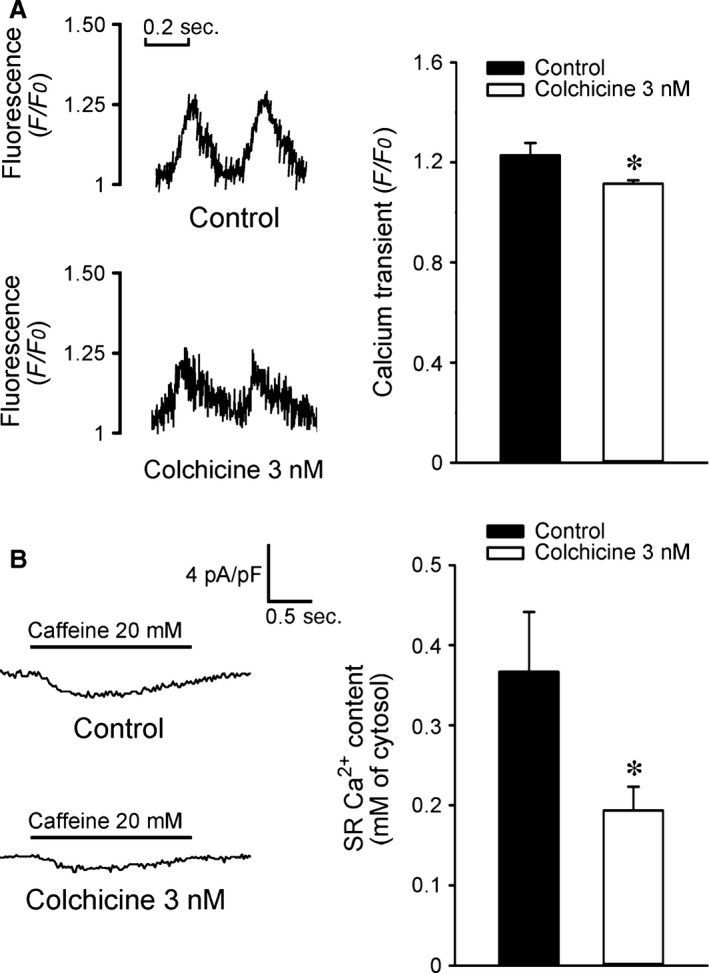
Calcium homeostasis of control and colchicine (3 nM)‐treated HL‐1 cells. (A) Tracings and average data from [Ca2+]i transients in control (n = 16) and colchicine (3 nM)‐treated (n = 16) HL‐1 cells. (B) Tracings and average data of caffeine‐induced Na+–Ca2+ exchanger (NCX) currents and sarcoplasmic reticulum Ca2+ contents from integrating NCX currents in control (n = 12) and colchicine (3 nM)‐treated (n = 13) HL‐1 cells. *P < 0.05 versus the control.
Figure 2.
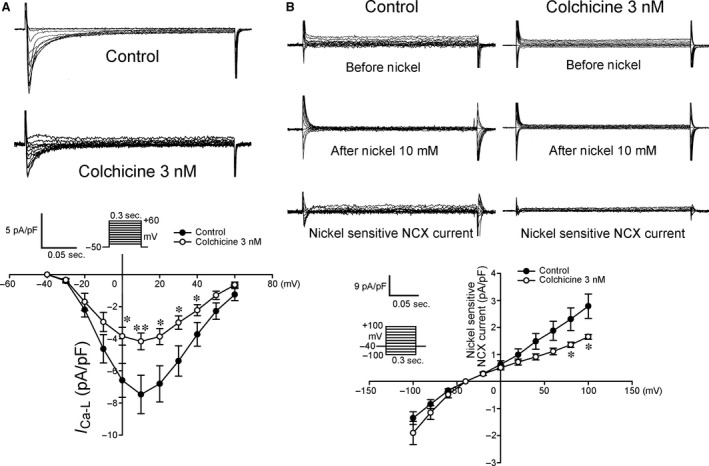
Current tracings and I–V relationship of the L‐type calcium current (ICa‐L) and Na+–Ca2+ exchanger (NCX) current in HL‐1 cells with and without colchicine (3 nM) treatment. (A) The ICa‐L had decreased amplitudes in colchicine‐treated HL‐1 cells (n = 14) than control HL‐1 cells (n = 18). (B) Colchicine‐treated HL‐1 cells (n = 8) exhibited a smaller current amplitude of the NCX current than control HL‐1 cells (n = 8). The inset in the current traces shows the clamp protocol.*P < 0.05 versus the control, **P < 0.01 versus the control.
Figure 3.
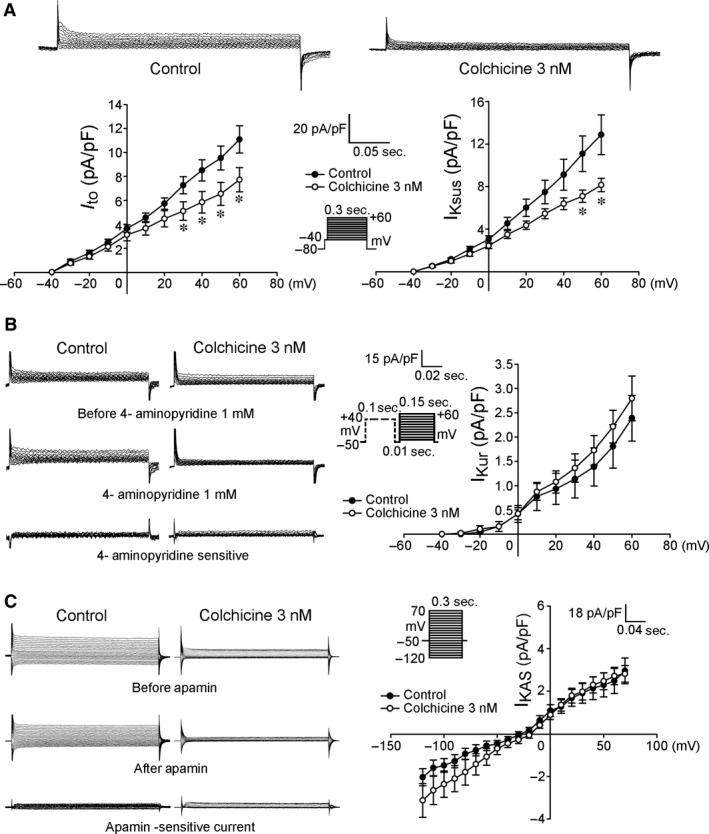
The transient outward potassium current (Ito), sustained outward potassium current (IKsus), ultra‐rapid delayed rectifier potassium current (IKur) and apamin‐sensitive small‐conductance Ca2+‐activated K+ current (IKAS) in HL‐1 cells with and without colchicine (3 nM) treatment. (A) Examples of tracings and I–V relationships of the Ito and IKsus from HL‐1 cells with (n = 14) and without (n = 15) colchicine treatment. (B) Examples of tracings and I–V relationships of the IKur from HL‐1 cells with (n = 8) and without (n = 7) colchicine treatment. (C) Examples of tracings and I–V relationship of the IKAS from HL‐1 cells with (n = 11) and without (n = 11) colchicine treatment. Insets in the current traces show the various clamp protocols. *P < 0.05 versus the control.
Effects of colchicine on Ca2+ regulatory proteins and Kv channel subunits
The expression of SERCA2a and Cav1.2 in the colchicine (3 nM)‐treated HL‐1 cells was lower by 14% and 11% than in the controls. Moreover, the expression of CaMKII, a multifunctional serine/threonine protein kinase that mediates Ca2+ handling, had also decreased by 15% in colchicine (3 nM)‐treated HL‐1 cells. The colchicine (3 nM)‐treated HL‐1 cells also had lower expressions of total PLB and PLB‐Thr17 by 18% and 23%, but had similar expressions of PLB‐Ser16 (Fig. 4A). The colchicine (3 nM)‐treated HL‐1 cells had lower expression of NCX by 9%, but had similar expressions of RyR, RyR‐2808, and RyR‐2814 compared with control cells. The colchicine (3 nM)‐treated HL‐1 cells had lower expressions of Kv1.4 and Kv1.5 by 10% and 14%, respectively. The expressions of Kv4.2 were similar in both the control and colchicine (3 nM)‐treated HL‐1 cells (Fig. 4B).
Figure 4.
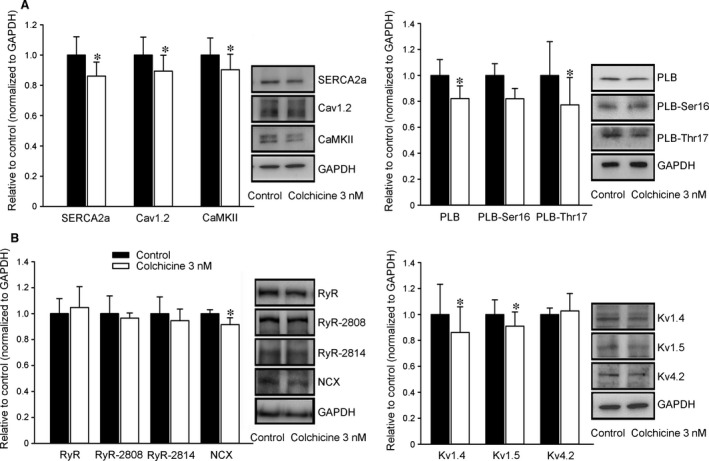
Effect of colchicine on the protein expression of calcium (Ca2+) regulatory proteins and potassium channel proteins. (A) Representative immunoblotting and average data of sarcoplasmic reticulum Ca2+ ATPase (SERCA2a), Cav1.2, Ca2+/calmodulin‐dependent protein kinase II (CaMKII), PLB, Ser16‐ and Thr17‐phosphorylated PLB (PLB‐Ser16 and PLB‐Thr17) from control and colchicine (3 nM)‐treated HL‐1 cells. (n = 9) (B) Representative immunoblotting and average data of RyR type 2, phosphorylation of RyR at S2808 and S2814 (RyR‐2808 and RyR‐2814), the Na+–Ca2+ exchanger (NCX), Kv1.4, Kv1.5 and Kv4.2 from control and colchicine (3 nM)‐treated HL‐1 cells (n = 7). *P < 0.05 versus the control. PLB, phospholamban; RyR, ryanodine receptor.
The effect of colchicine on microtubule structure and the AP morphology in collagen‐induced changes of HL‐1 cells
As shown in Figure 5A, a breakdown of the reticular microtubules was evident in the colchicine (3 nM)‐treated HL‐1 cells. When compared with the controls, the colchicine (3 nM)‐treated HL‐1 cells had longer APD90, APD50 and APD20, but had similar values for the APA and RMP. In contrast to the effects of colchicine, collagen (10 μg/ml) shortened the APD90 in HL‐1 cells and had more delayed afterdepolarizations (DADs; 20% versus 66.7%, P < 0.005). Furthermore, colchicine (3 nM) ameliorated APD changes in collagen (10 mg/ml)‐treated HL‐1 cells as shown in Figure 5B, and no DADs were noted.
Figure 5.
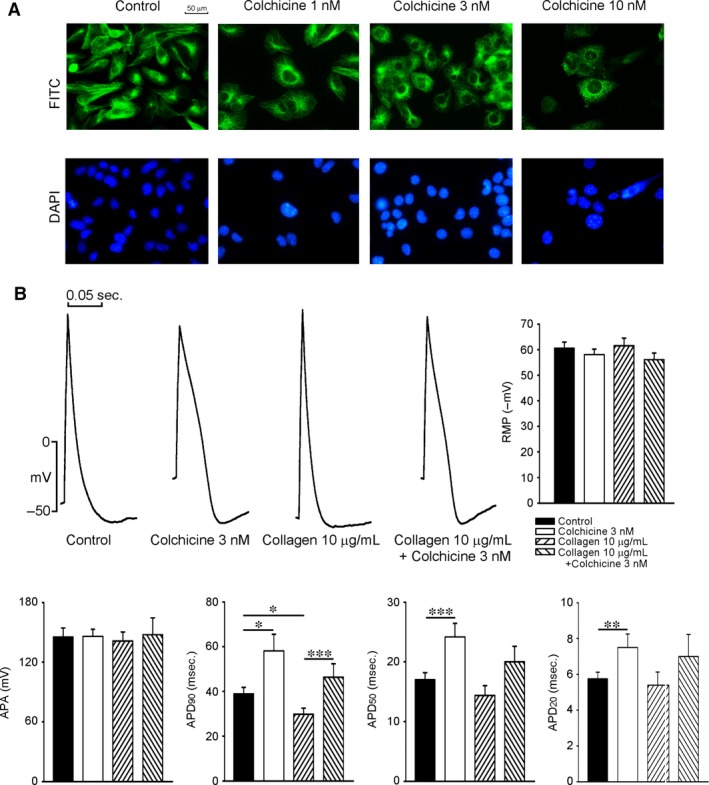
Immunofluorescent staining of microtubules and AP characteristics of HL‐1 cells with and without colchicine or collagen treatment. (A) The concentration (1, 3 and 10 nM) effect of colchicine on microtubule structure was studied in HL‐1 cells by confocal microscope. Upper panel shows that HL‐1 cells were labelled with α‐tubulin and then visualized with secondary antibodies conjugated to FITC (green), and lower panel shows that nuclei were stained with DAPI (blue). Colchicine (1 nM) did not have significant effect on microtubule structure, colchicine (3 nM) induced depolymerization with an uneven microtubule structure in HL‐1 cells, and colchicine (10 nM) caused patch aggregations of microtubule in cytoplasm and a decrease in the cell number. (B) Action potential (AP) characteristics of HL‐1 cells with and without colchicine (3 nM) or collagen (10 μg/ml) treatment. Examples and average data of APs from control (n = 12), colchicine‐treated (n = 14), collagen‐treated (n = 12) and colchicine and collagen‐treated (n = 15) HL‐1 cardiomyocytes. AP durations at 20%, 50% and 90% repolarization of the AP amplitude (APD20, APD50 and APD90, respectively) were measured at 1 Hz. *P < 0.05, **P < 0.01, ***P < 0.005. AP, action potential.
Discussion
Ca2+ homeostasis is essential for maintaining normal cardiac function and electrical activity. There is tight coupling between Ca2+ influx and Ca2+ release. Dysregulated Ca2+ handling can generate the triggered activity, commonly associated with AF development 33. Ca2+ overload may potentially produce arrhythmogenesis in atrial myocytes 34. In this study, colchicine was shown to reduce the amplitude of [Ca2+]i transients and SR Ca2+ content, which suggests colchicine decreases Ca2+ concentration in both cytoplasm and the SR. Accordingly, colchicine might directly modulate Ca2+ homeostasis in atrial myocytes and result in a decrease in the occurrence of AF. In contrast, previous studies on rat or chick embryos ventricular myocytes showed that microtubule disruption by colchicine (incubation for 10 min. or 2 hrs, 1~10 μM) increased the ICa‐L and [Ca2+]i transients, but did not modify SR Ca2+ content 10, 35, 36, 37. Additionally, other studies showed colchicine had no effect on APD and Ito in isolated mouse or dog ventricular myocytes 38, 39. This disparity may arise from different experimental settings (animal species, colchicine concentrations and incubation period) or the variance between HL‐1 cells and ventricular myocytes. As shown in Figure 5A, we have found that colchicine (3 nM) had significant effects on microtubule disruption, which supports the possibility that colchicine may modulate HL‐1 cell calcium homeostasis through microtubule disruption. A previous study showed that patients receiving colchicine had been given therapeutic doses that ranged from 0.5 to 3 μg/l (0.8~7 nM) 20. Therefore, the concentration used in this study was clinically relevant.
In addition to reductions in ICa‐L and reverse mode NCX in HL‐1 cells, colchicine can down‐regulate SERCA2a, total and PLB‐Thr17. A decrease in PLB phosphorylation at Thr17 can functionally reduce SERCA2a Ca2+ reuptake by increasing the effects of PLB on the inhibition of SERCA2a activity. All these changes can result in a decrease in SR Ca2+ content and Ca2+ transients. Colchicine decreased CaMKII expression, which is a key regulator of excitation‐contraction coupling in cardiomyocytes. CaMKII activation may enhance ICa‐L facilitation and increase Ca2+ influx, which accompanies Ca2+ overload. In addition, CaMKII can enhance Ca2+ leakage to generate triggered activity. The potential of CaMKII inhibition to reduce an arrhythmogenic outcome was demonstrated in AF 40. This action may result in the known effect of colchicine in decreasing Ca2+ sparks from a previous study 10. Therefore, colchicine decreases calcium overload through modulation of Ca2+ regulatory proteins, which may inhibit the occurrence of AF.
We found that colchicine can increase the APD, which may reduce the generation of re‐entrant circuits and AF maintenance. This prolongation of the APD may be caused by a reduction of reverse mode NCX, Ito, and IKsus, which can overcome the APD‐shortening effect by ICa‐L reduction. Thus, colchicine modulates atrial electrophysiological property by interfering Ca2+ and potassium currents, which may ameliorate AF occurrence. Similarly, cytoskeletal disruption was also reported to increase the APD in ventricular myocytes by decreasing the Ito 41. These findings suggest that interactions exist between the potassium channels and microtubules or associated cytoskeletal components. The decreased expressions of Kv1.4 and Kv1.5 may contribute to decreases in the Ito and IKsus mediated by colchicine.
The ECM of collagen causes atrial myocytes to exhibit a shorter APD by increasing the Ito and IKsus 34. In this study, it was found that colchicine reversed the APD‐shortening effects of collagen. Moreover, collagen was also shown to induce Ca2+ overload, which facilitates the occurrence of trigger activities in atrial and pulmonary vein myocytes. Colchicine normalized the APD in collagen‐treated HL‐1 cells, and this suggests that the ECM controls cytoskeletal mechanics in the development of cardiac remodelling, and colchicine may modulate the remodelling process and atrial arrhythmogenesis.
There are several limitations of our study that must be acknowledged. This study demonstrated the effect of colchicine on microtubule disruption in HL‐1 cells, an atrial cell line from mice. However, the ultrastructure of HL‐1 cells, including the cytoskeleton, and microtubule stability are different from adult naïve cardiomyocytes. 42, 43 Because of the immature phenotype of HL‐1 cells, our findings may not be fully applied to human atrial cells. In addition, we did not study the RyR or SERCA2a activity, and it is not clear whether factors other than those found in this experiment may also contribute to colchicine‐reducing SR content. Sorcin, a penta‐EF‐hand protein, interacts with intracellular target proteins, such as NCX, SERCA2a and ICa‐L 44, 45, 46, and may influence these current activities. Moreover, pulmonary vein cardiomyocyte plays an important role for triggering AF 47. Ca2+ dysregulation may increase arrhythmogenesis in pulmonary vein cardiomyocytes. Colchicine may reduce AF recurrence after pulmonary vein ablation 6. Theoretically, the electrophysiological effects of colchicine on pulmonary vein cardiomyocytes may possibly play a role in its anti‐AF potential.
In conclusion, our findings suggest that colchicine modulates atrial electrical activity and Ca2+ regulation, which may be responsible for preventing AF occurrence or recurrence. These changes also demonstrate that microtubule modulation may play an important role in the regulation of ionic channels and Ca2+ homeostasis, which contributes to the pathogenesis of AF.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Acknowledgements
This study was supported by grants NSC102‐2628‐B‐038‐002‐MY3, and 103‐2314‐B‐281‐005‐MY2 from the Ministry of Science and Technology of Taiwan; 103swf02, 103‐wf‐eva‐02, 104swf02, and 104‐wf‐eva‐01 from Taipei Medical University's Wan‐Fang Hospital, 103CM‐TMU‐07 from Chi‐Mei Medical Center and CGH‐MR‐A10219, CGH‐MR‐A10221, CGH‐MR‐A10222, CGH‐MR‐103‐05 from Cathay General Hospital.
References
- 1. Benjamin EJ, Wolf PA, D'Agostino RB, et al Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998; 98: 946–52. doi:10.1161/01.CIR.98.10.946. [DOI] [PubMed] [Google Scholar]
- 2. Go AS, Mozaffarian D, Roger VL, et al Executive summary: heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014; 129: 399–401. doi:10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- 3. Nabar A, Rodriguez LM, Timmermans C, et al Class IC antiarrhythmic drug induced atrial flutter: electrocardiographic and electrophysiological findings and their importance for long term outcome after right atrial isthmus ablation. Heart. 2001; 85: 424–9. doi:10.1136/heart.85.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wyse DG, Waldo AL, DiMarco JP, et al A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002; 347: 1825–33. doi:10.1056/NEJMoa021328. [DOI] [PubMed] [Google Scholar]
- 5. Imazio M, Brucato A, Ferrazzi P, et al Colchicine reduces postoperative atrial fibrillation: results of the Colchicine for the Prevention of the Postpericardiotomy Syndrome (COPPS) atrial fibrillation substudy. Circulation. 2011; 124: 2290–5. doi:10.1161/CIRCULATIONAHA.111.026153. [DOI] [PubMed] [Google Scholar]
- 6. Deftereos S, Giannopoulos G, Kossyvakis C, et al Colchicine for prevention of early atrial fibrillation recurrence after pulmonary vein isolation: a randomized controlled study. J Am Coll Cardiol. 2012; 60: 1790–6. doi:10.1016/j.jacc.2012.07.031. [DOI] [PubMed] [Google Scholar]
- 7. Millaire A, Ducloux G. Treatment of acute or recurrent pericarditis with colchicine. Circulation. 1991; 83: 1458–9. [PubMed] [Google Scholar]
- 8. Imazio M, Brucato A, Forno D, et al Efficacy and safety of colchicine for pericarditis prevention. Systematic Review and Meta‐Analysis. Heart. 2012; 98: 1078–82. doi:10.1136/heartjnl‐2011‐301306. [DOI] [PubMed] [Google Scholar]
- 9. Cocco G, Chu DC, Pandolfi S. Colchicine in clinical medicine. A guide for internists. Eur J Intern Med. 2010; 21: 503–8. DOI: 10.1016/j.ejim.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 10. Kerfant BG, Vassort G, Gomez AM. Microtubule disruption by colchicine reversibly enhances calcium signaling in intact rat cardiac myocytes. Circ Res. 2001; 88: E59–65. doi:10.1161/hh0701.090462. [DOI] [PubMed] [Google Scholar]
- 11. Lu YY, Chen YJ. A randomized trial of colchicine for acute pericarditis. N Engl J Med. 2014; 370: 780. doi:10.1056/NEJMc1315351#SA2. [DOI] [PubMed] [Google Scholar]
- 12. Molad Y. Update on colchicine and its mechanism of action. Curr Rheumatol Rep. 2002; 4: 252–6. [DOI] [PubMed] [Google Scholar]
- 13. Koyama T, Sekiguchi Y, Tada H, et al Comparison of characteristics and significance of immediate versus early versus no recurrence of atrial fibrillation after catheter ablation. Am J Cardiol. 2009; 103: 1249–54. doi:10.1016/j.amjcard.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 14. Heck JN, Ponik SM, Garcia‐Mendoza MG, et al Microtubules regulate GEF‐H1 in response to extracellular matrix stiffness. Mol Biol Cell. 2012; 23: 2583–92. doi:10.1091/mbc.E11‐10‐0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Putnam AJ, Schultz K, Mooney DJ. Control of microtubule assembly by extracellular matrix and externally applied strain. Am J Physiol Cell Physiol. 2001; 280: C556–64. [DOI] [PubMed] [Google Scholar]
- 16. Boldt A, Wetzel U, Lauschke J, et al Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004; 90: 400–5. doi:10.1136/hrt.2003.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsai CT, Tseng CD, Hwang JJ, et al Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor beta1. Cardiovasc Res. 2011; 89: 805–15. doi:10.1093/cvr/cvq322. [DOI] [PubMed] [Google Scholar]
- 18. Fernandes F, Ramires FJ, Ianni BM, et al Effect of colchicine on myocardial injury induced by Trypanosoma cruzi in experimental Chagas disease. J Card Fail. 2012; 18: 654–9. doi:10.1016/j.cardfail.2012.06.419. [DOI] [PubMed] [Google Scholar]
- 19. Sato H, Nagai T, Kuppuswamy D, et al Microtubule stabilization in pressure overload cardiac hypertrophy. J Cell Biol. 1997; 139: 963–73. DOI: 10.1083/jcb.139.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deftereos S, Gionnopoulos G, Papoutsidakis N, et al Colchicine and the heart: pushing the envelop. J Am Coll Cardiol. 2013; 62: 1817–25. doi:10.1016/j.jacc.2013.08.726. [DOI] [PubMed] [Google Scholar]
- 21. Chang SL, Chen YC, Yeh YH, et al Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J Cardiovasc Electrophysiol. 2011; 22: 1378–86. doi:10.1111/j.1540‐8167.2011.02126.x. [DOI] [PubMed] [Google Scholar]
- 22. Tsai WC, Lu YY, Chen YC, et al Ablation of androgen receptor gene triggers right ventricular outflow tract ventricular tachycardia. Int J Cardiol. 2015; 189: 172–81. doi:10.1016/j.ijcard.2015.04.080. [DOI] [PubMed] [Google Scholar]
- 23. Satoh H, Delbridge LMD, Blatter LA, et al Surface‐volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species‐dependence and developmental effects. Biophys J. 1996; 70: 1494–504. doi:10.1016/S0006‐3495(96)79711‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation‐contraction coupling in canine heart failure. Circ Res. 2001; 103: 1577–84. doi:10.1161/01.CIR.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 25. Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000; 87: 275–81. doi:10.1161/01.RES.87.4.275. [DOI] [PubMed] [Google Scholar]
- 26. Chen YJ, Chen YC, Wongcharoen W, et al Effect of K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br J Pharmacol. 2008; 153: 915–25. doi:10.1038/sj.bjp.0707564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claycomb WC, Lanson NA Jr, Stallworth BS, et al HL‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998; 95: 2979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brahmajothi MV, Campbell DL, Rasmusson RL, et al Distinct transient outward potassium current (Ito) phenotypes and distribution of fast‐inactivating potassium channel alpha subunits in ferret left ventricular myocytes. J Gen Physiol. 1999; 113: 581–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Udyavar AR, Chen YC, Chen YJ, et al Endothelin‐1 modulates the arrhythmogenic activity of pulmonary veins. J Cardiovasc Electrophysiol. 2008; 19: 285–92. doi:10.1111/j.1540‐8167.2007.01033.x. [DOI] [PubMed] [Google Scholar]
- 30. Nattel S, Matthews C, Blasio ED, et al Dose‐dependence of 4‐aminopyridine plasma concentrations and electrophysiological effects in dogs: potential relevance to ionic mechanisms in vivo . Circulaiton. 2000; 101: 1179–84. doi:10.1161/01.CIR.101.10.1179. [DOI] [PubMed] [Google Scholar]
- 31. Ting YK, Morikawa K, Kurata Y, et al Transcriptional activation of the anchoring protein SAP97 by heat shock factor (HSF)‐1 stabilizes Kv1.5 channels in HL‐1 cells. Br J Pharmacol. 2011; 162: 1832–42. doi:10.1111/j.1476‐5381.2011.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu YY, Chen YC, Kao YH, et al Extracellular matrix of collagen modulates arrhythmogenic activity of pulmonary veins through p38 MAPK activation. J Mol Cell Cardiol. 2013; 59: 159–66. doi:10.1016/j.yjmcc.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 33. Chen YJ, Chen SA, Chang MS, et al Arrhythmogenic activity of cardiac muscle in pulmonary veins of the dog: implication for the genesis of atrial fibrillation. Cardiovasc Res. 2000; 48: 265–73. doi:10.1016/S0008‐6363(00)00179‐6. [DOI] [PubMed] [Google Scholar]
- 34. Lu YY, Chen YC, Kao YH, et al Extracellular matrix of collagen modulates intracellular calcium handling and electrophysiological characteristics of HL‐1 cardiomyocytes with activation of angiotensin II type 1 receptor. J Card Fail. 2011; 17: 82–90. doi:10.1016/j.cardfail.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 35. Iribe G, Ward CW, Camelliti P, et al Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009; 104: 787–95. doi:10.1161/CIRCRESAHA.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calaghan SC, Le Guennec JY, White E. Modulation of Ca2+ signaling by microtubule disruption in rat ventricular myocytes and its dependence on the ruptured patch‐clamp configuration. Circ Res. 2001; 88: E32–7. doi:10.1161/01.RES.88.4.e32. [DOI] [PubMed] [Google Scholar]
- 37. Galli A, DeFelice LJ. Inactivation of L‐type Ca channels in embryonic chick ventricle cells: dependence on the cytoskeletal agents colchicine and taxol. Biophys J. 1994; 67: 2296–304. doi:10.1016/S0006‐3495(94)80715‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ozgen N, Lu Z, Boink GJJ, et al Microtubules and angiotensin II receptos contribute to modulation of repolarization induced by ventricular pacing. Heart Rhythm. 2012; 9: 1865–72. doi:10.1016/j.hrthm.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J, Gao J, Cohen IS, et al Angiotensin II type 1 receptor‐mediated electrical remodeling in mouse cardiac myocytes. PLoS ONE. 2015; 10: e0138711. doi:10.1371/journal.pone.0138711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heijman J, Voigt N, Wehrens XH, et al Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol. 2014; 5: 30. doi:10.3389/fphar.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang X, Salas PJ, Pham TV, et al Cytoskeletal actin microfilaments and the transient outward potassium current in hypertrophied rat ventriculocytes. J Physiol. 2002; 541: 411–21. doi:10.1113/jphysiol.2002.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guzun R, Karu‐Varikmaa M, Gonzalez‐Granillo M, et al Mitochondria‐cytoskeleton interaction: distribution of β‐tubulins in cardiomyocytes and HL‐1 cells. Biochim Biophys Acta. 2011; 1807: 458–69. doi:10.1016/j.bbabio.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 43. Belmadani S, Pous C, Fischmeister R, et al Post‐translational modifications of tubulin and microtubule stability in adult rat ventricular myocytes and immortalized HL‐1 cardiomyocytes. Mol Cell Biochem. 2004; 258: 35–48. [DOI] [PubMed] [Google Scholar]
- 44. Zamparelli C, Macquaide N, Colotti G, et al Activation of the cardiac Na+–Ca2+ exchanger by sorcin via the interaction of the respective Ca2+‐binding domains. J Mol Cell Cardiol. 2010; 49: 132–41. doi:10.1016/j.yjmcc.2010.03.003.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Collis LP, Meyers MB, Zhang J, et al Expression of a sorcin missense mutation in the heart modulates excitation‐contraction coupling. FASEB J. 2007; 21: 475–87. doi:10.1096/fj.06‐6292com. [DOI] [PubMed] [Google Scholar]
- 46. Matsumoto T, Hisamatsu Y, Ohkusa T, et al Sorcin interacts with sarcoplasmic reticulum Ca2+–ATPase and modulates excitation–contraction coupling in the heart. Basic Res Cardiol. 2005; 100: 250–62. doi:10.1007/s00395‐005‐0518‐7. [DOI] [PubMed] [Google Scholar]
- 47. Chen YJ, Chen SA, Chen YC, et al Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: implication in initiation of atrial fibrillation. Circulation. 2001; 104: 2849–54. doi:10.1161/hc4801.099736. [DOI] [PubMed] [Google Scholar]