

Abstract
Described is an efficient stereocontrolled route into valuable, densely functionalized fluorinated phosphonates that takes advantage of (i) a Clostridial enzyme to set the absolute stereochemistry and (ii) a new [3,3]-sigmatropic rearrangement of the thiono-Claisen variety that is among the fastest sigmatropic rearrangements yet reported. Here, a pronounced rate enhancement is achieved by distal fluorination. This rearrangement is completely stereoretentive, parlaying the enzymatically established β-C-O stereochemistry in the substrate into the δ-C-S stereochemistry in the product. The final products are of interest to chemical biology, with a platform for Zn-aminopeptidase A inhibitors being constructed here. The enzyme, Clostridium acetobutylicum (CaADH), recently expressed by our group, reduces a spectrum of γ,δ-unsaturated β-keto-α,α-difluorophosphonate esters (93–99% ee; 10 examples). The resultant β-hydroxy-α,α-difluorophosphonates possess the ‘L’-stereochemistry, opposite to that previously observed for the CaADH-reduction of ω-keto carboxylate esters (‘D’), indicating an unusual active site plasticity. For the thiono-Claisen rearrangement, a notable structure-reactivity relationship is observed. Measured rate constants vary by over three orders of magnitude, depending upon thiono-ester structure. Temperature-dependent kinetics reveal an unusually favorable entropy of activation (∆S‡ = 14.5 ± 0.6 e.u.). Most notably, a 400-fold rate enhancement is seen upon fluorination of the distal arene ring, arising from favorable enthalpic (∆∆H‡ = −2.3 kcal/mol) and entropic (∆∆S‡ = 4 e.u., i.e. 1.2 kcal/mol at RT) contributions. The unusual active site plasticity seen here is expected to drive structural biology studies on CaADH,, while the exceptionally facile sigmatropic rearrangement is expected to drive computational studies to elucidate its underlying entropic and enthalpic basis.
TOC Graphic
INTRODUCTION
In modern asymmetric synthesis and its translation into process chemistry applications on scale, we have entered an age in which the careful melding of sophisticated enzymatic chemistry with sophisticated organic chemistry can present state-of-the-art solutions. In thinking about this evolution, Turner has suggested that we rethink how we analyze synthetic challenges to include a dimension of “biocatalytic retrosynthesis.”1 In so doing, it is important to move beyond the mindset that enzymes are solely useful for the delivery of small molecule building blocks, and to more frequently embrace the notion of challenging enzymes with substrates of higher carbon count and/or that are decorated with a more complex array of functionality. One of the best examples of such a sophisticated hybrid asymmetric synthesis is the current Merck process for the synthesis of sitagliptin in which an engineered transaminase sets the absolute stereochemistry in acting upon an advanced 16-carbon substrate bearing 12 heteroatoms.2
In developing the synthetically useful biocatalytic chemistry that will form the basis of the retrosynthetic transform arrows3 that Turner’s commentary envisions, three broad approaches may be pursued: (i) panning the biosphere for wild type enzymes of utility in synthesis,4 (ii) engineering enzymes from natural protein templates through directed evolution5 or (iii) de novo design of active sites with the aid of modern computational techniques.6 Whichever approach is taken, the optimal solution will lead to enzymes that not only are able to set absolute stereochemistry in synthetically complex substrates, but ideally will also show significant substrate generality and, as such, can be treated much as a “privileged” chiral organic or organometallic catalyst would be.7
We describe herein such an approach that melds a new stereoselective enzymatic transformation to set the absolute stereochemistry for a C-O bond in a densely functionalized fluorinated phosphonate environment, with a new type of hetero-Claisen rearrangement that parlays the established β-C-O bond stereochemistry into δ-C-S bond stereochemistry, with stereochemical fidelity and remarkable facility, via formal allylic transposition. The enzymatic transformation avails itself of a Clostridial alcohol dehydrogenase (CaADH) recently expressed and characterized by our group,8 but identifies a previously unexplored substrate class for this enzyme, or any dehydrogenase for that matter; namely β-keto-α,α-difluorophosphonates, thereby generating a retrosynthetic transform arrow not previously available in the biocatalytic toolbox. Perhaps most importantly, these studies reveal a sort of “active site plasticity”9 rarely seen, in which two distinct substrate classes are each reduced with high enantioselectivity in the same active site, but with opposite facial selectivity. Molecular modeling is used to examine how these two substrate classes bind with high facial bias for hydride transfer, but in quite different three-dimensional arrangements in the CaADH active site. The chemical step introduces a rearrangement of the [3,3]-sigmatropic variety that rivals even the classic Evans-Golob anion-accelerated oxy-Cope rearrangement in terms of rate10 and that is worthy of note (i) for its unusually favorable entropy of activation, (ii) for its stereoretentive nature and (iii) for the remarkable 400-fold gain in rate that is seen upon fluorination of the flanking aromatic ring in the thionocarbonate system (vide infra).
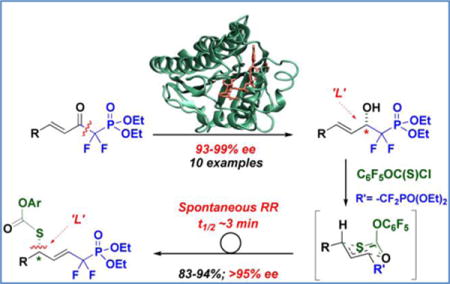
As is illustrated in Figure 1, the melded process constitutes a new retrosynthetic approach to the stereocontrolled synthesis of δ-thio-α,α-difluorinated phosphonates that both possess an intervening functionalizable alkene and that may be decorated with additional functionality, as desired for chemical biological purposes. In the case at hand, we provide proof of principle, by utilizing this hybrid biocatalytic/pericyclic sequence to construct a platform for potential zinc-aminopeptidase A (Zn-APA) inhibitor development. Zn-APA converts angiotensin II into angiotensin III in the brain and is an emerging target in hypertension.11
Figure 1.
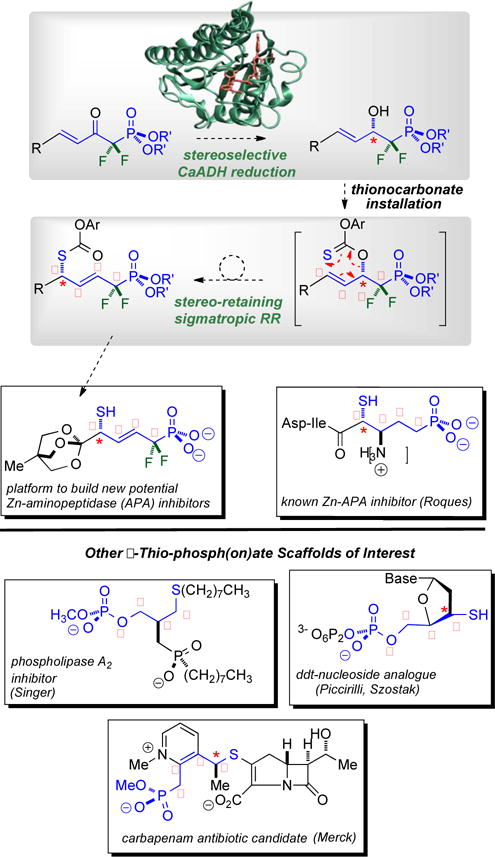
Stereocontrolled route into densely functionalized δ-thio-α,α-difluorophosphonates of interest to chemical biology. Top: CaADH is challenged with a new ketophosphonate substrate class to set the absolute stereochemistry, and a new [3,3]-sigmatropic RR is employed to parlay the enzymatically established β-C-O stereochemistry into the δ-C-S center; Bottom: Mapping the δ-thio-α,α-difluorophosphonate motif onto lipid, carbohydrate and β-lactam scaffolds of interest to chemical biology.
Phosphonates12 bearing varying degrees of α-fluorination are of considerable value to medicinal chemistry13 and chemical biology.14 To be sure, we15,16 and others17 have had a longstanding interest in synthetic routes into this compound class, dating back to the early proposition by Blackburn18 and McKenna,19 that α,α-difluoroalkyl phosphonates are “isopolar” analogues of the corresponding phosphate esters. Importantly, these “teflon phosphates” are inert to digestive phosphatase enzymes, and therefore serve as useful tools in chemical biology. For example, we were the first to synthesize the CF2-phosphonate analogues of phosphoserine and phosphothreonine,16 and these have proven to be very useful chemical mimics for the “constitutive phosphorylation” phenotype in studying kinase regulation in the cell.20
The CaADH reduction/sigmatropic rearrangement sequence here constitutes a new approach into densely functionalized, α,α-difluorinated phosphonates, bearing β,γ-unsaturation and δ-thio substitution. As is illustrated in Figure 1, this substitution pattern maps onto thio-substituted peptide,21 β-lactam,22 phospholipid23 scaffolds of interest in chemical biology, as well as thio-nucleic acid systems exploited by Piccirilli,24 and Szostak.25 In none of these systems are the corresponding difluorinated phosphonates yet known, as all bear phosphate esters or simple CH2-phosphonate functionalities δ to the sulfur.
As alluded to above, to demonstrate proof of principle, the focus here is on a platform related to the δ-thio-phosphonate embedded within a β-amino acid motif developed by Roques21 that serves as the key residue in tripeptide inhibitors of Zn-aminopeptidase A (APA; potential new target for antihypertensives; Figure 1). Note that the thiol here is critical for Zn2+ coordination and that compound 3e, reported herein (vide infra), represents a very useful platform upon which to build APA inhibitor candidates, as it bears three of the four requisite functional groups (phosphonate, thiol and carboxylic acid) all properly positioned, with α,α-difluorination. Thus successful synthetic entry into this platform sets the stage for future studies directed at library generation, particularly through functionalization of the intervening alkene.
To put the subsequent sigmatropic rearrangement step into perspective, the Claisen rearrangement has been in the domain of synthetic chemistry for over a century,26 and is also in Nature’s repertoire of enzymatic transformations. Most notably, the enzyme chorismate mutase catalyzes a Claisen rearrangement as a key step the biosynthesis of the aromatic amino acids. Recently, the groups of Tanner27 and Schmidt28 have discovered that Nature apparently exploits other [3,3]-sigmatropic rearrangements, an aromatic Cope and Claisen rearrangement, respectively, in formal biosynthetic prenylation reactions, as well. In the domain of chemical biology as well, great efforts have been expended to understand and mimic chorismate mutase, including two of the earliest examples of catalytic antibodies from Hilvert29 and Schultz,30 and contemporary studies of protein dynamics.31
The non-enzymatic reaction generally proceeds at elevated temperature and requires an organized transition state, prompting considerable effort at developing synthetic catalysts, including a supramolecular, tetrahedral M4L6 assembly32 and an elegant spiroligozyme system.33 The former promotes an aza-Claisen rearrangement by pre-organizing an otherwise entropically unfavorable reaction leading to a small entropic driver for the catalyzed process (∆S‡uncat = −8 e.u.; ∆S‡cat = + 2 e.u.). The latter achieves 58-fold acceleration of an aromatic Claisen rearrangement, presumably via H-bonding catalysis.
There are precedents for what are formally thermal and metal-catalyzed [3,3]-sigmatropic rearrangements of the thiono-Claisen variety (vide infra). However, these transformations generally require elevated temperatures and significant reaction times. For example, thionocarbonate rearrangements reported by Crich and coworkers required refluxing in benzene for 4 h.34 There are also early reports from Nakai35 and Zaim36 about simple thionocarbonate and thiono chloroformate-type rearrangements, respectively, though these communications are accompanied with very limited experimental detail. The former gives a measured kobs corresponding to a half-life of over 104 minutes at 80 °C for a thionocarbamate rearrangement, and claims that the corresponding xanthate is significantly faster (data not given; trend is opposite to the data reported herein). The latter reports on very reactive chloroformates, generated in situ from thiophosgene, that are unfunctionalized, difficult to handle, and of limited synthetic utility. Stereochemical control does not enter into these studies. A single kobs is reported, corresponding to a half-life of 60 min at RT.
RESULTS AND DISCUSSION
α,α-Difluoro-β-keto-phosphonate Synthesis and Asymmetric CaADH Reduction
As is illustrated in Scheme 1, the requisite keto phosphonates were prepared by condensation of LiCF2P(O)(OEt)2 with methyl cinnamate and related β-aryl-α,β-unsaturated esters. A two step procedure proved to be more efficient for systems bearing aliphatic substituents at the β-position. In such cases, the β-aryl-α,β-unsaturated aldehyde was condensed with LiCF2P(O)(OEt)2, followed by oxidation with the Dess-Martin periodinane.
Scheme 1. Synthesis of β-Ketophosphonate Substrates.
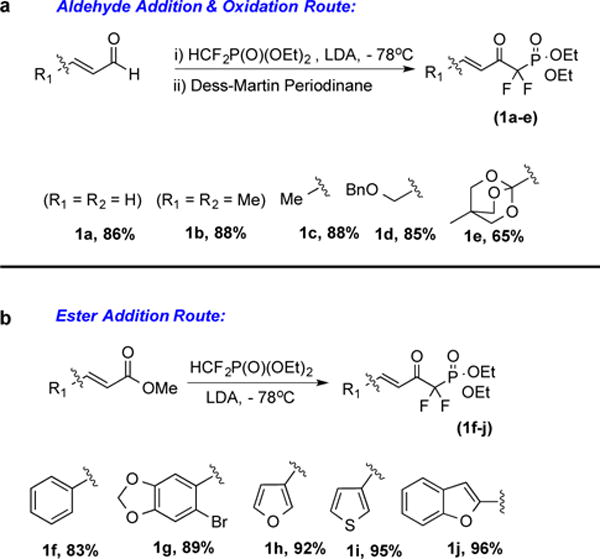
a –aldehyde addition/DMP oxidation route; yields reflect isolated yield following two-step procedure; b- direct ester addition route-all yields are for isolated pure compounds.
The Berkowitz laboratory has a longstanding interest in the creative use of enzymes in asymmetric synthesis,8,37 and recently we described an alcohol dehydrogenase from Clostridium acetobutylicum (CaADH) that is particularly effective for the stereoselective reduction of ω-ketoesters.8 In seeking to expand the substrate repertoire for CaADH, we set out to challenge this enzyme with a battery of β-ketophosphonates as a new and highly value-added substrate class. Indeed, we are delighted to report here that CaADH enantioselectively reduces an expansive array of α,α-difluorinated β-ketophosphonates bearing β,γ-unsaturation (Note: E:Z ratio > 20:1 in all cases) in good yield and with excellent enantioselectivity. The results are displayed in Scheme 2. The expensive nicotinamide cofactor was regenerated with glucose dehydrogenase, allowing D-glucose to serve as the stoichiometric carbonyl reductant.
Scheme 2. Substrate Scope-CaADH-Mediated Reductions.
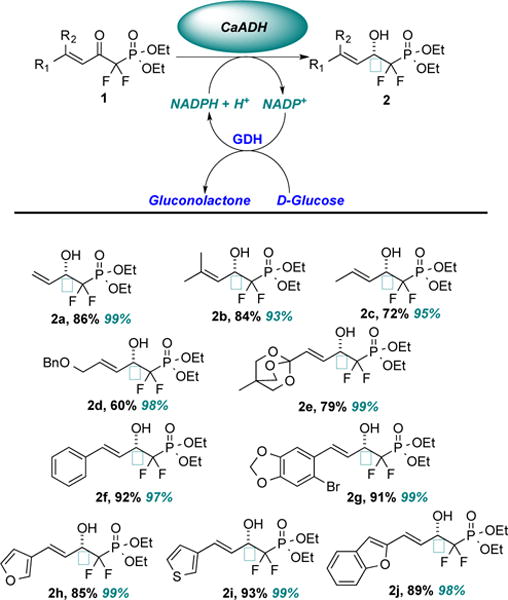
Yields (in black) represent isolated quantities of pure secondary alcohols; enantiomeric excesses (in teal) determined by chiral phase-HPLC; D-glucose employed as terminal reductant throughout, utilizing NADP+-dependent D-glucose DH from Thermoplasma acidophilum.
To our knowledge, these represent the first examples of a dehydrogenase-based entry into valuable enantioenriched α,α-difluorinated-β-hydroxyphosphonates. Stereoselective approaches to β-hydroxyphosphonates, particularly those bearing α-fluorination, have been quite limited so far. A handful of enzymatic approaches based upon kinetic resolution exist, but have been limited to simple systems with limited functionality.38 Chemical routes include the asymmetric reduction of β-keto phosphonates with pinene-derived chiral organoboron reagents, a borohydride/chiral auxiliary combination or via Ru(II)-BINAP-mediated hydrogenation.39 Of these, only the latter asymmetric hydrogenation approach of Kitamura and Noyori provides significant stereoinduction.39
CaADH Active Site Plasticity: High Enantioselectivity, Yet Opposite Facial Selectivity for α-Difluorinated β-Keto-Phosphonate Substrates vs. ω-Keto-Carboxylate Substrates
In order to determine the absolute stereochemistry of the CaADH-derived alcohols obtained here, the Mosher ester method was initially applied, utilizing alcohols 2d and 2f. According to the Kakisawa model, as is shown in Figure 2 for the case of 2f, the alcohol stereochemistry is predicted to be L.40 Pleasingly, X-ray crystallographic structural determination for alcohols 2f and 2h utilizing anomalous dispersion served to confirm the L-stereochemistry (Figure 2-bottom).
Figure 2.
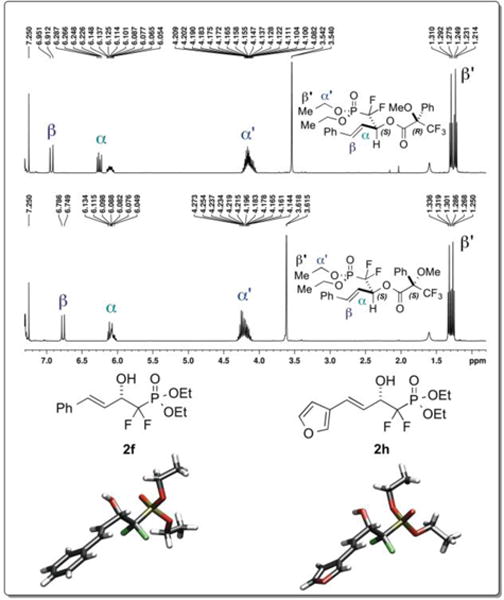
Determination of the absolute stereochemistry of the CaADH-derived products. Top: Mosher ester analysis for 2f. Bottom: X-ray crystallographic structures for 2f and 2h.
Molecular Modeling: Exploring the Origins of CaADH Active Site Plasticity
CaADH has proven to be an exceptionally versatile enzyme from the point of view of asymmetric synthesis, being able to process substrates that range from (i) a broad variety of ω-keto carboxylate esters8 to (ii) the significant spectrum of α,α-difluorinated β-keto phosphonate esters examined here (Table 2). That said, the facial selectivity observed for the CaADH reduction of β-keto-phosphonate esters here is opposite to that previously observed for the reduction of β-keto-carboxylate esters8 (Figures 2 and 3). To investigate this further, computational docking experiments were performed in the CaADH active site with a representative β-keto ester substrate, ethyl benzoylacetate, and with a typical α,α-difluoro-β-ketophosphonate 1f (Figure 4; Alignment: Clustal W;41 Homology Modeling: Modeller;42 Molecular Dynamics: GROMACS;43 Docking: Autodock 4;44 Graphics/Rendering: VMD45-see SI for details). The result suggests that both substrates occupy the same binding pocket of the enzyme, even though the facial preference for hydride transfer is reversed. Ethyl benzoylacetate docks with its keto group coordinated to S140, a typical binding motif for short-chain dehydrogenases.46 This orientation gives rise to the D-stereoisomer. However, the keto-phosphonate 1f docks with its keto group inverted, driven by a favorable H-bonding interaction with K207. This difference in keto orientation would give rise to the L-stereoisomer, as observed. This type of substrate inversion contrasts with typical cases of reversed enantioselectivity in enzyme active sites, which normally arise from alteration of substituent size that, in turn, transposes the occupants of medium and large active site pockets.47
Figure 3.
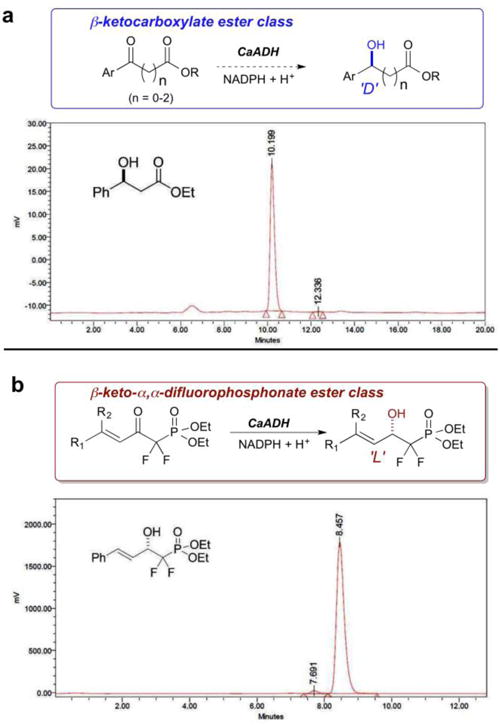
CaADH: Substrate promiscuity with stereochemical fidelity. Top: β-keto carboxylate ester reduced with D-stereochemistry as evidenced by HPLC on Chiralcel OD vs. racemic standard (90:10 – hexane/i-PrOH; 1 mL/min); Bottom: β-keto-α,α-difluorophosphonate ester reduced with L-stereochemistry; also Chiralcel OD-HPLC (95:5-hexane/i-PrOH).
Figure 4.
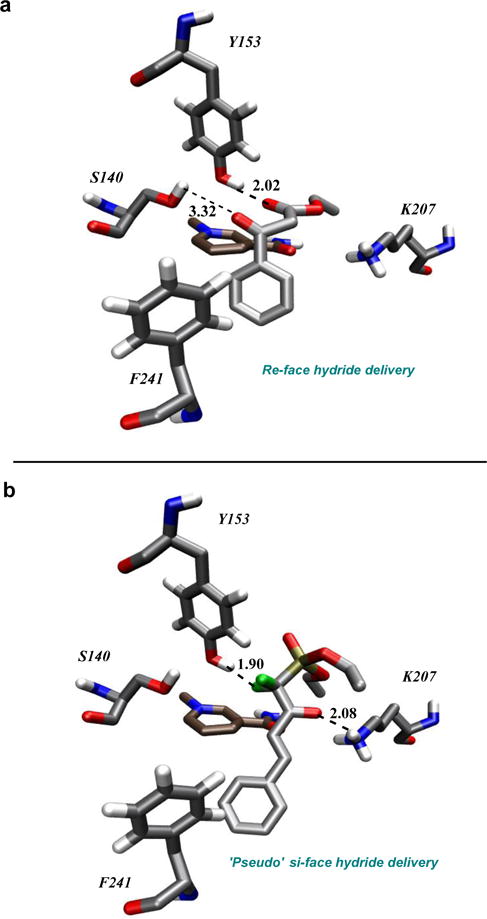
Active site plasticity: a view from molecular modeling. The observed switch in facial selectivity as a function of substrate class with preservation of high facial selectivity is evidence of unusual active site plasticity in CaADH. Note: The coordinates of the NADPH-cofactor (background) were imported from PDB-1XG5.
The model suggested by the experimental results and the supporting docking studies here will drive future experimentation. Note that the docked, chain-extended conformer shown in Figure 4 was selected by visual inspection as the most reasonable member of a set of nineteen conformers in the lowest energy cluster identified by molecular docking with Autodock 4.44 Factors that were considered in inspecting cluster members were NADPH-C4’-substrate carbonyl distance and positioning with respect to hydride transfer, as well as consistency with the known stereochemical outcome of hydride transfer. There is some variation among docked conformers in the rotamer distribution about the Cα-Cβ bond, with some deviation from the anti-arrangement of the phosphonate and Cγ when sighting down this bond. This is perhaps to be expected given the known tendency of α-fluorine substitution to elicit Thorpe-Ingold-like effects in some systems.48 To shed light on the precise molecular details of enzyme-substrate recognition here, going forward, there will be a serious effort to crystallize CaADH, both as the holoenzyme and in ketone-bound states. Ultimately, such structural biological studies will be invaluable in further addressing the question of how CaADH catalyzes the reduction of these two substrate classes. It should be noted that there are really three structural changes seen in moving from the D- to the L-selective substrates depicted in Figure 4. (i) While perhaps the most notable change is the replacement of the carboxylate ester with a phosphonate ester, (ii) α-fluorination is also introduced and (iii) a double bond is also inserted (phenyl to styrenyl). Future studies will be directed at mapping how the observed switch in enantioselectivity evolves across this set of structural changes. All told, this is a notable example of substrate promiscuity with stereochemical fidelity, but with an interesting twist; namely, antipodal facial preference based upon compound class (Figure 4ab).47 CaADH displays re-face hydride delivery (D-product) to β-keto carboxylate esters, but pseudo-si-face hydride delivery (L-product) to β-keto fluorinated phosphonate esters. That the CaADH apparently exhibits complementary modes of binding two different substrate classes, and yet processes each with high efficiency and stereochemical fidelity, speaks to the unusual active site plasticity of this enzyme.
Thiono-Claisen Rearrangements: Structure-Reactivity Studies
With the difluorinated-β-hydroxyphosphonates, in hand, we next set out to investigate the desired [3,3]-sigmatropic rearrangement. Toward this end, the cinnamyl substrate, 2f, was chosen, and four distinct thiono-esters were synthesized; namely (i) the xanthate; (ii) the phenyl carbonate; (iii) the pentafluorophenyl thionocarbonate and (iv) the N-phenyl thionocarbamate (Figure 4). To study the rearrangement of each, advantage was taken of the difluorinated phosphonate moiety permitting the kinetics of rearrangement to be conveniently followed by 19F NMR (Scheme 3). It quickly became clear that the xanthate, though able to undergo the targeted rearrangement at room temperature, did so at a very slow rate, with the observed half-time being approximately 2.5 days. On the other hand, replacing the SMe group with the more electronegative OPh, NHPh and OC6F5 groups led to a rather dramatic structure-reactivity relationship (SRR) with half-time decreasing significantly across this series.
Scheme 3. Structure-Reactivity Relationships Across a Family of Thiono-Claisen Rearrangements.
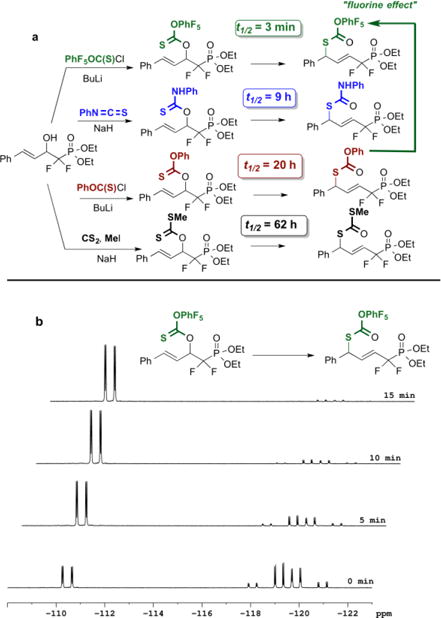
a Synthetic schemes and measured half-times for the four thiono-Claisen rearrangements studied here. b 19F NMR stack plot for the rearrangement of the pentafluorophenyl thionocarbonate ester @ RT. Note: The t = 0 point is arbitrarily labeled; this rearrangements proceeds too fast to capture a true starting time point.
Most notable is the pronounced effect of distal fluorination upon rate, whereby the simple expedient of moving from an allylic phenyl thionocarbonate to the corresponding pentafluorophenyl thionocarbonate accelerates the sigmatropic rearrangement by a factor of 400 [half-time goes from 20 h to 3 min (0.05 h)]. Note that we use the term “distal” in consideration that all substituent effects seen here are for substitution upon an aromatic nucleus that is not in direct resonance with the 6-membered transition state, as would be the case for a thionobenzoate system, for example. Rather, here an oxygen atom serves as a bridge between the arene ring and the atoms of the 6-centered system for the sigmatropic rearrangement. Moreover, given the electronically opposing inductive and resonance effects of fluorine substitution, the significant rate enhancement seen here by fluorination of this distal aromatic nucleus is really quite notable and could not have been anticipated prior to conducting these studies.
Indeed, a survey of the literature reveals that the rate observed for this allylic pentafluorophenyl thionocarbonate rearrangement (t1/2 ~ 3 min @ RT) is among the fastest seen for a neutral (hetero)Claisen rearrangement (see the Supporting Information for comprehensive tables). Earlier studies49 noted that properly situated alkoxy groups could accelerate the parent Claisen rearrangement by up to two orders of magnitude. The most facile rearrangement seen in those studies has a half-time of 45 min @ room temperature (alkoxy substitution; cyclopentenyl ether). Similarly, the Ireland Claisen rearrangement (silyloxy substituent) proceeds with t1/2 = 1–210 min @ 42 °C.50 Probably the most well studied Claisen variant with an additional heteroatom, the allylic imidate rearrangement, proceeds with t1/2 = 45 min at 140 °C.51
Of course, charge-accelerated [3,3]-sigmatropic rearrangements have drawn great attention in the synthetic and mechanistic chemistry communities, as these are widely regarded as among the most facile such rearrangements known, with enormous rate enhancements reported for oxy-anionic variants of the Cope and Claisen rearrangement, for example.10,52 Probably the key finding here is that in terms of absolute rate, the title allylic pentafluorophenyl thionocarbonate rearrangement (t1/2 = 3 min @ 23 °C) is on par with (i) the anionic oxy-Cope rearrangement reported by Evans and Golob (t1/2 = 8 min @ 0 °C; K+ counterion, 18-Cr-6),10 and (ii) the anionic oxy-Claisen rearrangement discovered by Koreeda a decade later (t1/2 = 6 min @ −23 °C; K+ counterion).52 In all three cases, a sigmatropic rearrangement that normally occurs at elevated temperature is converted to a room temperature reaction by judicious placement of an oxyanionic substituent in the earlier cases, and by judicious introduction of fluorination into an O-aryl substituent here.
Thiono-Claisen Rearrangement: Scope and Stereochemical Course
To examine scope and stereochemical fidelity, the rearrangement of pentafluorophenyl allylic thionocarbonates was next examined across the battery of structurally diverse, enantiomerically enriched substrates generated via CaADH-mediated β-keto-phosphonate reduction. As is depicted in Scheme 4, the desired [3,3]-sigmatropic rearrangement proceeds seamlessly across the series, with essentially complete conservation of ee in transposing the β-hydroxy stereocenters in the educts into a δ-thio stereocenter in the products. Enantiomeric excess was determined by chiral phase HPLC analysis, and absolute stereochemistry of the rearrangement product was unambiguously established in the case of product 3f via chemical correlation. Namely, ozonolytic processing of 3f was followed by comparison of the measured optical rotation obtained for the methyl (S)-thiomandelate oxidation/esterification product thereby obtained to the literature value (see SI for details). Diverse functionality is tolerated, both in the active site and in the subsequent rearrangement from aromatic heterocycles and fluorinated phosphonates (all cases) to benzyl ether and orthobicyclo[2.2.2]octyl (OBO) ester protecting groups. Indeed, especially noteworthy is the ability to carry a bulky OBO ester through both the enzymatic reduction and sigmatropic rearrangement steps. The OBO ester neither perturbs the exquisite facial selectivity observed in the CaADH active site nor blocks the rearrangement, although the sizeable orthoester is adjacent to the C-S bond-forming center. As noted at the outset, the resultant product, 3e, presents an ideal platform upon which to build a focused library of Zn-aminopeptidase inhibitor candidates.21 The essentially complete conservation of ee likely results from a pericyclic process that proceeds through a chair-like transition state, with both the α,α-difluorinated phosphonate ester and the R-group at the δ-position disposed pseudo-equatorially. However, given the precedent for boat-like transition states in certain Claisen-type rearrangements,53 most notably of the Ireland variety, at this stage one cannot rule out a boatlike transition state in which the δ-R group is disposed in the bowsprit position, for example.
Scheme 4. Substrate Scope for the New Allylic Pentafluorophenyl Thionocarbonate Rearrangement.
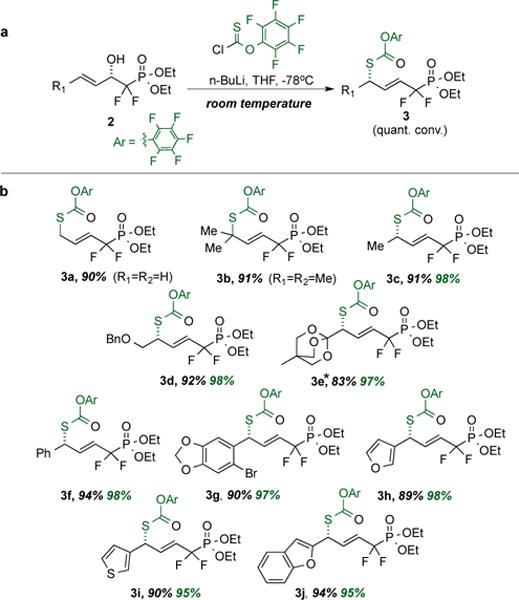
Yields (in black) represent isolated quantities of pure secondary alcohols; enantiomeric excesses (in green) determined by chiral phase-HPLC. *This RR requires mild heating (50°C)-all others proceed smoothly at RT.
Eyring Analysis and Solvent Dependence
While the low activation barrier, functional group tolerance, and exquisite stereocontrol observed constitute the practical advantages of the title transformation, perhaps even more important from a reaction design standpoint is the observation that one gains two to three orders of magnitude in rate enhancement via distal fluorine substitution here, as discussed. This remarkable SRR prompted us to investigate how fluorination leads to a more favorable free energy of activation (ΔΔG‡ ≈ 3.6 kcal/mol) for this type of sigmatropic rearrangement. Accordingly, Eyring analysis was performed on three different classes of thiono-ester to obtain the enthalpic and entropic contributions for each rearrangement (Figure 5). The results yield an observed enthalpy of activation (ΔH‡) for rearrangement ranging from 24–27 kcal/mol for all three thiono-Claisen rearrangements. This is consistent with values observed for other [3,3]-sigmatropic rearrangements.51,54,55
Figure 5.
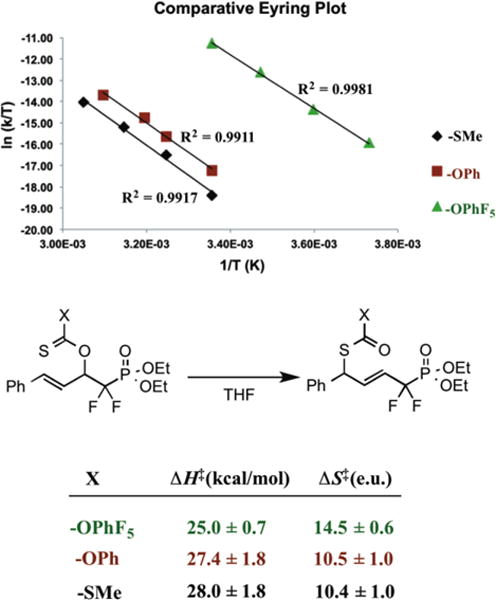
Eyring analysis yields compartitive enthalpies and entropies of activation for several thiono-Claisen RR types
However, the entropy of activation (ΔS‡) was found to be highly favorable for both thionocarbonates examined and for the xanthate. This is quite unusual. As noted, for sigmatropic rearrangements in general, ΔS‡ is almost always negative, presumably reflecting the need for a precisely organized transition state. Indeed, studies of the Claisen and related rearrangements reveal just this: (i), cyclopentenyl allyl ether (−21 e.u);49 (ii) (uncatalyzed) chorismate rearrangement (−13 e.u.);56 (iv) allylic imidate rearrangement (−18 e.u.); (v) allylic thionobenzoate rearrangement (−10 e.u.).55 Prior to these findings, we are aware of only a couple of cases in which a favorable entropy of activation has been observed for a neutral [3,3]-sigmatropic rearrangement in the absence of a catalyst, the most notable involving a system in which a the terminal alkene is masked as a cyclopropane,54 i.e. technically not a bonafide example of a [3,3]-sigmatropic rearrangement.
To further understand the transition state, the rearrangement of 2f was carried across a number of solvents. A rate enhancement is observed as the solvent dielectric constant increases as shown in Figure 6. This solvent dependence upon rate trends similarly, but appears to be somewhat more pronounced than that observed in the aforementioned thionobenzoate system.57 Interestingly then, by moving from THF to acetonitrile, one accelerates the title rearrangement still further, with t1/2 ~ 1.7 min @ RT in the latter solvent. This solvent dependence, when combined with the stereochemical course discussed above, suggests that the transition state for the title rearrangement is significantly more polarized than the ground state, i.e. partially dissociative as depicted in Figure 6, yet still chair-like, as depicted in Scheme 5.
Figure 6.
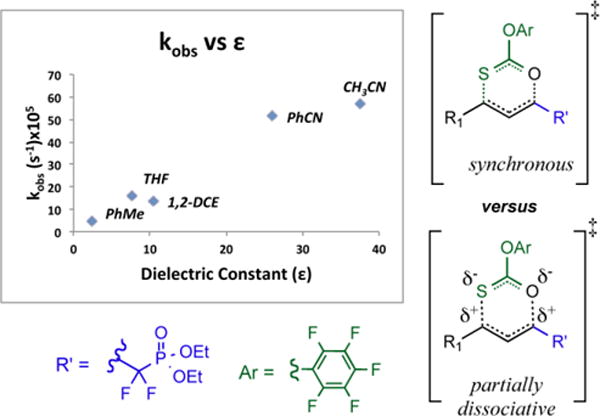
Rate dependence upon solvent-rearrangement kinetics of 2f. The increase in kobs as a function of solvent dielectric is suggestive of a polarized, partially dissociative transition state (lower right) as opposed to a symmetrical, synchronous one (upper right).
Scheme 5. Transition State Geometry for the Stereospecific [3,3]-Sigmatropic Rearrangement.
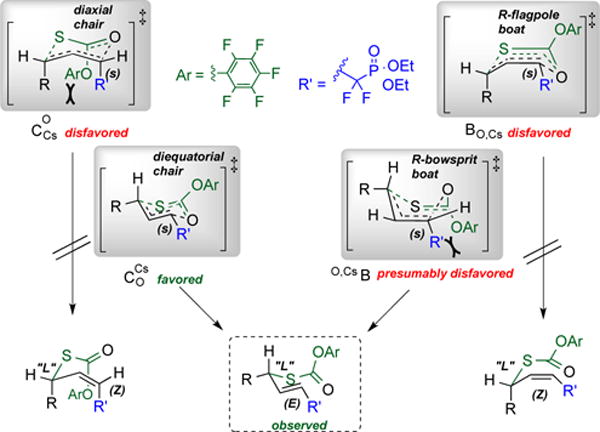
Consideration of possible transition state conformations for the facile, stereospecific sigmatropic RR of allylic pentafluorophenyl thionocarbonates described herein. Note: All conformations shown are consistent with the observed stereochemistry at the newly formed C-S bond, but only the O,CsB and CsCO transition states also give the correct alkene geometry.
CONCLUSIONS
As was mentioned at the outset, we have entered an age in which the state-of-the-art solution for complex synthesis, chemical biology or process chemistry may involve the careful melding of sophisticated enzymatic chemistry with sophisticated organic chemistry.58 This study describes the melding of a dehydrogenase enzyme from Clostridium acetobutylicum (CaADH), that exhibits remarkable active site plasticity with a new allylic pentafluorophenyl thionocarbonate rearrangement that is among the fastest sigmatropic rearrangements yet reported (t1/2 = 1.7 min at RT in MeCN). The combination of (i) LiCF2P(O)(OR)2/ester condensation; (ii) highly stereoselective CaADH-mediated α,α-difluoro-β-keto-phosphonate reduction and (iii) pentafluorophenyl thionocarbonate sigmatropic rearrangement provides a highly efficient, stereocontrolled entry into densely functionalized fluorinated phosphonate synthons of value to chemical biology. In particular, a platform for the construction of a focused library of zinc aminopeptidase A-directed inhibitor candidates is constructed here. Importantly, this example also illustrates the value in challenging enzymes with complex, advanced substrates. In this case, the enzymatic substrate features 12 carbon atoms, an additional 10 heteroatoms, and includes an unusual bridged oxabicyclo[2.2.2]octyl ring system; yet, it is processed efficiently by the CaADH enzyme (79% isolated yield, 99% ee).
As for the enzymology, in the Introduction, it was pointed out that from the point of view of asymmetric synthesis, one really seeks biocatalysts that show both high enantioselectivity and broad substrate generality,59,60 whether these enzymes be “engineered” through Darwinian evolution in the biosphere,4 through “directed evolution” in the laboratory,5,59,61 or through de novo design in silico.6,62 Only then can one associate that enzyme with a truly reliable “biocatalytic retrosynthetic1 transform arrow.”3 To put the observed “active site plasticity” into perspective, it is well to compare the CaADH enzyme to literature precedents for enzymes of value to synthesis. For example, Kroutil and coworkers describe a dehydrogenase from Ralstonia that also shows the ability to process distinct carbonyl compound classes and does so with a switch in stereofacial preference, suggestive of distinct binding modes, one for aryl alkyl ketones, another for α- or β-keto esters.63 Yet, with this plasticity, there is drop-off in enantioselectivity in going from enzymatic reduction of the former substrate class (99% ee, 6 examples) to the latter (96%, 64%, 37% ee-3 examples). Thus, the fact that high stereoselectivity is seen in both the binding mode for ω-keto carboxylate esters (90–99% ee), producing L-products,8 and also for β-keto-α,α-difluorophosphonate esters (93–99% ee), producing D-products, attests to a particularly fortuitous plasticity in this CaADH active site. Based upon molecular docking, a model is put forth here to account for these complementary binding modes (i.e. switch of key catalytic residues for carbonyl protonation from S140 to K207) and will drive structural biology studies to better understand this very promising biocatalyst.
For the new sigmatropic rearrangement, Eyring analysis indicates a favorable entropy of activation for the title transformation (14 e.u.) that contrasts with values reported for most [3,3]-sigmatropic rearrangements. The highly stereo-retentive nature of the rearrangement points to an organized, pericyclic transition state (Scheme 3), and the preference for polar solvents suggests that this transition state is also polarized and asynchronous (Figure 6). That said, the large positive entropy of activation observed cannot be accounted for solely by a polarized transition state that must remain organized to seamlessly transmit stereochemistry from educt to product. One could imagine that another source of entropy gain must be at play here, such as release of a solvent molecule or relaxation of solvent structure along the reaction coordinate as one approaches the transition state. Clearly, the findings reported here will drive future computational studies to better understand the geometry and charge distribution in the transition state for the title rearrangement.
Perhaps the most notable observation here is that this exceptionally facile sigmatropic rearrangement displays a distal fluorine effect that includes both a significant enthalpic component (Δ∆H‡ = −2.4 kcal/mol) and a significant entropic component (ΔΔS‡ = 4 e.u.; 1.2 kcal/mol @ RT). These enthalpic and entropic effects arise exclusively from fluorination of the distal heteroatom-bridged arene in the thionocarbonate substrate. While one can imagine that such fluorination might help to dissipate charge in the likely polarized transition state, one cannot easily intuit how this would parse into enthalpic and entropic components; computational interrogation will be needed. That said, these observations speak to the power of fluorine substitution to drive this particular thiono-Claisen rearrangement, and raise the possibility that such a strategy may be able to drive a broader suite of pericyclic reactions in the future.
EXPERIMENTAL SECTION
Ester Addition Route into β-Keto-α,α-Difluorophosphonates (Aryl Systems)
Aryl-linked α,α-difluoro-β-keto phosphonates were efficiently synthesized by the addition of lithiated α,α-difluorophosphonate to the α,β-unsaturated methyl esters. The methyl ester substrates were synthesized via Horner-Wadsworth-Emmons condensation chemistry. A typical procedure for the PCF2-C-bond forming reaction, using 1j as an example, is as follows. To diisopropylamine (297 μL, 2.12 mmol, 2 equiv.) in THF at −78 °C under N2 atmosphere was added n-BuLi (1.33 mL, 2.12 mmol, 2 equiv., 1.6 M in hexanes), dropwise via syringe. The resulting solution was allowed to warm to 0 °C for 25 min and then cooled to −78 °C, followed by dropwise addition of diethyl α,α-difluoromethylphosphonate (298 mg, 1.59 mmol, 1.5 equiv.) dissolved in THF. After 30 min at −78 °C, the benzofuran-substituted α,β-unsaturated methyl ester (215 mg, 1.06 mmol, 1 equiv.) in THF was added slowly to the reaction mixture via syringe down the wall of the flask. The reaction mixture was left for 1–2 h at the same temperature and then quenched with few drops of conc. acetic acid. The reaction mixture was gradually brought to room temperature, diluted with NH4Cl (aq) and extracted with EtOAc. The combined organics were dried over sodium sulfate, filtered and concentrated. Purification by column chromatography (typically eluent gradient 10–30% EtOAc in hexanes) yielded the product 1j (365 mg, 96%)
Aldehyde Addition/DMP Oxidation Route into β-Keto-α,α-Difluoro-phosphonates (Alkyl Systems)
The general procedure for the two step synthesis of the β-keto-α,α-difluoro-phosphonate substrates proceeded as follows (using 1c as an example): To diisopropylamine (2.80 mL, 20.0 mmol, 2 equiv.) in THF at −78 °C under nitrogen atmosphere was added n-BuLi (12.5 mL, 20.0 mmol, 2 equiv., 1.6 M in hexanes), dropwise via syringe. The resulting solution was allowed to warm to 0 °C for 25 min and then cooled to −78 °C, followed by dropwise addition of diethyl α,α-difluoromethylphosphonate (3.76 g, 20.0 mmol, 2 equiv.) dissolved in THF. After 30 min at −78 °C, crotonaldehyde (700 mg, 9.98 mmol, 1 equiv.) in THF was added slowly to the reaction mixture via syringe down the wall of the flask. The reaction mixture was left for 2 h at the same temperature and then quenched with aqueous NH4Cl solution and gradually brought to room temperature. The reaction mixture was extracted with EtOAc, The combined organics were dried over sodium sulfate, filtered and concentrated. If necessary, the α,α-difluoro-β-hydroxyphosphonate intermediate was purified by column chromatography. The β-hydroxyphosphonate obtained (2.59 g, 9.98 mmol, 1 equiv.) was dissolved in dry dichloromethane under nitrogen atmosphere and was cooled to 0 °C, followed by the addition of Dess-Martin periodinane (5.50 g, 12.97 mmol, 1.3 equiv.) in one portion. The reaction mixture was stirred at the same temperature for 15 min, brought to room temperature and stirred for an additional 2 h. Progress of the reaction was monitored by TLC. The reaction mixture was diluted with sat’d. sodium bicarbonate solution and extracted with dichloromethane. The organic layer was dried with sodium sulfate, filtered and concentrated. In this case, purification by column chromatography (typically eluent gradient 10–30% EtOAc in hexanes) yielded β-keto–α,α-difluorophosphonate 1c in (2.25 g, 88%)
Enzyme Expression, Purification and Standardization
CaADH was expressed and purified as previously described. CaADH was standardized by monitoring the reduction of benzaldehyde by NADPH at 340 nm. One unit is defined as the amount of enzyme needed to reduce one μmol of benzaldehyde to benzyl alcohol in one minute.
Enzymatic β-Keto-α,α-Difluoro-phosphonate Reductions
The general procedure for the enzymatic reductions (using 2f as an example) is as follows. A solution of NADP+ (17 mg, 0.02 mmol, 0.01 eq.), D-glucose (4.0 g, 22.3 mmol, 10 eq.), CaADH (10 units) and glucose dehydrogenase (2 units)(Thermoplasma acidophilum) in KPO4 buffer (100 mM, pH 7.0) was shaken at 37°C and 250 rpm. Keto-phosphonate 1f (730 mg, 2.29 mmol, in DMSO) was added so that the final DMSO concentration was 10% (v/v). Reaction progress was monitored by TLC until conversion was complete. Product was extracted with ethyl acetate and dried over sodium sulfate. Following vacuum filtration and concentration, purified by column chromatography (typically eluent gradiet 20–40% EtOAc in hexanes) yielded product 2f (675 mg, 92%). Alcohol products were further characterized by NMR and chiral HPLC (see Supporting Information for details).
Thiono-Claisen Sigmatropic Rearrangement
A typical thiono-Claisen RR is carried out as follows, using 2j as an example. To alcohol 2j (191 mg, 0.53 mmol, 1 equiv.) in THF at −78 °C was added n-BuLi (364 μL, 0.58 mmol, 1.1 equiv., 1.6 M in hexanes) under inert atmosphere (N2). After 15 min, pentafluorophenyl thionochloroformate (119 μL, 0.742 mmol. 1.4 equiv.) was added dropwise, via syringe, and the reaction was allowed to run for 30–60 min. The reaction was quenched with aqueous, saturated NH4Cl solution. The mixture was extracted with Et2O. The combined organics were dried (Na2SO4), filtered, concentrated and left at room temperature, if necessary, until the rearrangement was complete. The rearrangement is easily followed by TLC, as one generally observes conversion of the thionocarbonate intermediate to a more polar thiocarbonate product. The product thiocarbonate was purified by column chromatography (typically eluent gradient 10–30% EtOAc in hexanes) to yield thiocarbonate 3j (292 mg, 94%).
Supplementary Material
Acknowledgments
The authors thank Douglas R. Powell (U. of Oklahoma) for x-ray crystal structure determination and the NSF (1214019) for support. This research was facilitated by the IR/D (Individual Research and Development) program associated with DBB’s appointment at the NSF. The authors thank the NIH (SIG-1-510-RR-06307) and NSF (CHE-0091975, MRI-0079750) for NMR instrumentation support and the NIH (RR016544) for facilities renovation.
Footnotes
active site plasticity, substrate promiscuity, Clostridium acetobutylicum dehydrogenase, enantioselective carbonyl reduction, stereochemical fidelity, sigmatropic rearrangement, positive entropy of activation, thiono-Claisen rearrangement, stereoretentive rearrangement, fluorinated phosphonates
Supporting Information
Detailed experimental procedures; characterization of new compounds, including copies of NMR spectra; kinetic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.Turner NJ, O’Reilly E. Nat Chem Biol. 2013;9:285–288. doi: 10.1038/nchembio.1235. [DOI] [PubMed] [Google Scholar]
- 2.Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]
- 3.(a) Corey EJ, Long AK, Lotto GI, Rubenstein SD. Recl Trav Chim Pays-Bas. 1992;111:304–10. [Google Scholar]; (b) Corey EJ, Jorgensen WL. J Am Chem Soc. 1976;98:203–9. [Google Scholar]
- 4.Koeller KM, Wong C-H. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 5.Bornscheuer U, Huisman G, Kazlauskas R, Lutz S, Moore J, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 6.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon TP, Jacobsen EN. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 8.Applegate GA, Cheloha RW, Nelson DL, Berkowitz DB. Chem Commun. 2011;47:2420–2422. doi: 10.1039/c0cc04585c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Obexer R, Studer S, Giger L, Pinkas DM, Gruetter MG, Baker D, Hilvert D. ChemCatChem. 2014;6:1043–1050. [Google Scholar]; (b) Bacik J-P, Whitworth GE, Stubbs KA, Vocadlo DJ, Mark BL. Chem Biol. 2012;19:1471–1482. doi: 10.1016/j.chembiol.2012.09.016. [DOI] [PubMed] [Google Scholar]; (c) Muralidhara BK, Sun L, Negi S, Halpert JR. J Mol Biol. 2008;377:232–245. doi: 10.1016/j.jmb.2007.12.068. [DOI] [PubMed] [Google Scholar]; (d) Todd AE, Orengo CA, Thornton JM. Trends Biochem Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 10.Evans DA, Golob AM. J Am Chem Soc. 1975;97:4765–6. [Google Scholar]
- 11.(a) Balavoine F, Azizi M, Bergerot D, Mota N, Patouret R, Roques BP, Llorens-Cortes C. Clin Pharmacokinet. 2014;53:385–395. doi: 10.1007/s40262-013-0125-y. [DOI] [PubMed] [Google Scholar]; (b) Marc Y, Gao J, Balavoine F, Michaud A, Roques BP, Llorens-Cortes C. Hypertension. 2012;60:411–418. doi: 10.1161/HYPERTENSIONAHA.112.190942. [DOI] [PubMed] [Google Scholar]; (c) Padia SH, Kemp BA, Howell NL, Fournie-Zaluski M-C, Roques BP, Carey RM. Hypertension. 2008;51:460–465. doi: 10.1161/HYPERTENSIONAHA.107.103242. [DOI] [PubMed] [Google Scholar]; (d) Fournie-Zaluski M-C, Fassot C, Valentin B, Djordjijevic D, Reaux-Le Goazigo A, Corvol P, Roques BP, Llorens-Cortes C. Proc Natl Acad Sci U S A. 2004;101:7775–7780. doi: 10.1073/pnas.0402312101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fei X, Holmes T, Diddle J, Hintz L, Delaney D, Stock A, Renner D, McDevitt M, Berkowitz DB, Soukup JK. ACS Chem Biol. 2014;9:2875–2882. doi: 10.1021/cb500458f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Loranger MW, Forget SM, McCormick NE, Syvitski RT, Jakeman DL. J Org Chem. 2013;78:9822–9833. doi: 10.1021/jo401542s. [DOI] [PubMed] [Google Scholar]; (b) Diab SA, De Schutter C, Muzard M, Plantier-Royon R, Pfund E, Lequeux T. J Med Chem. 2012;55:2758–2768. doi: 10.1021/jm201694y. [DOI] [PubMed] [Google Scholar]
- 14.(a) Batra VK, Pedersen LC, Beard WA, Wilson SH, Kashemirov BA, Upton TG, Goodman MF, McKenna CE. J Am Chem Soc. 2010;132:7617–7625. doi: 10.1021/ja909370k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nieschalk J, O’Hagan D. J Chem Soc, Chem Commun. 1995:719–720. [Google Scholar]
- 15.Berkowitz DB, Bose M, Pfannenstiel TJ, Doukov T. J Org Chem. 2000;65:4498–4508. doi: 10.1021/jo000220v. [DOI] [PubMed] [Google Scholar]
- 16.Berkowitz DB, Eggen M, Shen Q, Shoemaker RK. J Org Chem. 1996;61:4666–4675. doi: 10.1021/jo9604752. [DOI] [PubMed] [Google Scholar]
- 17.(a) Romanenko VD, Kukhar VP. Chem Rev. 2006;106:3868–3935. doi: 10.1021/cr051000q. [DOI] [PubMed] [Google Scholar]; (b) Lopin C, Gautier A, Gouhier G, Piettre SR. J Am Chem Soc. 2002;124:14668–14675. doi: 10.1021/ja027850u. [DOI] [PubMed] [Google Scholar]; (c) Lequeux TP, Percy JM. J Chem Soc, Chem Commun. 1995:2111–2112. [Google Scholar]
- 18.Blackburn GM, England DA, Kolkmann F. J Chem Soc, Chem Commun. 1981:930–932. [Google Scholar]
- 19.McKenna CE, Shen P-D. J Org Chem. 1981;46:4573–4576. [Google Scholar]
- 20.(a) Panigrahi K, Eggen M, Maeng J-H, Shen Q, Berkowitz DB. Chem Biol. 2009;16:928–936. doi: 10.1016/j.chembiol.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Szewczuk LM, Tarrant MK, Sample V, Drury WJ, Zhang J, Cole PA. Biochemistry. 2008;47:10407–10419. doi: 10.1021/bi801189d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David C, Bischoff L, Meudal H, Mothe A, De Mota N, DaNascimento S, Llorens-Cortes C, Fournie-Zaluski M-C, Roques BP. J Med Chem. 1999;42:5197–5211. doi: 10.1021/jm9903040. [DOI] [PubMed] [Google Scholar]
- 22.Christensen BG, Schmitt SM, Johnston DBR. Application: EP-1985-108135. 1986:185. 167139. [Google Scholar]
- 23.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. J Biol Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 24.(a) Sengupta RN, Herschlag D, Piccirilli JA. ACS Chem Biol. 2012;7:294–299. doi: 10.1021/cb200202q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun S, Yoshida A, Piccirilli JA. RNA. 1997;3:1352–1363. [PMC free article] [PubMed] [Google Scholar]; (c) Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Nature. 1993;361:85–8. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 25.Meena, Sam M, Pierce K, Szostak JW, McLaughlin LW. Org Lett. 2007;9:1161–1163. doi: 10.1021/ol070147w. [DOI] [PubMed] [Google Scholar]
- 26.Claisen L. Ber Dtsch Chem Ges. 1913;45:3157–66. [Google Scholar]
- 27.Luk LYP, Qian Q, Tanner ME. J Am Chem Soc. 2011;133:12342–12345. doi: 10.1021/ja2034969. [DOI] [PubMed] [Google Scholar]
- 28.McIntosh JA, Donia MS, Nair SK, Schmidt EW. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilvert D, Nared KD. J Am Chem Soc. 1988;110:5593–4. [Google Scholar]
- 30.Jackson DY, Jacobs JW, Sugasawara R, Reich SH, Bartlett PA, Schultz PG. J Am Chem Soc. 1988;110:4841–2. [Google Scholar]
- 31.Pervushin K, Vamvaca K, Voegeli B, Hilvert D. Nat Struct Mol Biol. 2007;14:1202–1206. doi: 10.1038/nsmb1325. [DOI] [PubMed] [Google Scholar]
- 32.Fiedler D, Bergman RG, Raymond KN. Angew Chem Int Ed. 2004;43:6748–6751. doi: 10.1002/anie.200461776. [DOI] [PubMed] [Google Scholar]
- 33.Parker MFL, Osuna S, Bollot G, Vaddypally S, Zdilla MJ, Houk KN, Schafmeister CE. J Am Chem Soc. 2014;136:3817–3827. doi: 10.1021/ja409214c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crich D, Krishnamurthy V, Brebion F, Karatholuvhu M, Subramanian V, Hutton TK. J Am Chem Soc. 2007;129:10282–10294. doi: 10.1021/ja072969u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakai T, Ari-Izumi A. Tetrahedron Lett. 1976;17:2335–2338. [Google Scholar]
- 36.Zaim O. Tetrahedron Lett. 1999;40:8059–8062. [Google Scholar]
- 37.(a) Friest JA, Broussy S, Chung WJ, Berkowitz DB. Angew Chem Int Ed. 2011;50:8895–8899. doi: 10.1002/anie.201103365. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Friest JA, Maezato Y, Broussy S, Blum P, Berkowitz DB. J Am Chem Soc. 2010;132:5930–5931. doi: 10.1021/ja910778p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Broussy S, Cheloha RW, Berkowitz DB. Org Lett. 2009;11:305–308. doi: 10.1021/ol802464g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dey S, Powell DR, Hu C, Berkowitz DB. Angew Chem Int Ed. 2007;46:7010–7014. doi: 10.1002/anie.200701280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu H-w. Angew Chem Int Ed. 2002;41:4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (b) Attolini M, Iacazio G, Peiffer G, Maffei M. Tetrahedron Lett. 2002;43:8547–8549. [Google Scholar]
- 39.Kitamura M, Tokunaga M, Noyori R. J Am Chem Soc. 1995;117:2931–2. [Google Scholar]
- 40.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–6. [Google Scholar]
- 41.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 43.Hess B, Kutzner C, van der Spoel D, Lindahl EJ. Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 44.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.Kallberg Y, Oppermann U, Jornvall H, Persson B. Eur J Biochem. 2002;269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- 47.Mugford PF, Wagner UG, Jiang Y, Faber K, Kazlauskas RJ. Angew Chem Int Ed. 2008;47:8782–8793. doi: 10.1002/anie.200705159. [DOI] [PubMed] [Google Scholar]
- 48.Urbina-Blanco CA, Skibiński M, O’Hagan D, Nolan SP. Chem Commun. 2013;49:7201–7203. doi: 10.1039/c3cc44312d. [DOI] [PubMed] [Google Scholar]
- 49.Coates RM, Rogers BD, Hobbs SJ, Curran DP, Peck DR. J Am Chem Soc. 1987;109:1160–70. [Google Scholar]
- 50.Ireland RE, Mueller RH, Willard AK. J Am Chem Soc. 1976;98:2868–77. [Google Scholar]
- 51.Overman LE. J Am Chem Soc. 1976;98:2901–10. [Google Scholar]
- 52.Koreeda M, Luengo JI. J Am Chem Soc. 1985;107:5572–3. [Google Scholar]
- 53.(a) Gul S, Schoenebeck F, Aviyente V, Houk KN. J Org Chem. 2010;75:2115–2118. doi: 10.1021/jo100033d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen C-L, Namba K, Kishi Y. Org Lett. 2009;11:409–412. doi: 10.1021/ol8027225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harano K, Kiyonaga H, Hisano T. Chem Pharm Bull. 1987;35:1388–96. [Google Scholar]
- 55.McMichael KD. J Am Chem Soc. 1967;89:2943–7. [Google Scholar]
- 56.Jackson DY, Liang MN, Bartlett PA, Schultz PG. Angew Chem Int Ed. 1992;31:182–3. [Google Scholar]
- 57.Smith SG. J Am Chem Soc. 1961;83:4285–4287. [Google Scholar]
- 58.(a) Kohler V, Turner NJ. Chem Commun. 2015;51:450–464. doi: 10.1039/c4cc07277d. [DOI] [PubMed] [Google Scholar]; (b) Heidlindemann M, Rulli G, Berkessel A, Hummel W, Groeger H. ACS Catal. 2014;4:1099–1103. [Google Scholar]; (c) Reetz MT. J Am Chem Soc. 2013;135:12480–12496. doi: 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]; (d) Dunsmore CJ, Carr R, Fleming T, Turner NJ. J Am Chem Soc. 2006;128:2224–2225. doi: 10.1021/ja058536d. [DOI] [PubMed] [Google Scholar]
- 59.Bornscheuer UT, Kazlauskas RJ. Angew Chem Int Ed. 2004;43:6032–6040. doi: 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- 60.Carr R, Alexeeva M, Enright A, Eve TSC, Dawson MJ, Turner NJ. Angew Chem Int Ed. 2003;42:4807–4810. doi: 10.1002/anie.200352100. [DOI] [PubMed] [Google Scholar]
- 61.(a) Wu Q, Soni P, Reetz MT. J Am Chem Soc. 2013;135:1872–1881. doi: 10.1021/ja310455t. [DOI] [PubMed] [Google Scholar]; (b) Daugherty AB, Govindarajan S, Lutz S. J Am Chem Soc. 2013;135:14425–14432. doi: 10.1021/ja4074886. [DOI] [PubMed] [Google Scholar]; (c) Abrahamson MJ, Vazquez-Figueroa E, Woodall NB, Moore JC, Bommarius AS. Angew Chem Int Ed. 2012;51:3969–3972. doi: 10.1002/anie.201107813. [DOI] [PubMed] [Google Scholar]; (d) Reetz MT. Angew Chem, Int Ed. 2011;50:138–174. doi: 10.1002/anie.201000826. [DOI] [PubMed] [Google Scholar]; (e) Musa MM, Phillips RS. Catal Sci Technol. 2011;1:1311–1323. [Google Scholar]; (f) Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M. J Am Chem Soc. 2010;132:9144–9152. doi: 10.1021/ja1030479. [DOI] [PubMed] [Google Scholar]; (g) Toscano MD, Woycechowsky KJ, Hilvert D. Angew Chem Int Ed. 2007;46:3212–3236. doi: 10.1002/anie.200604205. [DOI] [PubMed] [Google Scholar]; (h) Kazlauskas RJ. Curr Opin Chem Biol. 2005;9:195–201. doi: 10.1016/j.cbpa.2005.02.008. [DOI] [PubMed] [Google Scholar]; (i) Qian Z, Lutz SJ. Am Chem Soc. 2005;127:13466–13467. doi: 10.1021/ja053932h. [DOI] [PubMed] [Google Scholar]; (j) May O, Nguyen PT, Arnold FH. Nat Biotechnol. 2000;18:317–320. doi: 10.1038/73773. [DOI] [PubMed] [Google Scholar]
- 62.(a) Kries H, Blomberg R, Hilvert D. Curr Opin Chem Biol. 2013;17:221–228. doi: 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]; (b) Blomberg R, Kries H, Pinkas DM, Mittl PRE, Gruetter MG, Privett HK, Mayo SL, Hilvert D. Nature. 2013;503:418–421. doi: 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- 63.Lavandera I, Kern A, Ferreira-Silva B, Glieder A, de Wildeman S, Kroutil W. J Org Chem. 2008;73:6003–6005. doi: 10.1021/jo800849d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.