

Abstract
Cysteine S-nitrosation and S-sulfination are naturally occurring post-translational modifications (PTMs) on proteins induced by physiological signals and redox stress. Here we demonstrate that sulfinic acids and nitrosothiols react to form a stable thiosulfonate bond, and leverage this reactivity using sulfinate-linked probes to enrich and annotate hundreds of endogenous S-nitrosated proteins. In physiological buffers, sulfinic acids do not react with iodoacetamide or disulfides, enabling selective alkylation of free thiols and site-specific analysis of S-nitrosation. In parallel, S-nitrosothiol-linked probes enable enrichment and detection of endogenous S-sulfinated proteins, confirming that a single sulfinic acid can react with a nitrosothiol to form a thiosulfonate linkage. Using this approach, we find that hydrogen peroxide addition increases S-sulfination of human DJ-1 (PARK7) at Cys106, whereas Cys46 and Cys53 are fully oxidized to sulfonic acids. Comparative gel-based analysis of different mouse tissues reveals distinct profiles for both S-nitrosation and S-sulfination. Quantitative proteomic analysis demonstrates that both S-nitrosation and S-sulfination are widespread, yet exhibit enhanced occupancy on select proteins, including thioredoxin, peroxiredoxins, and other validated redox active proteins. Overall, we present a direct, bidirectional method to profile select redox cysteine modifications based on the unique nucleophilicity of sulfinic acids.
Introduction
The cysteine sulfhydryl group is a key target of redox stress, and depending on the abundance and type of redox-active species, is covalently modified to one of a series of distinct chemical moieties1. Reactive nitrogen species induce formation of S-nitrosocysteine (R-SNO)2, and reactive oxygen species induce reversible disulfides and S-sulfenylcysteine (R-SOH), as well as irreversible S-sulfinylcysteine (R-SO2H) and S-sulfonylcysteine (R-SO3H)3. Aberrant redox modifications are implicated in the pathology of many diseases, including inflammation4, stroke5, and neurodegeneration6.
S-nitrosation (R-SNO) of proteins can reversibly mask functional cysteines or alter protein dynamics to affect cellular function in both normal and diseased states7. Cysteine S-nitrosation is initiated by nitric oxide, which can directly react with cysteine thiyl radicals, undergo secondary oxidation and react with cysteine thiolates8, or form through reactions with dinitrosyliron complexes9. Importantly, S-nitrosation is reversible, primarily by trans-nitrosation10 of cellular thiols. S-nitrosation has been extensively profiled using the biotin-switch assay and its variants11, which captures cysteine residues sensitive to ascorbate reduction. This indirect approach relies on the chemical orthogonality of ascorbate, which is known to reduce weak disulfides12 and other labile redox modifications13. S-nitrosated proteins can also be enriched with organomercury resin, followed by performic acid oxidation for release and downstream analysis14. Both methods have been used for large-scale mass spectrometry profiling of S-nitrosated proteins, both in normal and diseased states. While the majority of mass spectrometry proteomics studies focus on the effects of nitric oxide donors, more recent analyses have identified nearly 1000 endogenous S-nitrosated proteins in tissues15. In addition, various S-nitrosothiol selective phosphine-based probes have been introduced with significant promise for large-scale proteomic analysis16, and have already been demonstrated as selective reagents to label and quantify S-nitrosated metabolites17. While each method has contributed important biological revelations regarding S-nitrosation, new simplified, non-toxic, direct, and selective approaches remain in high demand.
Reactive oxygen species primarily oxidize cysteine to form disulfide bonds, which first proceed from a S-sulfenylcysteine (R-SOH) intermediate18. In the absence of a resolving thiol, additional oxidation leads to formation of cysteine sulfinic acid (Cys-SO2H)19. S-sulfination is generally irreversible, with the exception of peroxiredoxins, which employ sulfiredoxin to reverse accumulated S-sulfination20. Additionally, S-sulfination of the PARK7 (DJ-1) is enigmatically critical for the protein’s redox chaperone activity21. While there are no reported methods for mass spectrometry profiling of S-sulfination, recently reported substituted aryl-nitroso probes suggest S-sulfination is widespread, and may play a broader role in protein structure, redox homeostasis, and cellular regulation22.
Here we explore the cross reactivity of S-sulfination with S-nitrosation, which react to form a stable thiosulfonate. Biotin conjugated probes enable reciprocal detection, enrichment, and analysis of each redox post-translational modification in cell and tissue homogenates, and suggests a broader role for sulfinic acids in redox regulation.
Results and Discussion
Sulfinic acids react selectively with S-nitrosothiols
While exploring the interplay of cysteine post-translational modifications, we identified a reported reaction between phenylsulfinic acid and S-nitrosocysteine, leading to thiosulfonate formation in physiological buffers at room temperature23. Phenylsulfinic acid reacts rapidly with N-acetyl-S-nitrosocysteine methyl ester to yield a thiosulfonate product (Figure 1a). More than 99% of the thiosulfonate product remained after 5 hours at pH ≤ 7, demonstrating robust stability in physiological buffers (Figure S1). Moreover, we did not observe any additional products formed using a photodiode array detector, suggesting a direct conversion of reactants to products.
Figure 1.

Sulfinic acid reactivity in phosphate buffer. (a) Phenylsulfinic acid (2, 20 mM) reacts with N-acetyl S-nitroso cysteine methyl ester (1, 5 mM) to form thiosulfonate 3. Absorbance was measured at 283 nm. (b) No additional peaks are observed when phenylsulfinic acid is incubated with iodoacetamide in PBS for 30 minutes. Absorbance was measured at 291 nm. (c) Phenylsulfinic acid, 2, does not react with the activated disulfide DTNB, 5. Absorbance was measured at 291 nm for phenylsulfinic acid and 265 nm for DTNB and the reaction mixture.
Thiosulfonates are readily exchangeable with thiols, serving as the basis for the cysteine capping agent methyl methanethiosulfonate (MMTS)24. To prevent such exchange, we found that sulfinic acids do not react with iodoacetamide (IAM) (Figure 1b and Figure S2a) or cysteine (Figure S2b) in aqueous buffers, enabling orthogonal alkylation of thiols without perturbing nitrosothiols or sulfinic acids. Interestingly, a large number of reported S-nitrosation studies alkylate thiols with MMTS25, which releases methylsulfinic acid upon reaction with cysteine. Any released methylsulfinic acid may proceed to react with S-nitrosothiols, potentially reducing S-nitrosation detection26.
Approximately 6–10% of all cellular thiols are oxidized and engaged in a disulfide bonds, which can rise to >15% upon oxidative stress27. We find that phenylsulfinic acid does not react with biologically relevant disulfides such as cystine (Figure S2c) or activated disulfides such as 5,5′-dithiobis-(2-nitrobenzoic acid), DTNB (Figure 1c). However, the highly activated disulfide dipyridyldisulfide (aldrithiol) forms an insoluble species when mixed with equimolar phenylsulfinic acid, but does partially react overnight in a 50% DMSO/PBS solution (data not shown). Finally, we observe no reaction between phenylsulfinic acid and benzaldehyde (Figure S2d) or with pyrrolidinone sulfenamide (Figure S3). Overall, sulfinic acids do not react with free thiols, biological disulfides, aldehydes or sulfenamides, highlighting an unappreciated chemoselective reaction with S-nitrosothiols. Furthermore, after initial iodoacetamide alkylation of free thiols, sulfinic acid probes could be leveraged to label and enrich S-nitrosothiols for analysis.
Reaction analysis
To characterize the reaction of nitrosothiols and sulfinic acids, we assayed thiosulfonate formation by measuring the loss of S-nitroso-glutathione (GSNO) absorbance at 340 nm after phenylsulfinic acid addition (Figure S4). Increasing amounts of sodium phenylsulfinate were titrated to a 2 mM solution of S-nitrosoglutathione in the dark. The reported pKa of a sulfinic acid is ~2.828, yet the reaction proceeds similarly at pH 1, suggesting the sulfur lone pair acts as the nucleophile, independent of the protonation state. Sulfinic acids are ambident nucleophiles29, where the soft sulfur atom is the attacking species, and the oxygen charge state should not significantly affect sulfur nucleophilicity. At pH 1, 4, and 7, the reaction rate is approximately first order and proceeds at 3 x 10−2 M−1s−1. At neutral pH, the concentration dependence is slightly hyperbolic, suggesting a small contribution from an alternate reaction mechanism, and no reaction occurs under basic conditions.
A potential reaction mechanism was recently reported requiring two sulfinic acids; one to attack the nitrogen of the nitrosothiol, and a second sulfinic acid to react with the sulfur to displace N-hydroxysulfonamide and the thiosulfonate in stoichiometric amounts30. This is in contrast to our initial reaction analysis, which did not reveal any additional products. Standard curves were then derived from isolated chemical standards to allow detailed analysis of the reaction between sodium 4-methyl-phenyl sulfinate and GSNO (Figure S5a–d). This analysis reveals sub-stoichiometric formation of N-hydroxybenzenesulfonamide, which is only detected in the presence of significant excess sulfinic acid (Figure 2b), and thus not in our initial near-stoichiometric HPLC analysis.
Figure 2.
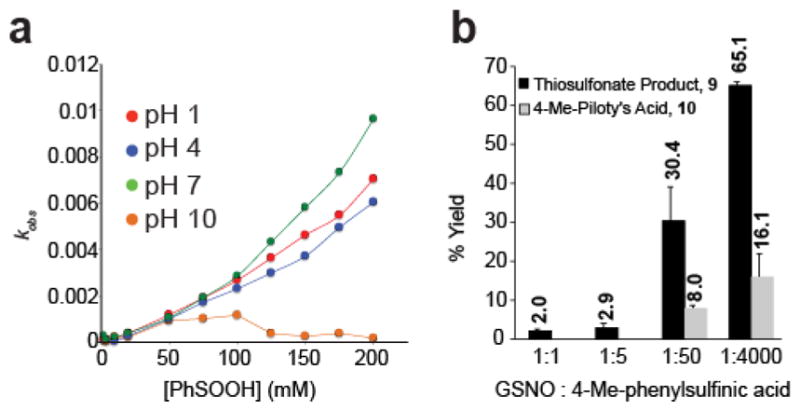
Reaction kinetics and by-product analysis. (a) Reaction rate between phenylsulfinic acid and GSNO at various pHs. (b) Percent yield of the thiosulfonate product and the 4-methyl-Piloty’s acid product.
N-hydroxysulfonamides are prone to degradation at higher pH, decomposing to release sulfinic acid and nitroxyl (HNO)31. To investigate whether the sub-stoichiometric formation of the N-hydroxysulfonamide product can be attributed to loss through degradation, we assayed the stability of N-hydroxy-4-methylbenzenesulfonamide at pH 1, 4, 7 and 10 (Figure S5e). Our findings demonstrate that N-hydroxy-4-methylbenzenesulfonamide is stable at pH 1, and degrades slowly at pH 4. At pH 7, N-hydroxy-4-methylbenzenesulfonamide decomposes with a half-life of ~4 hours. Furthermore, since the decomposition of N-hydroxy-4-methylbenzenesulfonamide generates nitroxyl and a sulfinic acid, excess sulfinic acid will slow decomposition by mass action32. After taking degradation into account, N-hydroxysulfonamide formation is not stoichiometric with formation of the thiosulfonate product. Overall, the reaction between sulfinic acids and nitrosothiols proceeds by an alternative mechanism without concomitant N-hydroxysulfonamide formation.
Biotin-SO2H detects S-nitrosated proteins
Next, we examined the reactivity of reporter linked sulfinic acids with S-nitrosated proteins. Biotin and fluorescein N-hydroxysuccinimide (NHS) esters were directly coupled to the sulfinic acid metabolite hypotaurine (Biotin-SO2H) or the sulfonic acid metabolite taurine (Biotin-SO3H) in de-gassed water, purified by preparative HPLC, and stored in single-use aliquots at −80 °C to prevent oxidation. Biotin-SO2H is stable during the duration of labeling (< 1 hour), but is fully oxidized to biotin-SO3H after ~5 hours in atmospheric oxygen.
Each probe was incubated with mammalian cell lysates denatured in 6 M urea in phosphate-buffered saline (PBS), pre-alkylated with excess iodoacetamide, and analyzed by non-reducing SDS-PAGE and streptavidin-Cy5 blot detection. Biotin-SO2H, but not biotin-SO3H, labeled a rich profile of proteins (Figure 3a), confirming the reaction is dependent on the nucleophilic sulfinic acid. Biotin-SO2H labeling is enhanced at higher probe concentrations (Figure S6a) or by pre-incubation with the nitric oxide donor methylamine hexamethylene me-thylamine NONOate (MAHMA-NONOate) (t1/2 = 3 min) (Figure 3b and S6b). Furthermore, labeling is eliminated by pretreatment with excess hypotaurine (Figure S6c), demonstrating saturated labeling above 5 mM. In addition, 365 nm ultraviolet photolysis of S-nitrosothiols or pretreatment with ascorbate eliminates biotin-SO2H labeling (Figure 3c–d). Importantly, all biotin-SO2H labeling is all labeling is thiol-dependent, and reversed by post-incubation with tris(2-carboxyethyl)phosphine (TCEP).
Figure 3.

Sulfinic acid probes selectively label S-nitrosothiols in HEK293T cell lysates. Unless otherwise noted, all lysates are denatured in 6 M urea supplemented with 50 mM iodoacetamide (IAM). (a) Biotin-SO2H, but not biotin-SO3H, labels S-nitrosothiols in lysates. (b) Biotin-SO2H labeling increases following pre-treatment with MAHMA NONOate, a nitric oxide donor before IAM addition. (c) UV photolysis (365 nm) pre-treatment eliminates biotin-SO2H labeling. (d) Biotin-SO2H labeling is eliminated by pre-treatment with ascorbate. (e) The products of biotin-SO2H labeling are sensitive to post-treatment by the reductant TCEP. (f) The sulfenic acids probe dimedone does not reduce biotin-SO2H labeling. (g) Denaturing buffers or ascorbate reduce dimedone-alkyne labeling of sulfenic acids. Following a 1 hour incubation with dimedone alkyne, lysates were chloroform/methanol precipitated and mixed with TBTA, CuSO4, TCEP, and TAMRA-azide for 1 hour in PBS before gel analysis. (h) MMTS and IAM both react with free thiols, but MMTS liberates methane sulfinic acid and interferes biotin-SO2H labeling of S-nitrosated proteins.
Sulfenic acids (R-SOH) describe the meta-stable oxidation of thiols to a highly reactive intermediate, which are typically resolved by a secondary thiol during disulfide formation18a, 33. Based on the electrophilic nature of sulfenic acids, we next examined if sulfinic acids react with sulfenic acids. The sulfenic acid sensitive probe dimedone is widely used to covalently trap sulfenic acids in complex proteomes and in living systems3, 34. In order to explore this potential cross-reactivity, we found that standard working concentrations of dimedone had no effect on biotin-SO2H labeling in cell lysates, confirming dimedone does not significantly interfere with S-nitrosothiols. Interestingly, dimedone-alkyne labeling was largely eliminated after denaturing proteins in 6 M urea. Thus, the denaturing biotin-SO2H assay conditions remove any sulfenic acids, limiting any concerns about cross-reactivity. In addition, ascorbate eliminates nearly all dimedone labeling, providing further evidence in the absence of denaturing buffers, sulfenic acids may also be analyzed when using the biotin-switch method for detecting S-nitrosation. In addition, sodium meta-arsenite has been reported as a selective sulfenic acid reductant35. We do not observe any arsenite-dependent effects on biotin-SO2H labeling (Figure S7), providing further support for orthogonal labeling between sulfinic acids and nitrosothiols.
The common alkylating agent, methyl methane thiosulfonate (MMTS) reacts with free thiols to form a disulfide bond, releasing methane sulfinic acid. Surprisingly, if we block thiols with MMTS instead of iodoacetamide, we observe a complete loss in biotin-SO2H labeling (Figure 3h and Figure S8a). Based on this analysis, sufficient methyl sulfinic acid is released through MMTS alkylation of thiols to react with nitro-sothiols26. This effect could similarly be caused by trans-nitrosation between thiol contaminants formed through slow disproportionation of sulfinic acids36. This is unlikely, since removing MMTS prior to biotin-SO2H does not restore labeling (Figure S8b). These findings warrant a careful reinterpretation of the biotin-switch S-nitrosation enrichment method, which typically begins with MMTS alkylation of free thiols. Once nitrosothiols react with methyl sulfinic acid, the resulting thiosulfonate can be slowly reduced by ascorbate, liberating a free thiol for further enrichment. Overall, iodoacetamide effectively blocks all thiols in our experiments, as well as sites of persulfidation37, providing a robust platform for the detection of endogenous S-nitrosothiols.
Next, biotin-SO2H was used to profile the S-nitrosation of purified recombinant human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a known S-nitrosated protein38. GAPDH was dissolved in PBS and then treated under different conditions, followed by incubation with iodoacetamide and biotin-SO2H to primarily evaluate the specificity of sulfinic acid probes for nitrosothiols using a purified protein. As expected, no S-nitrosation was observed until treatment with MAHMA NONOate, which was reduced by ascorbate pre-treatment (Figure 4a). S-nitroso-glutathione (GSNO) is reported to S-nitrosate GAPDH via trans-nitrosation10. GAPDH is only labeled by biotin-SO2H after GSNO addition. Labeling is reversed by incubation with dithiothreitol, which cleaves the thiosulfonate linkage (Figure 4b). Further high-resolution mass spectrometry analysis of denatured and labeled GAPDH tryptic peptides confirmed thiosulfonate formation (Figure S9).
Figure 4.

Labeling of recombinant human GAPDH with biotin-SO2H. (a) GAPDH labeling is observed only in the presence of MAHMA NONOate, and eliminated by pre-treatment with ascorbate. (b) GAPDH is labeled by the trans-nitrosation donor GSNO, and eliminated by pre-treatment with the reducing agent DTT.
Biotin-GSNO detects S-sulfinated proteins
We next asked what would happen if we reversed our detection scheme, by applying S-nitrosothiol-linked probes to detect endogenous S-sulfination. Similar reactivity was recently reported using an aryl-nitroso probe for conjugation to sulfinic acid standards to form a stable N-sulfonylbenzisoxazolone22. While both approaches use nitroso moieties to react with sulfinic acids, each follow different mechanistic pathways.
Accordingly, biotin-GSNO was synthesized in one step from biotin-NHS and GSNO in de-gassed phosphate buffer in the dark. After HPLC purification, the probe was stored as single-use aliquots at −80 °C to minimize formation of oxidized biotin-GSSG-biotin, or used immediately for best results. Mammalian cell lysates were first denatured in 6 M urea in PBS, and treated with dithiothreitol (DTT) to reduce disulfides (Figure S10) without affecting sulfinic acids21a, 39. This is essential as to prevent exchange with any de-nitrosated glutathione in the probe stock. The reduced lysate was then incubated with iodoacetamide to block free thiols, and precipitated to remove any residual reactants. After solubilization in 6 M urea, biotin-GSNO was added to label S-sulfinated proteins for gel-based analysis. Labeling is not affected by pretreatment with S-methyl glutathione (Figure 5a), but competed by excess GSNO (Figure 5b). Together, these controls confirm nitrosothiol-dependent labeling of iodoacetamide resistant protein modifications with no bias towards specific sites. Similarly, dimedone had no effect on biotin-GSNO labeling, confirming there is no cross reactivity with thiosulfonates, sulfinic acids, or that any residual sulfenic acids are present after denaturation (Figure 5c). Additionally, all labeling is eliminated by post-incubation with TCEP, which reduces the thiosulfonate product (Figure 5d). Pretreatment of iodoacetamide labeled lysates with hydrogen peroxide (50 mM) prevented any reaction with biotin-GSNO, presumably by oxidizing sulfinic acids to sulfonic acids (Figure 5e). Finally, oxidized glutathione (GSSG) was coupled to biotin-NHS to examine if any non-specific labeling occurred by disulfide exchange. Even though disulfides are reduced and alkylated at the beginning of the assay, incomplete alkylation could lead to significant false positives if the biotin-GSNO probe decomposes to the oxidized disulfide. Under these assay conditions, biotin-GSSG does not label any proteins (Figure 5f), confirming selective conjugation of between nitrosothiols and sulfinic acids is selective and bidirectional.
Figure 5.

Biotin-GSNO (1 mM) labels a unique profile of S-sulfinated proteins in HEK293T cell lysates. (a) Biotin-GSNO labeling is not competed by S-methylglutathione (1 mM). (b) GSNO competes with biotin-GSNO for labeling native S-sulfinated proteins. (c) Biotin-GSNO labeling of S-sulfination is unaffected by dimedone (1 mM). (d) Proteins first labeled with biotin-GSNO are then lost after TCEP (5 mM) addition. (e) Peroxide pre-treatment in iodoacetamide alkylated lysates eliminates biotin-GSNO labeling, suggesting terminal oxidation of sulfinic acids to non-reactive sulfonic acids. (f) Biotin-GSSG, a putative contaminant in biotin-GSNO, does not label any proteins.
The redox chaperone DJ-1 (PARK7) readily forms a stable sulfinic acid at Cys10621a, 21d, which is critical for suppressing redox stress21b. DJ-1 also reduces nitrosative stress21b, 21c, 40, suggesting that oxidized DJ-1 may react directly with cellular nitrosothiols forming a thiosulfonate, which is readily reduced by cellular thiols like glutathione. Here, we demonstrate that under non-denaturing oxidative conditions, DJ-1 reacts with N-acetyl-S-nitrosocysteine methyl ester and GSNO (Figure 6). After peroxide and iodoacetamide treatment, native DJ-1 was incubated with N-acetyl-S-nitrosocysteine methyl ester and digested with trypsin for high-resolution LC-MS analysis. High resolution MS/MS analysis unambiguously confirmed thiosulfonate formation at Cys106 (Figure S11). Furthermore, peptides lacking a cysteine were unaffected by up to 200 mM peroxide treatment. In the absence of peroxide, DJ-1 Cys106 is fully alkylated by iodoacetamide and no labeling with GSNO is detectable. After incubation with 10 mM peroxide, Cys106 is detected primarily as the thiosulfonate conjugate, with partial conversion to the unreactive sulfonic acid. Oxidation is further enhanced after treatment with 200 mM peroxide, which eliminates all Cys106-SO2H and prevents thiosulfonate formation. Both Cys46 and Cys53 did not react with GSNO after peroxide treatment, as they were directly converted to the unreactive sulfonic acid.
Figure 6.

Analysis of thiosulfonate formation on recombinant human DJ-1. Purified, recombinant DJ-1 was treated with buffer, 10 mM hydrogen peroxide, or 200 mM peroxide. Samples were treated with iodoacetamide (IAM) to block free thiols, and excess reagents were removed by gel filtration before incubating with GSNO. The relative abundance of each of the modified peptide was measured by mass spetrometry of trypic peptides. The peptide abundances were normalized to reflect relative changes within each condition. Error bars represent standard deviations of from three replicates. The control peptide E64–K89 showed no peroxide-dependent changes.
Profiling native S-nitrosation and S-sulfination
Immortalized mammalian cell lines are adapted to atmospheric oxygen, potentially augmenting the profile of oxidative modifications. Therefore, we isolated mouse tissues for immediate processing and gel-based analysis. Surprisingly, we detect defined tissue-specific patterns of both S-nitrosation (Figure 7a) and S-sulfination (Figure 7b), demonstrating orthogonal protein targets of each modification in vivo.
Figure 7.
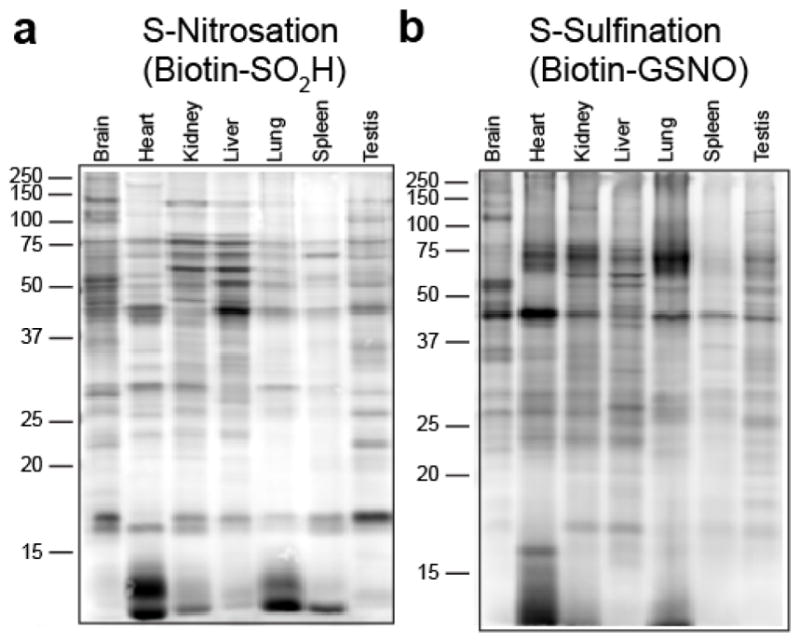
Profiling native S-nitrosation and S-sulfination in mouse tissues. (a) S-nitrosation profile of mouse tissues labeled with biotin-SO2H. (b) S-sulfination profile of mouse tissues labeled with biotin-GSNO using matched protein loading, fluorescence detection, and image settings.
To annotate the endogenous targets of each redox modification in cells, both biotin-SO2H and biotin-GSNO conjugates were profiled using stable-isotope labeling with amino acids in cell culture (SILAC) for quantitative mass spectrometry proteomics. For profiling S-nitrosation, heavy or light cell 293T cell pellets were lysed by sonication in 6 M urea, treated with excess iodoacetamide, and incubated with either biotin-SO2H or biotin-SO3H. After chloroform/methanol precipitation, the two proteomes were combined for streptavidin enrichment, trypsin digestion, and high resolution mass spectrometry analysis. Through a combination of 4 biological replicates, each with 2 technical replicates, a total of 992 proteins were identified with SILAC ratios > 5 (biotin-SO2H/biotin-SO3H), quantified in ≥ 3 replicates, and represented by ≥ 3 quantified peptides (Table S1). After elution of tryptic peptides, resin-bound biotinylated peptides were eluted with TCEP and alkylated with maleimide, enabling detection of an additional 98 sites of S-nitrosation by mass spectrometry (Table S2). These lists include nearly all previously annotated S-nitrosated proteins, including ion channels, chaperones, peroxiredoxins, p53, HDACs, hundreds of metabolic enzymes, as well as a rich set of novel proteins.
To profile S-sulfination, heavy or light cell 293T cell lysates were separately treated with DTT, alkylated with excess iodoacetamide, and treated with either biotin-GSNO or free biotin. After enrichment and mass spectrometry analysis, nearly 300 S-sulfinated proteins were specifically enriched, including DJ-1 (PARK7), phosphatases, metabolic enzymes, and a partially overlapping set of oxidized proteins (Table S3). Resin-bound peptides were eluted using TCEP reduction, enabling identification of an additional 30 specific sites of S-sulfination (Table S4), altogether providing the first large-scale analysis of this redox modification.
Next, to evaluate the relative occupancy of each redox modification, we quantified both S-nitrosation and S-sulfination enrichment in comparison with relative protein abundance. Unenriched 293T proteomes were analyzed using label-free quantification (Table S5)41 based on the integrated ion intensity of the top 3 most intense ions for each protein. Individual protein values from the probe enrichment were divided by their corresponding relative abundance, providing a distribution of ratios reflecting proportionally higher modification occupancy (Figure 8a and Table S6). This analysis is critical to identify not just abundant proteins with fractional oxidation, but to highlight proteins with greater representative modification occupancy. Importantly, this is not an absolute stoichiometry. This value reflects the relative enrichment efficiency as compared to the estimated relative abundance, and helps identify proteins that may be low abundance, but are highly modified.
Figure 8.

Relative comparative protein-level occupancy of redox modifications. (a) Histogram of calculated relative occupancy ratios of S-nitrosated proteins (left) compared to S-sulfinated proteins (right), derived from label-free quantiation. (b) Comparative analysis of relative occupancy ratios of both S-nitrosated and S-sulfinated proteins reveals inherent preferences towards each redox modification. Arbitrary lines and color boundaries are presented, separating abundant, low occupancy proteins (grey), from highly S-nitrosated (blue), highly S-sulfinated (green), and proteins with enhanced occupancy for each modification (red).
For both redox modifications, the majority of proteins were observed with low ratios, signifying poor relative enrichment and low modification stoichiometry characteristic of abundant proteins, including several heat shock, cytoskeletal, and ribosomal proteins. In contrast, S-nitrosated proteins with large ratios signify high relative occupancy, including several metabolic enzymes and proteins with metal coordination sites, such as HDAC1 and carbonic anhydrase. Interestingly, S-sulfinated proteins with large ratios include validated oxidation prone enzymes such as peroxiredoxins, thioredoxin, pyruvate kinase, and triosephosphate isomerase. Approximately 175 proteins were selectively enriched with both probes, revealing inherent preferences for each redox modification (Figure 8b). Interestingly, DJ-1 was found to be both S-nitrosated and S-sulfinated. Both Cys46 and C53 are surface exposed and established sites of S-nitrosation42 and the tryptic peptide containing Cys53 was identified in our endogenous site-specific analysis. In contrast, Cys106 is primarily S-sulfinated21a, 21b, 21d, demonstrating how a single endogenous protein can harbor more than one distinct redox modifications.
Conclusions
Overall, this approach highlights proteins with enhanced susceptibility to distinct redox modifications, opening new opportunities for multiplexed profiling of disease-dependent mutually competitive cysteine modifications. This simplified, direct approach bypasses the hazards of mercury-based affinity reagents, and avoids complex disulfide chemistry commonly used for ascorbate-dependent reduction strategies. While the thiosulfonate linkage is not ideal for stable enrichment, we find that after proper alkylation of free thiols, thiosulfonates are sufficiently stable for non-reducing gel-based analysis and mass spectrometry profiling. Importantly, these findings establish that sulfinic acids possess intrinsic reactivity that may contribute to cellular redox regulation. As observed for human DJ-1, once Cys106 is oxidized to a sulfinic acid, it readily converts S-nitrosated thiols to an easily exchangeable thiosulfonate. Future studies will explore if enzymes, once exposed to oxidative stress, form nucleophilic sulfinic acids that aid cells from accumulating further oxidative damage.
Supplementary Material
Acknowledgments
Support for these studies was provided by the National Institutes of Health R00 CA151460, DP2 GM114848, T32 CA140044 Proteome Informatics of Cancer Training Grant (A.M.K), F31 EB019320 (C.T.M.B.T.), the National Science Foundation (Graduate Research Fellowship to A.M.K.), the American Heart Association 14POST20420040 (J.D.M.), and the University of Michigan.
Footnotes
Supporting Information. Additional figures, tables, compound characterization, and detailed methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Gruhlke MCH, Slusarenko AJ. Plant Physiol Biochem. 2012;59:98. doi: 10.1016/j.plaphy.2012.03.016. [DOI] [PubMed] [Google Scholar]; (b) Giles GI, Jacob C. Biol Chem. 2002;383:375. doi: 10.1515/BC.2002.042. [DOI] [PubMed] [Google Scholar]; (c) Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majmudar JD, Martin BR. Biopolymers. 2014;101:173. doi: 10.1002/bip.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman S, Nolin J, McMillan D, Wouters E, Janssen-Heininger Y, Reynaert N. J Cell Biochem. 2015;116:884. doi: 10.1002/jcb.25017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Andersson A, Hultberg B, Lindgren A. Atherosclerosis. 2000;151:535. doi: 10.1016/s0021-9150(99)00418-9. [DOI] [PubMed] [Google Scholar]; (b) Maron BA, Tang SS, Loscalzo J. Antioxid Redox Signal. 2013;18:270. doi: 10.1089/ars.2012.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Circ Res. 2013;112:382. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Chung KKK. Neurosignals. 2006;15:307. [Google Scholar]; (b) Fox JH, Connor T, Stiles M, Kama J, Lu Z, Dorsey K, Liebermann G, Sapp E, Cherny RA, Banks M, Volitakis I, DiFiglia M, Berezovska O, Bush AI, Hersch SM. J Biol Chem. 2011;286:18320. doi: 10.1074/jbc.M110.199448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Majmudar JD, Martin BR. Biopolymers. 2014;101:173. doi: 10.1002/bip.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Broillet MC. Cell Mol Life Sci. 1999;55:1036. doi: 10.1007/s000180050354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stamler JS. Cell. 1994;78:931. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]; (d) Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]; (e) Zhang YH, Hogg N. Free Rad Biol Med. 2005;38:831. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]; (f) Seth D, Stamler JS. Curr Op Chem Biol. 2011;15:129. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jourd’heuil D, Jourd’heuil FL, Feelisch M. J Biol Chem. 2003;278:15720. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 9.(a) Tonzetich ZJ, McQuade LE, Lippard SJ. Inorg Chem. 2010;49:6338. doi: 10.1021/ic9022757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsai ML, Tsou CC, Liaw WF. Acc Chem Res. 2015;48:1184. doi: 10.1021/ar500459j. [DOI] [PubMed] [Google Scholar]
- 10.Barnett DJ, McAninly J, Williams DLH. J Chem Soc Perkin Trans 2. 1994;1131 [Google Scholar]
- 11.(a) Jaffrey SR, Snyder SH. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]; (b) Foster MW. Biochim Biophys Acta - General Subjects. 2012;1820:675. doi: 10.1016/j.bbagen.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R. Nitric Oxide-Biol Ch. 2008;19:252. doi: 10.1016/j.niox.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 13.(a) Monteiro G, Horta BB, Pimenta DC, Augusto O, Netto LES. Proc Natl Acad Sci USA. 2007;104:4886. doi: 10.1073/pnas.0700481104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM. FEBS J. 2013;280:6150. doi: 10.1111/febs.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doulias P-T, Raju K, Greene JL, Tenopoulou M, Ischiropoulos H. Methods. 2013;62:165. doi: 10.1016/j.ymeth.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doulias P-T, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Sci Sig. 2013;6 doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Biggs TD, Weerasinghe L, Park CM, Xian M. Tetrahedron Lett. 2015;56:2741. doi: 10.1016/j.tetlet.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li S, Wang H, Xian M, Whorton AR. Nitric Oxide-Biol Ch. 2012;26:20. doi: 10.1016/j.niox.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR. J Am Chem Soc. 2013;135:7693. doi: 10.1021/ja401565w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Toennies G. J Biol Chem. 1937;122:27. [Google Scholar]; (b) Rehder DS, Borges CR. Biochemistry. 2010;49:7748. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simonsen DG. J Biol Chem. 1933;101:35. [Google Scholar]
- 20.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. J Biol Chem. 2004;279:50994. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 21.(a) Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. J Biol Chem. 2009;284:6476. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. Proc Natl Acad Sci USA. 2004;101:9103. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM. J Neurosci Res. 2009;87:123. doi: 10.1002/jnr.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Biochem Biophys Res Commun. 2004;317:722. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 22.(a) Lo Conte M, Lin J, Wilson MA, Carroll KS. ACS Chem Biol. 2015;10:1825. doi: 10.1021/acschembio.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo Conte M, Carroll KS. Angew Chemie. 2012;51:6502. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Hart TW. Tetrahedron Lett. 1985;26:2013. [Google Scholar]; (b) Hart TW, Vine MB, Walden NR. Tetrahedron Lett. 1985;26:3879. [Google Scholar]
- 24.Smith DJ, Kenyon GL. J Biol Chem. 1974;249:3317. [Google Scholar]
- 25.(a) Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Sci Signal. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Nat Cell Biol. 2001;3:193. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]; (c) Forrester MT, Foster MW, Stamler JS. J Biol Chem. 2007;282:13977. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 26.Reeves BD, Joshi N, Campanello GC, Hilmer JK, Chetia L, Vance JA, Reinschmidt JN, Miller CG, Giedroc DP, Dratz EA, Singel DJ, Grieco PA. Org Biomol Chem. 2014;12:7942. doi: 10.1039/c4ob00995a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen RE, Roth D, Winther JR. Proc Natl Acad Sci USA. 2009;106:422. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Defilipp D, Momicchi F. Tetrahedron. 1969;25:5733. [Google Scholar]
- 29.Baidya M, Kobayashi S, Mayr H. J Am Chem Soc. 2010;132:4796. doi: 10.1021/ja9102056. [DOI] [PubMed] [Google Scholar]
- 30.Reeves BD, Hilmer JK, Mellmann L, Hartzheim M, Poffenberger K, Johnson K, Joshi N, Singel DJ, Grieco PA. Tetrahedron Lett. 2013;54:5707. doi: 10.1016/j.tetlet.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DuMond JF, King SB. Antioxid Redox Signal. 2011;14:1637. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Hughes MN. Biochim Biophys Acta-Bioenergetics. 1999;1411:263. doi: 10.1016/s0005-2728(99)00019-5. [DOI] [PubMed] [Google Scholar]; (b) Williams DLH. Nitrosation Reactions and the Chemistry of Nitric Oxide. 1. Elsevier; 2004. [Google Scholar]
- 33.(a) Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Biochemistry. 1999;38:15407. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]; (b) Poole LB, Karplus PA, Claiborne A. Annu Rev Pharmacol Toxicol. 2004;44:325. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 34.(a) Poole LB, Zeng BB, Knaggs SA, Yakubu M, King SB. Bioconjugate Chem. 2005;16:1624. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]; (b) Reddie KG, Carroll KS. Curr Op Chem Biol. 2008;12:746. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]; (c) Yang J, Gupta V, Tallman KA, Porter NA, Carroll KS, Liebler DC. Nat Protoc. 2015;10:1022. doi: 10.1038/nprot.2015.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Saurin AT, Neubert H, Brennan JP, Eaton P. Proc Natl Acad Sci USA. 2004;101:17982. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Eaton P. Free Rad Biol Med. 2006;40:1889. doi: 10.1016/j.freeradbiomed.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 36.(a) Babbs CF, Gale MJ. Anal Biochem. 1987;163:67. doi: 10.1016/0003-2697(87)90093-5. [DOI] [PubMed] [Google Scholar]; (b) Scaduto RC. Free Rad Biol Med. 1995;18:271. doi: 10.1016/0891-5849(94)e0139-a. [DOI] [PubMed] [Google Scholar]; (c) Wudl F, Lightner DA, Cram DJ. J Am Chem Soc. 1967;89:4099. [Google Scholar]
- 37.Pan J, Carroll KS. ACS Chem Biol. 2013;8:1110. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Padgett CM, Whorton AR. Am J Physiol - Cell Ph. 1995;269:C739. doi: 10.1152/ajpcell.1995.269.3.C739. [DOI] [PubMed] [Google Scholar]
- 39.(a) Fuangthong M, Helmann JD. Proc Natl Acad Sci USA. 2002;99:6690. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. J Biol Chem. 2006;281:14400. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]; (c) Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. J Biol Chem. 2002;277:38029. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 40.Kahle PJ, Waak J, Gasser T. Free Rad Biol Med. 2009;47:1354. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Silva JC, Gorenstein MV, Li GZ, Vissers JPC, Geromanos SJ. Mol Cell Proteomics. 2006;5:144. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.(a) Ito G, Ariga H, Nakagawa Y, Iwatsubo T. Biochem Biophys Res Commun. 2006;339:667. doi: 10.1016/j.bbrc.2005.11.058. [DOI] [PubMed] [Google Scholar]; (b) Chen YJ, Lu CT, Su MG, Huang KY, Ching WC, Yang HH, Liao YC, Chen YJ, Lee TY. Nucleic Acids Res. 2015;43:D503. doi: 10.1093/nar/gku1176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee TY, Chen YJ, Lu CT, Ching WC, Teng YC, Huang HD, Chen YJ. Bioinformatics. 2012;28:2293. doi: 10.1093/bioinformatics/bts436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.