

Abstract
The challenge for Glycogen Synthase Kinase-3 (GSK-3) inhibitor design lies in achieving high selectivity for one isoform over the other. The therapy of certain diseases, such as acute myeloid leukemia (AML) may require α-isoform specific targeting. The scorpion shaped GSK-3 inhibitors developed by our group achieved the highest GSK-3α selectivity reported so far, but suffered from insufficient aqueous solubility. This work presents the solubility-driven optimization of our isoform-selective inhibitors using a scorpion shaped lead. Among 15 novel compounds, compound 27 showed high activity against GSK-3α/β with the highest GSK-3α selectivity reported to date. Compound 27 was profiled for bioavailability and toxicity in a zebrafish embryo phenotype assay. Selective GSK-3α targeting in AML cell lines was achieved with compound 27, resulting in a strong differentiation phenotype and colony formation impairment, confirming the potential of GSK-3α inhibition in AML therapy.
Keywords: Acute myeloid leukemia, drug discovery, glycogen synthase kinase-3, inhibitors, zebrafish
Introduction
Glycogen Synthase Kinase-3 (GSK-3) is a constitutively active, ubiquitous serine/threonine kinase, which takes part in a number of physiological processes ranging from glycogen metabolism to apoptosis.1–3 GSK-3 is a key mediator of various signalling pathways, such as the Wnt and the Insulin/AKT signalling pathways.1 Therefore, dysregulation of GSK-3 has been linked to various human diseases, such as cancer, diabetes and neurodegenerative diseases.1–2, 4–7 Two related isoforms of GSK-3 exist in mammals, GSK-3α and β, which share a sequence identity within their catalytic domains of 98%.2–3, 8 Beyond the catalytic domains they show significant differences.2, 8 Although these isoforms are structurally related, they are not functionally equivalent, and one cannot compensate for loss of the other.6 The debate on the respective contributions of the isoforms GSK-3α and GSK-3β on the pathogenesis of different diseases is ongoing.9 Various studies indicate that the therapies of certain diseases benefit from specific targeting of GSK-3α and GSK-3β.6, 10–14 GSK-3α was recently identified as a differentiation target in acute myeloid leukemia (AML).10 AML is a haematopoietic malignancy defined by uncontrolled proliferation and disrupted myeloid differentiation.15–16 AML is the second most common form of leukaemia in adults.16 The current treatment of AML with conventional chemotherapy is very aggressive yet ineffective for the majority of patients with the disease.15 Thus, alternative targeted treatment approaches for AML are highly desirable.10, 15 GSK-3α recently emerged as a potential target in this disease.10
Many molecular approaches for the inhibition of GSK-3 have been reported. However, most of these inhibitors lack selectivity for GSK-3 over other related kinases. This selectivity problem can challenge the clinical utility of these inhibitors leading to dose-limiting toxicities. Moreover, due to the structural similarity between the two ATP binding pockets of GSK-3α and GSK-3β most published inhibitors do not differ effectively between the GSK-3 isoforms. Therefore, our research focuses on the development of isoform-selective, ATP-competitive inhibitors using a scorpion shaped lead (Figure 1A).9 Compound 1 (Figure 1) shows high kinase inhibitory activity and an impressive selectivity for GSK-3α.9 However, compound 1 could not be tested in vivo due to insufficient solubility. Therefore, it was chosen as lead structure for the design of improved GSK-3 inhibitors. Here we report the synthesis and optimization of scorpion shaped GSK-3 inhibitors with improved solubility. The most promising compounds were evaluated in a wild-type zebrafish embryo assay and in AML cell lines.
Figure 1.

A) Compound 1 was used as lead structure for systematic modification. B) Docking study of compound 1 using MOE 2014.09 and the known GSK-3β crystal structure (PDB: 3F88). C) From the docking study (B) Resulted pharmacophore hypothesis is used for screening. The pharmacophore is represented by an H-bond acceptor (blue), aromatic rings (orange) and hydrophobic centroids (yellow). D) Selection of compounds that match the docking model of 1.17
Optimization Strategy
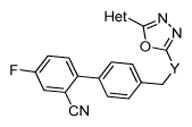
Optimization of compound 1 was performed to improve its pharmacokinetics and cell penetration. In addition, our intention was to increase the selectivity towards GSK-3α. Unfortunately, the structure of GSK-3α has not been solved. Therefore, target-oriented synthesis of isoform-specific inhibitors can inform the mechanism behind α-selectivity of our scorpion shaped GSK-3 inhibitors. All structures share the oxadiazole moiety, as it provides a high inhibitory activity and isoform-selectivity.9 Different substitution patterns at the biphenylic scaffold were explored in order to enhance GSK-3α selectivity and to concurrently improve solubility.
We next designed 140 lead-like compounds by the systematic modification of scaffold elements: the heteroaromatic head group (Figure 1A: blue), the spacer between the oxadiazole and the biphenylic scaffold (Figure 1A: yellow), the first aromatic ring of the biphenylic scaffold (Figure 1A: green) and the terminal aromatic ring (Figure 1A: red). To find promising compounds all lead-like structures were evaluated by molecular docking. Initially, the essential ligand-receptor interactions of compound 1 with GSK-3β had to be determined to make it possible to compare the candidates with the lead structure and the respective docking hypothesis. Docking of compound 1 into the GSK-3β active site (PDB: 3F88) was done by the software MOE 2014.09.17 Afterwards the docking poses were rescored by the DSX rescoring function18 with the aim to identify the best binding mode of compound 1 (Figure 1C). The resulting conformations agree with already published findings from our group.9 The head group (dihydrobenzodioxine and oxadiazole) of compound 1 is oriented to the hinge region where it forms hydrophobic interactions with Tyr134. The oxadiazole ring is located between Val70 and Cys199. The biphenylic tail group establishes π-π-stacking interaction with Phe67 and H-π-interactions with Gln185. In addition, the cyano moiety builds an H-bond to Thr138. From this model a phamacophore was generated. It consists of three aromatic features fulfilled by the oxadiazole ring and two phenyl rings (Figure 1B: orange), one hydrophobic centroid covered by one phenyl ring (Figure 1B: yellow) and one H-bond acceptor pharmacophore feature fulfilled by one nitrogen atom of the oxadiazole ring (Figure 1B: blue). With the intention to identify the best candidates of 140 lead-like compounds, docking was performed with phamacophore placement. Then the resulted docking poses were again rescored by the DSX rescoring function.18 Thereby the best rated docking poses reproduce the overall orientation of the lead structure compound 1 (Figure 1D).
Based on the results of the docking simulations the best candidates were evaluated further. The next filtering was set by limitation of the ClogP value to be less than or equal to 4.5. This ensured that the number of hits were limited to more polar compounds than 1. This filtering step reduced the number of hits to 94. The analysis of possible ways for derivatization and chemical accessibility led to 15 promising candidates, which were synthesized in the next steps as outlined below.
Chemistry
The reference Compound 1 and some of the screening hits were synthesized according to the published procedure.9 The building blocks of the head group and the tail group were prepared in a converging synthesis route.9
The synthesis of the head group building blocks 4a–b was accomplished starting from the carboxylic acids 2a–b. The carboxylic acids 2a–b were esterified to the methyl esters 3a–b. The oxadiazole derivates 4a–b resulted from 3a–b after reaction with hydrazine monohydrate followed by cyclization of the resulting hydrazides with carbondisulfide under basic conditions (Scheme 1).9
Scheme 1.

Reagents and conditions: (a) MeOH, SOCl2, 0 – 50 °C, 96 – 99%; (b) NH2NH2·H2O, EtOH, reflux, then CS2, Et3N, EtOH, reflux, 11 – 45%.
The synthesis of the biphenylic scaffold starts either with a Suzuki coupling of the commercially available boronic acid 6 with various bromides 5a–d,g or (hydroxyl)boronic acid 11 with bromines 10e,f,h (Scheme 2). For the cases of intermediates 7a–d,g a radical bromination with NBS and AIBN was performed to obtain bromides 8a–d,g. The resulting benzylic alcohols 9e,f,h, however, were treated with phosphorous tribromide, to convert them into the benzylic bromides 8e,f,h (Scheme 2). Coupling of the biphenylic bromides 8a–h to the mercaptanes 4a–b gave the final compounds 12a, 13b–h.
Scheme 2.

Reagents and conditions: (a) Pd(PPh3)4, 2N aq. Na2CO3, toluene/EtOH 1:1, 80 °C, 27 – 99%; (b) NBS, AIBN, CCl4, reflux, 52 – 64%; (c) PBr3, toluene, reflux, (d) 4a–b, 4N aq. NaOH, DMF, rt, 12 – 66%.
Suzuki coupling of the fluoroboronic acids 15a–c with bromines 14a–c followed by radical bromination gave the benzylic bromides 17a–c, which were coupled under basic conditions to the mercaptane 4b to yield the final compounds 18a–c (Scheme 3).
Scheme 3.

Reagents and conditions: (a) Pd(PPh3)4, 2N aq. Na2CO3, toluene/EtOH 1:1, 80 °C, 87 – 99%; (b) NBS, AIBN, CCl4, reflux, 22 – 45%; (c) 4b, 4N aq. NaOH, DMF, rt, 31 – 37%.
The ortho-substituted product 21 was obtained by a nucleophilic aromatic substitution of 2,5-difluorobenzonitrile (20) and methylpiperidine-4-carboxylate (19) with cesium carbonate (Scheme 4). The reduction of the methoxy ester 21 by lithium borohydride provided the primary alcohol 22. Appel reaction of 22 gave the bromide 23, which was coupled under basic conditions to the mercaptane 4b to yield the final compound 24.
Scheme 4.

Reagents and conditions: (a) Cs2CO3, DMSO, 100 °C, 42%; (b) LiBH4, THF, reflux, 98%; (c) CBr4, PPh3, acetonitrile, rt, 35%; (d) 4b, K2CO3, DMF, 80 °C, 71%.
The sulfur atom of the previously synthesized Compound 1 was oxidized by mCPBA to the final compounds (rac)-25 and 26 (Scheme 5). The final compound 27 was obtained by the partial hydrolysis of the cyano group of 1 under acidic conditions (Scheme 5).
Scheme 5.
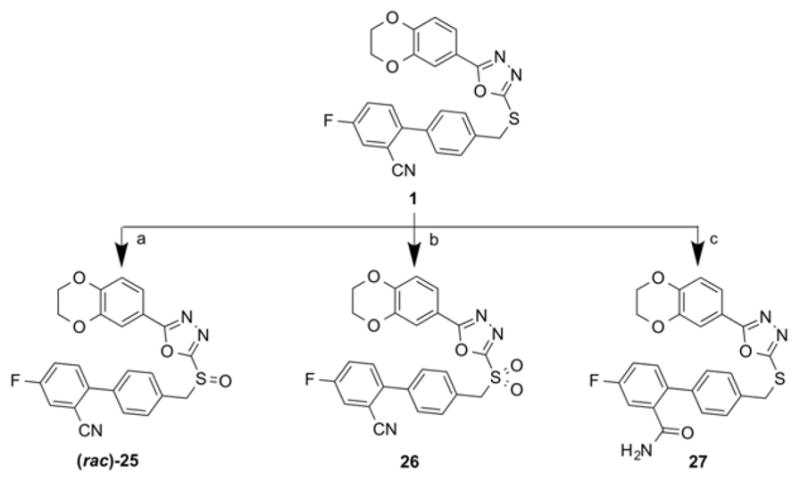
Reagents and conditions: (a) mCPBA, DCM, 0° C – RT, 79%; (b) mCPBA, DCM, 0 – 30 °C, 68%; (c) H2SO4, TFA, reflux, 78%.
In vitro Phamacology and Structure-Activity Relationship
The obtained compounds were tested for their inhibitory activity against GSK-3α and GSK-3β in a commercial in vitro assay.19–20 The assay was performed at an inhibitor concentration of 1 μM (100 nM for 26), using isolated GSK-3α and GSK-3β from porcine brain and staurosporine as control.19–20 The results are expressed as percentage of control specific activity.19–20 The structure-activity relationship was analyzed by systematic modification of scaffold moieties.
Initially, the hinge binding dihydrobenzodioxine moiety of the lead structure 1 was replaced by a benzimidazole group as potential hinge binder. This lead to a massive decreased inhibitory activity against both isoforms as compared to the lead structure 1 (Table 1). Consequently, the benzimidazole scaffold was abandoned. For all further modification steps the dihydrobenzodioxine scaffold was unmodified.
Table 1.
Inhibitory activity against GSK-3α and GSK-3β.
| ||||||
|---|---|---|---|---|---|---|
| Compound | Het | Y | ClogPa | tPSAa | GSK-3α Inhibition [%]b | GSK-3β Inhibition [%]b |
|
| ||||||
| 1 μM of compound | ||||||
| 1 |
|
S | 4.60 | 76.2 | 80 | 45 |
| 12a |
|
S | 3.98 | 82.1 | 36 | 0 |
| (rac)-25 |
|
SO | 3.40 | 93.3 | 48 | 42 |
| 26 |
|
SO2 | 3.11 | 110.3 | 0 (100 nM) | 0 (100 nM) |
Calculated by ChemBioDraw Ultra (version 13.0.2).21
The results are expressed as a percent of control specific activity = 100 − ((measured specific activity/control specific activity)*100), at a concentration of 1 μM (100 nM for 26) of the test compound. The measurements were performed in duplicates.
In the next step, the impact of the spacer between the oxadiazole and the biphenylic scaffold was investigated. The oxidation of the thioether 1 to the racemic sulfoxide (rac)-25 resulted in a decrease of inhibitory activity, especially against GSK-3α (Table 1). Due to the heavy loss of isoform selectivity the racemic product was not separated into the pure enantiomers, as neither of the enantiomeric sulfoxides is as active as the sulfide. The further oxidation of 1 to the sulfone 26 led to an even stronger decrease of inhibitory activity (Table 1). The introduction of oxygen atoms on the linking sulfur apparently results in repulsive interactions, presumably as the biphenylic scaffold is displaced and the important π-stacking interaction with Phe67 or with Gln185 cannot be formed.
The influence of the biphenylic scaffold was investigated next keeping the hinge binding moiety and the spacer constant. The introduction of a non-aromatic piperidine in 24 resulted in an immense drop of inhibitory potency (Table 2), because the π-stacking interaction with Phe67 or with Gln185 can no longer be formed.
Table 2.
Inhibitory activity against GSK-3α and GSK-3β.
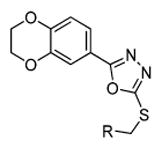
| |||||
|---|---|---|---|---|---|
| Compound | R | ClogPa | tPSAa | GSK-3α Inhibition [%]b | GSK-3β Inhibition [%]b |
|
| |||||
| 1 μM of compound | |||||
| 1 |
|
4.60 | 76.2 | 80 | 45 |
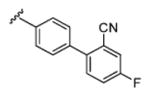
| 13b |
|
5.17 | 52.4 | 13 | 6 |
| 13c |
|
3.97 | 64.8 | 86 | 84 |
| 13d |
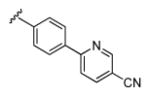
|
3.37 | 88.6 | 86 | 44 |
| 13e |

|
4.24 | 64.8 | 86 | 88 |
| 13f |
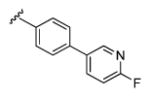
|
3.76 | 64.8 | 95 | 78 |
| 13g |
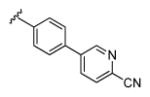
|
3.36 | 88.6 | 58 | 21 |
| 13h |
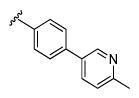
|
4.03 | 64.8 | 99 | 89 |
| 18a |
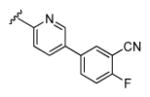
|
3.15 | 88.6 | 21 | 0 |
| 18b |
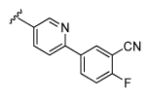
|
3.36 | 88.6 | 49 | 28 |
| 18c |
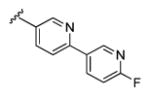
|
2.55 | 77.1 | 93 | 75 |
| 24 |

|
3.88 | 79.4 | 20 | 10 |
| 27 |

|
3.39 | 95.5 | 100 | 97 |
Calculated by ChemBioDraw Ultra (version 13.0.2).21
The results are expressed as a percent of control specific activity = 100 − ((measured specific activity/control specific activity)*100), at a concentration of 1 μM of the test compound. The measurements were performed in duplicates.
The impact of the cyano substituent on the activity and α-selectivity of 1 was addressed next (Table 2). The complete removal of the cyano substituent in 13b (Table 2) resulted in a substantial loss of inhibitory activity.
However, the exchange of the cyano substituent for the amide substituent in Compound 27 resulted in the most potent compound (Table 2). Compound 27 inhibits GSK-3α and GSK-3β at a concentration of 1 μM at 100 percent. The substitution by 3-fluoropyridine in 13c and 2-fluoropyridine in 13f led to an improved inhibitory potency against GSK-3β (Table 2). The inhibitory potency against GSK-3α increased slightly. However, the increase of the inhibitory potency against the α-isoform is stronger for 13f than for 13c. Consequently, the pyridine in 13f was changed back to a phenyl with a cyano substituent in ortho position to the fluorine. Due to the chemical accessibility the first phenyl ring of the biphenylic scaffold had to be changed simultaneously to a 2-pyridine in 18b and a 3-pyridine in 18a. In both cases the modifications led to an immense drop of inhibitory potency (Table 2). Moreover, a total loss of activity against GSK-3β was observed for 18a. We assumed that the ortho-cyano substituent in 18b and 18a was responsible for the sharp drop of activity. This hypothesis was confirmed by the introduction of a second 2-pyridine (18c) into 13f, where 18c shows the same activity like 13f (Table 2). In the last step the impact of the fluoro substituent within the pyridines of 13f and 13c on the activity and α-selectivity was explored. The introduction of electron-donating methyl substituents in 13e and 13h leads to a slight improvement of inhibitory potency against GSK-3β (Table 2). However, the inhibitory potency against GSK-3α was not affected. Consequently, the differentiation between the isoforms was reduced. The introduction of an electron-withdrawing cyano-substituent in 13d and 13g led to a decrease of inhibitory potency against GSK-3α (Table 2). However, both compounds showed the best discrimination between the isoforms with α-selectivity towards GSK-3α. In the case of the inhibition of the β-isoform both compounds behave differently. 13d showed the same inhibitory activity against GSK-3β as 1. However, in the case of 13g a striking decrease of inhibitory activity was observed against the GSK-3β. The docking of 13d and 13g into the GSK-3β active site suggested that both cyano substituents contribute to the inhibitor activity by establishing a H-bond to the amino acid Arg144. The different activities between both compounds can be explained by the rotation of the biphenylic residue. The biphenylic residue of 13g is rotated by about 60 degree compared to 13d. Consequently, 13g can not form a π-stacking interaction with Gln185. Due to the π-stacking interaction with Gln185, 13d interacts better with the active site. Accordingly, 13d shows a higher activity against GSK-3β than 13g.
The IC50 values of the lead compound 1 and the most potent compound 27 were determined for detailed analysis (Table 3). With our novel inhibitor 27 the activity and selectivity against GSK-3α/β could be increased further. With an IC50 value of 42 nM for GSK-3α and 140 nM for GSK-3β, compound 27 inhibits GSK-3α/β stronger than the lead compound 1.
Table 3.
Inhibitory activity against GSK-3α and GSK-3β IC50 [μM].
The evaluation of the activity in GSK-3α (h)19 and GSK-3β (h)20 was performed by the company Cerep using a published procedure by Greengard et al. (Supporting Info).22 The IC50 values were determined by measurement of eight concentrations (0.03 nM, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM, 1 μM).
With the intention to elucidate the difference between both isoforms of GSK-3 a sequence alignment of GSK-3α (UniProtKB: P49840) on the 3D-structure of GSK-3β (PDB: 3F88) was performed. The sequence identity between GSK-3β and GSK-3α was calculated to be 86% by MOE 2014.09.17 However, all differences between the two isoforms are located away from the ATP-binding site. This results in a similarity of 98% within the active site. The SAR study of 13d is best suited to illustrate the reasons for isoform-selectivity (Figure 2A). It shows that the cyano substituent contributes to the inhibitor selectivity by establishing an H-bond to the amino acid Arg144 (GSK-3β) and within the α-isoform to Arg207 (GSK-3α). Arg144 and Arg207 are located in the Arg-rich loop. However, the neighboring amino acids within the Arg-rich loop differ between the isoforms. Within GSK-3β there is Arg148 (green) while within GSK-3α there is Lys211 (pink). Within GSK-3β Arg144 interacts with the next located amino acid Arg148 over a coordinated water molecule (Figure 2A). However, within GSK-3α the adjacent amino acids, Arg207 and Lys211, do not interact with each other (Figure 2A). We suggest that the water-mediated H-bond between Arg144 and Arg148 is responsible for the isoform selectivity of 13d. The amino acids within the Arg-rich loop are likely solvent exposed and quite flexible. But the water-mediated H-bond between Arg144 and Arg148 may restrict the motion of the side chains, which would cause a conformational entropic penalty.23 This could cancel out any gain in binding energy from the interaction between ligand and protein.23 Consequently, 13d inhibits GSK-3β less potent than GSK-3α (Table 2). We therefore propose that the isoform selectivity of our scorpion shaped GSK-3 inhibitors is caused by the two different amino acids, GSK-3β: Arg148 (green), GSK-3α: Lys211 (pink), within the Arg-rich loop. Consequently, the para-substituent of the biphenylic moiety of our scorpion shaped GSK-3 inhibitors plays a key role in the selectivity of the inhibitor (Figure 2B). Obviously, this is a speculative hypothesis, which has to be confirmed by more advanced theoretical and experimental work as well as X-ray studies of GSK-3α.
Figure 2.

A) Docking study of compound 13d using MOE 2014.09 and the aligned structure of GSK-3α (UniProtKB: P49840) and GSK-3β (PDB: 3F88).17 Differing amino acids are highlighted, GSK-3β (green) and (GSK-3α) pink. B) Schematic overview of the selectivity hypothesis of our scorpion shaped GSK-3 inhibitors.
Aqueous solubility of selected compounds
The aqueous solubility of chemical agents affects the uptake of the substances into cells and their oral bioavailability. Consequently, a good water-solubility is essential for sufficient bioavailability, which must be considered for the performance of the compound within the organism. Solubility-driven optimization of 1 was the main intention of this work. The solubility values of the most active compounds 1, 13c–h, 18c and 27 are shown in Table 4. Some of the compounds (1, 13e, 13h, 18c) showed a poor water solubility of below 1 μg/mL, which makes them unsuitable for further development. 13d and 27 are not only the most potent compounds, but also the most soluble. The highest water solubility of 78 μg/mL (181 μM) was observed for 13d. The most potent compound 27 also has good aqueous solubility of 34 μg/mL (73 μM).
Table 4.
Aqueous solubility of selected compounds.
| Compound | Solubility [μg/mL]a | Solubility [μM]a |
|---|---|---|
| 1 | < 1 | 1 |
| 13c | dec | dec |
| 13d | 78 | 181 |
| 13e | < 1 | 1 |
| 13f | 7 | 17 |
| 13g | 21 | 50 |
| 13h | < 1 | < 1 |
| 18c | < 1 | 1 |
| 27 | 34 | 73 |
Related to the in vivo assay conditions all diluted samples contain water and 2% DMSO. The determination was done by the “Shake-Flask” method24 and HPLC measurements.
In vivo evaluation
The most soluble and most active candidates 13d and 27 were further profiled for in vivo activity in wild-type zebrafish (Danio rerio) embryos. The zebrafish embryo is a useful model to assess dose-dependent bioavailability and toxicity of compounds. The embryos were collected and maintained in E3 medium at ~26°C. The compounds were added 24 hours post fertilization (hpf) and the phenotypes compared at 96 and 120 hpf. Both compounds showed no lethality in our concentration range (< 60μM). Compound 13d showed no effects on the wild-type zebrafish embryos. At lower concentrations of compound 27 from 1 to 15 μM no abnormalities were observed (Figure 3). At concentrations of 27 higher than 30 μM all zebrafish embryos showed a stunted and crooked tailed phenotype (Figure 3). These observations support the absorption and cell penetration of compound 27. Furthermore, this zebrafish embryo assay demonstrates that compound 27 disturbs the zebrafish development. For embryonic development of the zebrafish the Wnt/β-catenin-signalling pathway plays an important role.25 The Wnt-signalling pathway involves the complex interplay of multiple proteins, including GSK-3β in this signalling network.26 The observed phenotype is consistent with activation of β-catenin, which is associated with inhibition of GSK-3β.25, 27 Compound 27 exhibits IC50 values of 42 nM for GSK-3α and 14 nM for GSK-3β inhibition. The concentrations in this experiment are too high to achieve isoform-selectivity. Consequently, compound 27 is no longer selective to GSK-3α at concentrations higher than 30 μM. It is likely that with this gross experiment isoform-selectivity cannot be detected. Therefore, we tested compound 27 in a cell assay at lower concentrations to investigate the effects on GSK-3α versus GSK-3β.
Figure 3.

In vivo effects on wt zebrafish embryos by compound 27 (A–F). Compound 27 was added 24 hours post-fertilization (hpf) to five embryos in duplicates, and the phenotypes were compared after 96 hpf. (A) Control embryo in 2% DMSO. Zebrafish embryos treated with compound 27 at increasing concentrations of (B) 1 μM, (C) 15 μM, (D) 30 μM, (E) 45 μM, (F) 60 μM.
In vitro effects in AML cell lines
To further investigate the activity of compound 27 in a disease-based model in vitro, we treated two AML cell lines (HL-60 and NB4) with increasing concentrations of compound 27. Compound 27 shows a specific on-target effect with a concentration-dependent decrease in Tyr279 GSK-3α phosphorylation without any relevant effect on Tyr216 GSK-3β phosphorylation up to 20 μM (Figure 4A). GSK-3α inhibition with compound 27 induces morphological (Figure 4B) and surface marker changes (Figure 4C) consistent with AML differentiation. Selective GSK-3α inhibition results in methylcellulose colony formation impairment (Figure 4D) in NB4 and HL-60 cell lines.
Figure 4.

In vitro effects of compound 27 in AML cell lines (HL-60 and NB4). Selective GSK-3α inhibition after 24h treatment with increasing concentrations of compound 27 in NB4. Histograms show a quantification of p-GSK-3α tyr279 and p-GSK-3β tyr216 relative to total GSK-3α and β respectively (A). Morphological changes (B: decreased nuclear to cytoplasmic ratio and increased vacuolization) and surface markers expression (CD11b, CD11c and CD14) consistent with differentiation after 6 days of treatment, histograms and means of fluorescence of 10 000 independent events are represented (C). Concentration-dependent colony formation impairment. *p<0,05 relative to corresponding DMSO. Error bars represent means +/− SEM (D).
Conclusion
Starting from the poorly soluble lead compound 1 a series of improved GSK-3 inhibitors was obtained by rational design. Optimization, primarily of the biphenylic scaffold of 1 led to the potent and α-selective GSK-3 inhibitor 27 with improved solubility. Compound 27 exhibits IC50 values of 42 nM for GSK-3α and 140 nM for GSK-3β. The homology model of GSK-3α/β and the in vitro activities of the isoform-selective compounds lead us to the hypothesis, that interactions to Arg148 (GSK-3β) and Lys211 (GSK-3α) within the Arg-rich loop are significant contributors for isoform-selectivity.
Our best compounds 13d and 27 were further profiled for efficacy and toxicity in the wt zebrafish embryo assay. Compound 27 showed reported phenotypes without lethality in vivo and proof of efficacy in vitro in two AML cell lines. A strong dose dependent differentiation phenotype and colony formation impairment through selective GSK-3α inhibition was observed in AML cell lines. The properties of 27 make it an excellent tool compound to study the different pathogenesis contributions of the isoforms GSK-3α and GSK-3β and a promising therapeutic candidate in AML.
Experimental Section
General Information
All chemicals were purchased as reagent grade from commercial suppliers and used without further purification. All reactions under anhydrous conditions were carried out under argon atmosphere with dry solvents.
The 1H-NMR spectra were recorded on a Bruker DRX 300 spectrometer at 300 MHz and on a Bruker DRX 500 spectrometer at 500 MHz. The 13C-spectra were recorded on a Bruker DRX 300 spectrometer at 75 MHz and on a Bruker DRX 500 spectrometer at 125 MHz. Chemical shifts are expressed in parts per million (ppm) and calibrated from the used solvent: CHCl3 (δCDCl3 = 7.26 ppm, δCDCl3 = 77.16 ppm), DMSO-d6 (δ(CHD2)SO(CD3) = 2.50 ppm, δ (CD3)SO(CD3) = 39.52 ppm). The respective measuring frequency, the used solvent and the measuring temperature are added in brackets to the spectroscopic data. Fine structures of proton signals are labeled with the following abbreviations: s for singlet, d for doublet, t for triplet, q for quartet and m for multiplet. Coupling constants (J values) are given in hertz (Hz).
Mass spectrometry was performed on a MAT 95 double focusing sector field EI-MS. Detected ion masses (m/z) are expressed in u.
Analytical high performance liquid chromatographies (HPLC) were carried out on an Agilent 1100 (column: reversed phase, Synergi 4u Polar-RP 80A, 4.6×150 mm, Variable Wavelength Detector λ = 254 nm). As a standard method a solvent gradient of acetonitrile (B) and 0.1% trifluoroacetic acid in water (A) (70% A; 30% B (0 to 1 min) up to 90% B) with a flow rate of 1 mL/min and an elapsed time of 12 min was used.
Flash column chromatography was carried out using silica gel 60 (15 – 40 μm, Merck). The automated flash chromatography was carried out on a Combi Flash Rf from Teledyne Isco using a reversed phase C18 high performance column (RediSep) and a mixture of MeCN/H2O. Thin-layer chromatography was carried out using aluminium sheets precoated with silica gel 60 F254 (0.2 mm, Merck). UV light with a wavelength of 254 nm and 360 nm was used for detection.
All compounds that were evaluated in biological assays had >95% purity using HPLC method described above.
Methyl 1H-benzo[d]imidazole-5-carboxylate (3a)
3a was synthesized in a similar manner to that described for 4b in the literature procedure by Schmidt et al.9 HPLC: 83%, tR = 0.83 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 3.87 (s, 3H), 7.68 (d, J = 8.3 Hz, 1H), 7.84 (dd, J = 1.4 Hz, J = 8.3 Hz, 1H), 8.23 (s, 1H), 8.40 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 51.9, 112.1, 123.0, 123.3, 125.7, 137.9, 142.1, 144.6, 166.8.
Methyl 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylate (3b)
3b was synthesized according to the literature procedure by Schmidt et al.9
5-(1H-Benzo[d]imidazol-5-yl)-1,3,4-oxadiazole-2-thiol (4a)
4a was synthesized in a similar manner to that described for 4b in the literature procedure by Schmidt et al.9 HPLC: 90%, tR = 0.92 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 7.75 (dd, J = 1.6 Hz, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 0.8 Hz, 1H), 8.50 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 113.9, 116.2, 120.1, 123.2, 137.9, 140.0, 144.5, 163.3, 177.3.
5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazole-2-thiol (4b)
4b was synthesized according to the literature procedure by Schmidt et al.9
General Procedure A: Suzuki coupling reaction (7a–d,g; 9e,f,h; 16a–c).9
Under an argon atmosphere the aryl bromide (2.50 mmol, 1.00 eq) was dissolved in 7 mL of toluene/EtOH (1/1). To the solution Pd(PPh3)4 (0.07 mmol, 0.03 eq) and 3.75 mL of 2 N aqueous Na2CO3 were added and the two-phase system was stirred for 15 min. Then the boronic acid (3.00 mmol, 1.20 eq) was added and the reaction mixture was refluxed at 80 °C for 2 d. After confirming the completion of the reaction by HPLC the mixture was diluted with water and extracted with EtOAc. The organic layers were dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of cyclohexane/EtOAc.
4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-carbonitrile (7a)
7a was synthesized according to the literature procedure by Schmidt et al.9
4-Fluoro-4′-methyl-1,1′-biphenyl (7b)
Yield 99%, colorless solid. HPLC: 97%, tR = 8.12 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.34 (s, 3H), 7.26 (m, 4H), 7.53 (d, J = 8.07 Hz, 2H), 7.66 (m, 2H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 20.6, 115.6 (d, JC-F = 19 Hz, 2C), 126.4 (2C), 128.3 (d, JC-F = 8 Hz, 2C), 129.5 (2C), 136.2, 136.5 (d, JC-F = 3 Hz), 136.6, 161.6, (d, JC-F = 242 Hz).
5-Fluoro-2-(p-tolyl)pyridine (7c)
Yield 67%, beige solid. HPLC: 98%, tR = 6.17 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.35 (s, 3H), 7.29 (d, J = 7.9 Hz, 2H), 7.78 (td, J = 8.8 Hz, J = 3.0 Hz, 1H), 7.94 (d, J = 8.2 Hz, 2H), 7.99 (dd, J = 8.9 Hz, J = 4.4 Hz, 1H), 8.62 (d, J = 3.0 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 20.7, 121.2 (d, JC-F = 4 Hz), 124.0 (d, JC-F = 18 Hz), 126.3 (2C), 129.3 (2C), 135.0, 137.2 (d, JC-F = 24 Hz), 138.4, 152.7 (d, JC-F = 3 Hz), 158.4 (d, JC-F = 254 Hz).
6-(p-Tolyl)nicotinonitrile (7d)
Yield 85%, beige solid. HPLC: 96%, tR = 6.77 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.38 (s, 3H), 7.35 (d, J = 7.9 Hz, 2H), 8.08 (m, 2H), 8.16 (dd, J = 8.4 Hz, J = 0.8 Hz, 1H), 8.34 (dd, J = 8.4 Hz, J = 2.2 Hz, 1H), 9.06 (dd, J = 2.2 Hz, J = 0.8 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 20.9, 107.0, 117.3, 119.7, 127.1 (2C), 129.6 (2C), 134.2, 140.5, 140.8, 152.4, 159.1.
5-(p-Tolyl)picolinonitrile (7g)
Yield 75%, colorless solid. HPLC: 96%, tR = 6.71 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.37 (s, 3H), 7.35 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 8.2 Hz, 2H), 8.08 (d, J = 8.2 Hz, 1H), 8.31 (dd, J = 8.2 Hz, J = 2.3 Hz, 1H), 9.07 (d, J = 2.3 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 20.8, 117.7, 127.2 (2C), 129.1, 129.9 (2C), 130.8, 132.3, 134.9, 138.9, 139.2, 149.0.
(4-(5-Methylpyridin-2-yl)phenyl)methanol (9e)
Yield 62%, colorless solid. HPLC: 99%, tR = 0.83 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.33 (s, 3H), 4.55 (d, J = 5.7 Hz, 2H), 5.21 (t, J = 5.7 Hz, OH), 7.41 (d, J = 8.0 Hz, 2H), 7.67 (m, 1H), 7.83 (d, J = 8.1 Hz, 1H), 8.01 (d, J = 8.1 Hz, 2H), 8.48 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 17.6, 62.6, 119.4, 125.9 (2C), 126.7 (2C), 131.5, 137.1, 137.4, 1143.1, 149.7, 153.3.
(4-(6-Fluoropyridin-3-yl)phenyl)methanol (9f)
Yield 27%, colorless solid. HPLC: 99%, tR = 3.55 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.61 (s, 2H), 6.96 (dd, J = 8.5 Hz, J = 3.0Hz, 1H), 7.40 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 7.93 (ddd, J = 8.5 Hz, J = 2.6 Hz, J = 1.3 Hz, 1H), 8.31 (d, J = 2.3 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 63.0, 108.9 (d, JC-F = 36 Hz), 126.2 (2C), 126.9 (2C), 134.1 (d, JC-F = 4 Hz), 134.4, 139.1 (d, JC-F = 7 Hz), 141.7, 144.8 (d, JC-F = 15 Hz), 162.4 (d, JC-F = 239 Hz).
(4-(6-Methylpyridin-3-yl)phenyl)methanol (9h)
Yield 52%, colorless solid. HPLC: 93%, tR = 0.81 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.50 (s, 3H), 4.54 (d, J = 5.7 Hz, 2H), 5.23 (t, J = 5.7 Hz, OH), 7.32 (d, J = 8.0 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.94 (dd, J = 8.1 Hz, J = 2.3 Hz, 1H), 8.73 (d, J = 2.3 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 23.6, 62.6, 123.1, 126.3 (2C), 127.1 (2C), 132.6, 134.2, 135.5, 142.2, 146.7, 156.7.
2-Fluoro-5-(6-methylpyridin-3-yl)benzonitrile (16a)
Yield 87%, colorless solid. HPLC: 99%, tR = 1.75 min. 1H-NMR (CDCl3, 500 MHz, 300 K): δ (ppm) = 2.75 (2, 3H), 7.36 (t, J = 8.4 Hz, 1H), 7.41 (d, J = 7.9 Hz, 1H), 7.81 (dd, J = 11.9 Hz, J = 5.6 Hz, 2H), 7.91 (d, J = 7.9 Hz, 1H), 8.73 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 23.0, 102.9 (d, JC-F = 17 Hz), 113.5, 117.7 (d, JC-F = 23 Hz), 124.9, 132.0, 132.1, 133.7 (d, JC-F = 8 Hz), 134.2, 137.0, 145.0, 157.4, 163.3 (d, JC-F = 263 Hz).
2-Fluoro-5-(5-methylpyridin-2-yl)benzonitrile (16b)
Yield 91%, colorless solid. HPLC: 91%, tR = 2.61 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.34 (s, 3H), 7.61 (t, J = 9.0 Hz, 1H), 7.74 (dd, J = 8.6 Hz, 2.1 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 8.46 (ddd, J = 8.8 Hz, 5.3 Hz, 2.4 Hz, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.54 (dd, J = 6.3 Hz, 2.4 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 17.6, 100.6 (d, JC-F = 16 Hz), 113.9, 116.9 (d, JC-F = 21 Hz), 119.9, 131.3, 132.9, 133.6 (d, JC-F = 8 Hz), 136.1 (d, JC-F = 4Hz), 137.8, 149.9, 150.3, 162.6 (d, JC-F = 257 Hz).
6′-Fluoro-5-methyl-2,3′-bipyridine (16c)
Yield 99%, orange solid. HPLC: 92%, tR = 1.31 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.35 (s, 3H), 7.28 (dd, J = 8.8 Hz, 2.7 Hz, 1H), 7.74 (ddd, J = 8.1 Hz, 2.2 Hz, 0.7 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 8.53 (dd, J = 1.5 Hz, 0.7 Hz, 1H), 8.59 (m, 1H), 8.88 (d, J = 2.5 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 17.5, 109.5 (d, JC-F = 36 Hz), 119.5, 128.7 (d, JC-F = 10 Hz), 131.4 (d, JC-F = 16 Hz), 132.7, 132.8 (d, JC-F = 4 H), 137.7, 139.8 (d, JC-F = 8 Hz), 145.5 (d, JC-F = 16 Hz), 150.0, 163.2 (d, JC-F = 237 Hz).
General Procedure B: Radical bromination (8a–d,g; 17a–c)9
The substituted toluene (1.00 eq) was diluted in 20 mL of CCl4 and NBS (0.95 eq) and AIBN (5 mg per mmol) was added in portions over a period of 1 h. The reaction mixture was heated to reflux for 16 h. After cooling to rt the mixture was diluted with water and CCl4 was evaporated. The residue was extracted with EtOAc and washed with brine. The organic layers were dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography using a mixture of cyclohexane/EtOAc.
4′-(Bromomethyl)-4-fluoro-[1,1′-biphenyl]-2-carbonitrile (8a)
8a was synthesized according to the literature procedure by Schmidt et al.9
4-(Bromomethyl)-4′-fluoro-1,1′-biphenyl (8b)
Yield 52%, colorless solid. HPLC: 87%, tR = 8.39 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.76 (s, 2H), 7.29 (m, 2H), 7.53 (m, 2H), 7.64 (m, 2H), 7.72 (m, 2H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 34.2, 115.6, 115.8, 126.9 (2C), 128.6, 128.7, 129.9 (2C), 136.0 (d, JC-F = 2 Hz), 137.1, 139.0, 162.0 (d, JC-F = 245 Hz).
2-(4-(Bromomethyl)phenyl)-5-fluoropyridine (8c)
Yield 65%, colorless solid. HPLC: 61%, tR = 7.15 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.67 (s, 2H), 7.55 (d, J = 8.7 Hz, 2H), 7.82 (dd, J = 8.0 Hz, J = 3.0 Hz, 1H), 8.03 (d, J = 8.7 Hz, 2H), 8.03 (d, J = 8.0 Hz, 1H), 8.66 (d, J = 3.0 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 34.5, 122.2, 124.6 (d, JC-F = 24 Hz), 127.2, 127.8, 130.2, 132.7, 138.0 (d, JC-F = 24 Hz), 139.2, 152.6, 159.1 (d, JC-F = 245 Hz).
6-(4-(Bromomethyl)phenyl)nicotinonitrile (8d)
Yield 89%, pale yellow solid. HPLC: 73%, tR = 7.26 min.1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.78 (s, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 8.20 (m, 1H), 9.11 (m, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 34.2, 108.0, 117.6, 121.0, 128.2, 130.4, 132.9, 141.5, 144.5, 153.2, 158.4.
5-(4-(Bromomethyl)phenyl)picolinonitrile (8g)
Yield 64%, colorless solid. HPLC: 96%, tR = 7.17 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.78 (s, 2H), 7.62 (d, J = 8.3 Hz, 2H), 7.84 (m, 2H), 8.12 (d, J = 8.1 Hz, 1H), 8.36 (dd, J = 8.2 Hz, 2.3 Hz, 1H), 9.11 (d, J = 2.3 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 33.7, 117.6, 127.7 (2C), 129.8, 130.2 (2C), 131.3, 135.1, 135.4, 138.4, 139.3, 149.3.
5-(6-(Bromomethyl)pyridin-3-yl)-2-fluorobenzonitrile (17a)
Yield 22%, pale yellow solid. HPLC: 85%, tR = 6.77 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.70 (d, J = 63.6 Hz, 2H), 7.36 (t, J = 8.4 Hz, 1H), 7.61 (dd, J = 20.0 Hz, J = 8.4 Hz, 1H), 7.85 (m, 3H), 8.75 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 29.9, 113.5, 117.7 (d, JC-F = 21 Hz), 124.1, 132.2, 133.3, 133.8 (d, JC-F = 8 Hz),134.6 (d, JC-F = 4 Hz), 136.0, 147.4, 156.8, 163.3 (d, JC-F = 251 Hz).
5-(5-(Bromomethyl)pyridin-2-yl)-2-fluorobenzonitrile (17b)
Yield 39%, beige solid. HPLC: 85%, tR = 6.77 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.80 (s, 2H), 7.65 (t, J = 9.0 Hz, 1H), 8.00 (dd, J = 8.2 Hz, 2.3 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H), 8.50 (ddd, J = 8.7 Hz, 5.3 Hz, 2.4 Hz, 1H), 8.59 (dd, J = 6.3 Hz, 2.4 Hz, 1H), 8.76 (d, J = 2.1 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 30.6, 100.8 (d, JC-F = 25 Hz), 113.9, 117.1 (d, JC-F = 15 Hz), 120.5, 131.9, 133.8, 134.1 (d, JC-F = 15 Hz), 135.5, 138.3, 150.1, 152.7, 162.9 (d, JC-F = 256 Hz).
5-(Bromomethyl)-6′-fluoro-2,3′-bipyridine (17c)
Yield 45%, orange solid. HPLC: 76%, tR = 4.92 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.80 (s, 2H), 7.31 (dd, J = 8.2 Hz, J = 2.9 Hz, 1H), 8.00 (dd, J = 8.2 Hz, J = 2.3 Hz, 1H), 8.05 (dd, J = 8.2 Hz, J = 0.7 Hz, 1H), 8.63 (m, 1H), 8.76 (d, J = 1.9 Hz, 1H), 8.92 (d, J = 2.5 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 30.6, 109.7, 120.5, 132.8, 133.6, 138.2, 140.3, 146.1, 149.9, 150.1, 163.5 (d, JC-F = 245 Hz).
General Procedure C: Bromination of the benzylic alcohols (8e,f,g)
To a solution of the benzylic alcohol 9e,f,g (2.50 eq) in toluene (10 mL per mmol) was added phosphorous tribromide (1.00 eq). The reaction mixture was refluxed at 110 °C for 3 h. After cooling to rt the solvent was evaporated under a reduced pressure. The crude product was used for the next reaction without any purification.
General Procedure D: Preparation of thioethers by nucleophilic substitution (1; 12a;13b–h; 18a–c)
To a solution of the mercaptane (1.00 eq) in DMF (10 mL per mmol) was added 4N NaOH. The mixture was stirred at rt for 15 min. Afterwards the benzylic bromide (1.25 eq) was added and the reaction mixture was stirred at rt for another 6 h. The formed precipitate is filtered off, washed once with a small amount of DMF and then thoroughly with EtOH. The product was dried under reduced pressure.
Purification of compounds, which did not precipitate in DMF was performed as follows. The mixture was diluted with EtOAc. The separated organic layer was washed with brine several times, dried over MgSO4 and dried under reduced pressure. The crude product was purified by column chromatography using silica gel and a mixture of cyclohexane/EtOAc or by automated flash chromatography using a reversed phase C18 high performance column (RediSep) and a mixture of MeCN/H2O.
4′-(((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)-4-fluoro-[1,1′-biphenyl]-2-carbonitrile (1)
1 was synthesized according to the literature procedure by Schmidt et al.9
4′-(((5-(1H-Benzo[d]imidazol-6-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)-4-fluoro-[1,1′-biphenyl]-2-carbonitrile (12a)
Yield 13%, colorless solid. HPLC: 95%, tR = 5.53 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.68 (s, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.66 (m, 4H), 7.77 (d, J = 8.4 Hz, 1H), 7.83 (dd, J = 8.4 Hz, J = 1.4 Hz, 1H), 7.95 (m, 1H), 8.19 (s, 1H), 8.42 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.5, 111.6 (d, JC-F = 10 Hz), 116.6, 117.4 (d, JC-F = 27 Hz), 120.3 (d, JC-F = 7 Hz), 120.5, 121.0 (d, JC-F = 21 Hz), 122.6 (d, JC-F = 23 Hz), 125.5, 129.0 (2C), 129.4 (2C), 132.4 (d, JC-F = 8 Hz), 136.2, 137.5, 139.6, 140.8 (d, JC-F = 3 Hz), 144.3, 144.5, 160.8 (d, JC-F = 246 Hz), 162.6, 166.2. EI-MS: m/z (%): 427 (20, [M+]), 428 (5, [M+ + H]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(((4′-fluoro-[1,1′-biphenyl]-4-yl)methyl)thio)-1,3,4-oxadiazole (13b)
Yield 48%, colorless solid. HPLC: 99%, tR = 8.72 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.32 (m, 4H), 4.60 (s, 2H), 7.04 (d, J = 8.4, 1H), 7.27 (td, J = 6.7 Hz, 3.4 Hz, 2H), 7.39 (d, J = 2.1 Hz, 1H), 7.44 (dd, J = 8.4 Hz, 2.1 Hz, 1H), 7.54 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H), 7.69 (m, 2H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.0, 64.4, 115.0, 115.6, 115.7, 115.9, 118.1, 119.9, 126.8 (2C), 128.5, 128.6, 129.6 (2C), 135.9, 136.0, 138.5, 143.8, 146.7, 161.9 (d, JC-F = 245 Hz, 1C), 162.5, 164.9. EI-MS: m/z (%): 420 (16, [M+]), 421 (5, [M+ + H]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(4-(5-fluoropyridin-2-yl)benzylthio)-1,3,4-oxa-diazole (13c)
Yield 66%, colorless solid. HPLC: 96%, tR = 8.09 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.31 (dd, J = 12.6 Hz, J = 5.0 Hz 4H), 4.61 (s, 2H), 7.04 (d, J = 8.4 Hz, 1H), 7.43 (m, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.80 (td, J = 8.7 Hz, J = 2.9 Hz, 1H), 8.05 (m,3H), 8.63 (d, J = 2.9 Hz, 1H).13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 115.1, 115.9, 118.1, 119.9, 121.7 (d, JC-F = 5 Hz), 124.1 (d, JC-F = 18 Hz), 126.6 (2C), 129.5 (2C), 137.1, 137.3, 137.5 (d, JC-F = 6 Hz), 143.8, 146.7, 152.2 (d, JC-F = 4 Hz), 158.6 (d, JC-F = 254 Hz), 162.5, 165.0. EI-MS: m/z (%): 421 (20, [M+]), 422 (4, [M+ + H]).
6-(4-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-ylthio)methyl)phenyl)-nicotinonitrile (13d)
Yield 51%, colorless solid. HPLC: 95%, tR = 7.89 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.31 (dtd, J = 2.1 Hz, J = 3.5 Hz, 4.3 Hz, 4H), 4.63 (s, 2H), 7.04 (d, J = 8.5 Hz, 1H), 7.38 (d, J = 2.1 Hz, 1H), 7.43 (dd, J = 2.1 Hz, J = 8.4 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 8.14 (d, J = 8.4 Hz, 2H), 8.18 (dd, J = 0.6 Hz, J = 8.4 Hz, 1H), 8.37 (dd, J = 2.2 Hz, J = 8.4 Hz, 1H), 9.07 (dd, J = 0.6 Hz, J = 2.1 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.0, 64.4, 107.4, 115.0, 115.9, 117.2, 118.1, 119.9, 120.2, 127.4 (2C), 127.9 (2C), 136.3, 139.4, 140.9, 143.8, 146.7, 152.5, 158.6, 162.4, 164.6. EI-MS: m/z (%): 428 (38, [M+]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(4-(5-methylpyridin-2-yl)benzylthio)-1,3,4-oxadiazole (13e)
Yield 27%, colorless solid. HPLC: 98%, tR = 5.26 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 2.33 (s, 3H), 4.32 (ddd, J = 5.2 Hz, J = 4.5 Hz, J = 1.6 Hz, 4H), 4.61 (s, 2H), 7.04 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.44 (dd, J = 8.4 Hz, 2.1 Hz, 1H), 7.56 (d, J = 8.3 Hz, 2H), 7.68 (dd, J = 8.2 Hz, 1.7 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 8.02 (d, J = 8.3 Hz, 2H), 8.49 (d, J = 1.2 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 17.6, 35.8, 64.0, 64.4, 115.0, 115.9, 118.1, 119.6, 119.9, 126.4 (2C), 129.4 (2C), 131.9, 137.1, 137.5, 138.1, 143.8, 146.7, 149.7, 152.8, 162.5, 164.9. EI-MS: m/z (%): 417 (32, [M+]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-((4-(6-fluoropyridin-3-yl)benzyl)thio)-1,3,4-oxadiazole (13f)
Yield 58%, colorless solid. HPLC: 98%, tR = 8.10 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.32 (m, 4H),4.61 (s, 2H), 7.04 (d, J = 8.4 Hz, 1H), 7.27 (dd, J = 8.5 Hz, J = 2.8 Hz, 1H), 7.38 (d, J = 2.0 Hz, 1H), 7.44 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.59 (d, J = 8.3 Hz, 2H), 7.70 (d, J = 8.3 Hz, 2H), 8.27 (td, J = 8.2 Hz, J = 2.6 Hz, 1H), 8.53 (d, J = 2.6 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 109.6 (d, JC-F = 37 Hz), 115.1, 115.9, 118.1, 119.9, 127.0 (2C), 129.8 (2C), 133.6 (d, JC-F = 4 Hz), 135.2 (d, JC-F = 4 Hz), 136.9, 140.3 (d, JC-F = 9 Hz), 143.8, 145.4 (d, JC-F = 16 Hz), 146.7, 162.5,, 162.7 (d, JC-F = 263 Hz), 165.0. EI-MS: m/z (%): 421 (100, [M+]), 422 (26, [M+ + H]), 423 (8, [M+ + 2H]).
5-(4-(((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)phenyl) picolinonitrile (13g)
Yield 58%, colorless solid. HPLC: 99%, tR = 8.02 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.32 (dtd, J = 7.3 Hz, J = 3.5 Hz, J = 2.1 Hz, 4H), 4.63 (s, 2H), 7.04 (d, J = 8.5 Hz, 1H), 7.38 (d, J = 2.1 Hz, 1H), 7.43 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.82 (d, J = 8.3 Hz, 2H), 8.11 (d, J = 8.2 Hz, 1H), 8.33 (dd, J = 8.2 Hz, J = 2.3 Hz, 1H), 9.09 (d, J = 1.7 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.5, 64.1, 64.4, 115.1, 155.9, 117.6, 118.2, 119.9, 127.5 (2C), 129.1, 130.0 (2C), 131.2, 134.6, 135.3, 138.3, 138.4, 143.8, 146.7, 149.2, 162.5, 165.0. EI-MS: m/z (%): 428 (100, [M+]), 429 (28, [M+ + H]), 430 (8, [M+ + 2H]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(4-(6-methylpyridin-3-yl)benzylthio)-1,3,4-oxadiazole (13h)
Yield 12%, colorless solid. HPLC: 97%, tR = 5.17 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = CH3 fehlt, 4.32 (m, 4H), 4.61 (s, 2H), 7.04 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.44 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.57 (d, J = 8.3 Hz, 2H), 7.66 – 7.69 (m, 2H), 7.95 (dd, J = 8.1 Hz, J = 2.5 Hz, 1H), 8.73 (d, J = 2.2 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 24.1, 36.1, 64.6, 64.9, 114.9, 115.6, 116.4, 118.6, 120.4, 123.6, 127.2, 130.2, 132.6, 134.8, 136.9, 137.0, 144.3, 147.2, 157.4, 163.1, 165.5. EI-MS: m/z (%): 417 (100, [M+]), 418 (26, [M+ + H], 419 (8, [M+ + 2H]).
2-(6-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-ylthio)methyl)pyridin-3-yl)-5-fluorobenzonitrile (18a)
Yield 33%, colorless solid. HPLC: 98%, tR = 7.32 min. 1H-NMR (CDCl3, 500 MHz, 300 K): δ (ppm) = 4.31 (dddd, J = 6.8 Hz, J = 5.0 Hz, 3.4 Hz, 1.6 Hz, 4H), 5.03 (s, 2H), 6.95 (m, 1H), 7.46 (dd, J = 2.0 Hz, J = 6.9 Hz, 3H), 7.85 (ddd, J = 12.3 Hz, J = 4.7 Hz, J = 2.1 Hz, 2H), 8.32 (m, 2H), 8.87 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 29.9, 64.4, 64.8, 102.5 (d, JC-F = 6 Hz), 114.4, 116.1, 116.1, 117.7 (d, JC-F = 29 Hz), 118.3, 120.6, 122.9, 128.4, 131.2, 132.4 (d, JC-F = 7 Hz), 133.8 (d, JC-F = 8 Hz), 134.0, 138.4, 144.1, 144.4, 149.8, 151.8, 155.4, 157.8 (d, JC-F = 254 Hz), 165.0. EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
5-(5-(((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)pyridin-2-yl)-2-fluorobenzonitrile (18b)
Yield 37%, colorless solid. HPLC: 99%, tR = 7.58 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.32 (m, 4H), 4.64 (s, 2H), 7.04 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 2.1 Hz, 1H), 7.43 (dd, J = 8.5 Hz, 2.1 Hz, 1H), 7.65 (t, J = 9.1 Hz, 1H), 8.03 (dd, J = 8.3 Hz, 2.2 Hz, 1H), 8.07 (dd, J = 8.2 Hz, 0.5 Hz, 1H), 8.48 (ddd, J = 8.9 Hz, 5.3 Hz, 2.4 Hz, 1H), 8.58 (dd, J = 6.2 Hz, 2.3 Hz, 1H), 8.77 (d, J = 1.7 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 32.8, 64.1, 64.4, 95.2, 113.9, 115.1, 115.8, 117.0, 117.1, 118.1, 119.9, 120.3, 131.8, 132.8, 134.0 (d, JC-F = 8 Hz), 136.1, 138.0, 143.8, 146.7, 150.0, 152.2, 156.7, 163.6 (d, JC-F = 356 Hz). EI-MS: m/z (%): 446 (100, [M+]), 447 (27, [M+ + H]), 448 (8, [M+ + 2H]).
2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(((6′-fluoro-[2,3′-bipyridin]-5-yl)methyl)thio)-1,3,4-oxadiazole (18c)
Yield 31%, pale yellow solid. HPLC: 95%, tR = 6.69 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.31 (dtd, J = 7.3 Hz, J = 3.6 Hz, 2.1 Hz, 4H), 4.64(s, 2H), 7.04 (d, J = 2.1 Hz, 1H), 7.30 (dd, J = 8.6 Hz, J = 2.6 Hz, 1H), 7.37 (d, J = 2.1 Hz, 1H), 7.43 (dd, J = 8.5 Hz, J = 2.1 Hz, 1H), 8.01 – 8.05 (m, 2H), 8.61 (td, J = 8.3 Hz, J = 2.6 Hz, 1H), 8.77 (d, J = 1.7 Hz, 1H), 8.90 (d, J = 2.5 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 32.8, 64.0, 64.4, 109.6 (d, JC-F = 39 Hz), 115.1, 115.8, 118.1, 119.9, 120.3, 132.4 (d, JC-F = 7 Hz), 132.5, 137.9, 140.2 (d, JC-F = 9 Hz), 143.8, 145.8, 145.9, 146.7, 150.1, 151.9, 162.4, 163.6 (d, JC-F = 356 Hz). EI-MS: m/z (%): 422 (100, [M+]), 423 (36, [M+ + H]), 424 (11, [M+ + 2H]).
Methyl-1-(2-cyano-4-fluorophenyl)piperidine-4-carboxylate (21)
Methyl piperidine-4-carboxylate (0.62 g, 4.32 mmol, 1.20 eq) and 2,5-difluorobenzonitrile (0.50 g, 3.60 mmol, 1.00 eq) were dissolved in 10 mL DMSO. Cesium carbonate (1.41 g, 4.32 mmol, 1.2 eq) was added and the reaction mixture was stirred at 100°C for 18 h. After cooling to rt, the reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with saturated NaHCO3 solution and brine, dried over MgSO4 and concentrated under reduced pressure. Purification by column chromatography (cyclohexane/EtOAc 4:1) gave (0.39 g, 42%) of compound 20 as a colorless solid.. HPLC: 98%, tR = 6.93 min. 1H-NMR (CDCl3, 500 MHz, 300 K): δ (ppm) = 2.03 (m, 4H), 2.48 (m, 1H), 2.86 (m, 2H), 3.43 (ddd, J = 3.16 Hz, J = 6.95 Hz, J = 8.38 Hz, 2H), 3.71 (s, 3H), 7.00 (dd, J = 4.61 Hz, J = 9.02 Hz, 1H), 7.20 (ddd, J = 3.05 Hz, J = 7.79 Hz, J = 9.02 Hz, 1H), 7.26 (m, 1H). 13C-NMR (CDCl3, 125 MHz, 300 K): δ (ppm) = 28.5(2C), 40.5, 51.9(2C), 52.1, 56.7, 107.9 (d, JC-F = 9 Hz), 112.7 (d, JC-F = 8 Hz), 117.1, 120.5 (d, JC-F = 25 Hz), 120.8 (d, JC-F = 8 Hz), 121.1 (d, JC-F = 23 Hz), 153.1, 157.1 (d, JC-F = 243 Hz), 175.0.
5-Fluoro-2-(4-(hydroxymethyl)piperidin-1-yl)benzonitrile (22)
To a solution of 21 (0.34 g, 1.30 mmol, 1.00 eq) in anhydrous THF (15 mL) was added lithium borohydride (1.9 mL, 3.38 mmol, 2.60 eq). The mixture was heated to reflux for 16 h under argon atmosphere. After cooling to rt, the reaction mixture was diluted carefully with water and extracted with EtOAc. The combined organic layer was washed with brine and dried over MgSO4. After removing the solvent compound 22 (0.30 g, 98%) was obtained as a pale yellow solid. HPLC: 99%, tR = 4.94 min. 1H-NMR (CDCl3, 500 MHz, 300 K): δ (ppm) = 1.51 (ddd, J = 3.91 Hz, J = 12.42 Hz, J = 24.3 Hz, 2H), 1.60 – 1.69 (m, 1H), 1.87 (dd, J = 1.62 Hz, J = 12.42 Hz, 2H), 2.76 (td, J = 1.62 Hz, J = 12.42 Hz, 2H), 3.49 (d, J = 11.95 Hz, 2H), 3.56 (d, J = 6.47 Hz, 2H), 6.98 (dd, J = 4.43 Hz, J = 9.19 Hz, 1H), 7.18 (ddd, J = 2.95 Hz, J = 7.99 Hz, J = 9.19 Hz, 1H), 7.24 (dd, J = 2.95 Hz, J = 7.99 Hz, 1H). 13C-NMR (CDCl3, 125 MHz, 300 K): δ (ppm) = 29.2 (2C), 38.3, 52.8 (2C), 67.8, 107.6 (d, JC-F = 9 Hz), 117.3 (d, JC-F = 2 Hz), 120.3 (d, JC-F = 26 Hz), 120.7 (d, JC-F = 8 Hz, 1C), 121.1 (d, JC-F = 23 Hz), 153.5, 156.9 (d, JC-F = 243 Hz).
2-(4-(Bromomethyl)piperidin-1-yl)-5-fluorbenzonitrile (23)
Anhydrous conditions and inert gas are essential for this reaction! 22 (0.18 g, 0.77 mmol, 1.00 eq), tetrabromomethane (0.38 g, 1.15 mmol, 1.50 eq) and triphenylphosphine (0.40 g, 1.55 mmol, 2.00 eq) was added in a dried vessel and purged for 15 min with argon. Afterwards the educts were dissolved in dry acetonitrile. The reaction mixture was stirred at rt. The progression of the reaction was controlled by HPLC. After no further conversion was observed, the reaction was stopped by addition of 15% NaOH solution. The reaction mixture was extracted with DCM. The combined organic layer was washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography (cyclohexane/EtOAc 9:1) to give 23 (0.08 g, 35%) as a brown oil. HPLC: 97%, tR = 8.41 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 1.43 (qd, J = 3.80 Hz, J = 11.94 Hz, 2H), 1.73 – 1.81 (m, 1H), 1.89 (d, J = 12.25 Hz, 2H), 2.77 (td, J = 2.09 Hz, J = 11.94 Hz, 2H), 3.41 (d, J = 12.25 Hz, 2H), 3.54 (d, J = 6.13 Hz, 2H), 7.20 (dd, J = 4.73 Hz, J = 8.99 Hz, 1H), 7.47 (ddd, J = 0.66 Hz, J = 3.11 Hz, J = 8.73 Hz, J = 8.99 Hz, 1H), 7.68 (dd, J = 3.11 Hz, J = 8.32 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) =30.5 (2C), 37.2, 39.9, 51.7 (2C), 106.3 (d, JC-F = 11 Hz), 116.9 (d, JC-F = 2 Hz), 120.2 (d, JC-F = 26 Hz), 121.3 (d, JC-F = 8 Hz), 121.4 (d, JC-F = 23 Hz), 152.9 (d, JC-F = 3 Hz), 156.2 (d, JC-F = 240 Hz).
2-(4-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)methyl)piperidin-1-yl)-5-fluorbenzonitrile (24)
A mixture of 4b (0.03 g, 0.14 mmol, 1.00 eq) and potassium carbonate (0.02 g, 0.17 mmol, 1.20 eq) was stirred in dry DMF (3.6 mL) at rt for 15 min. 23 (0.02 g, 0.17 mmol, 1.50 eq) was added to the orange solution. Afterwards the reaction mixture was stirred for 3 h at 80°C under an argon atmosphere. The resolution mixture was diluted with water and EtOAc. The aqueous layer was extracted with EtOAc. The organic layer was washed with bine, successively dried over MgSO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography (cyclohexane/EtOAc 2:1) to give (0.05 g, 71%) of compound 24 as a colorless solid. HPLC: 99%, tR = 8.86 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) =1.66 (ddd, J = 3.86 Hz, J = 12.18 Hz, J = 15.67 Hz, 2H), 2.01 (m, 1H), 2.07 (d, J = 13.17 Hz, 2H), 2.84 (td, J = 1.75 Hz, J = 11.82 Hz, 2H), 3.28 (d, J = 6.82 Hz, 2H), 3.51 (d, J = 12.05 Hz, 2H), 4.29 – 4.34 (m, 4H), 6.96 (d, J = 8.32 Hz, 1H), 7.09 (dd, J = 4.60 Hz, J = 8.97 Hz, 1H), 7.21 (ddd, J = 2.99 Hz, J = 7.84 Hz, J = 8.97 Hz, 1H), 7.26 – 7.28 (m, 1H), 7.49 – 7.52 (m, 2H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) =31.7 (2C), 35.4, 38.6, 52.8 (2C), 64.4, 64.8, 107.9 (d, JC-F = 9 Hz), 116.1, 117.1, 118.2, 120.5, 120.6 (d, JC-F = 25 Hz), 121.1, 121.2 (d, JC-F = 12 Hz), 121.3, 144.0, 146.9, 151.8, 158.3, 164.8 (d, JC-F = 232 Hz), 167.9. EI-MS: m/z (%): 452 (100, [M+]), 453 (28, [M+ + H]).
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-sulfinyl)methyl)-4-fluorbiphenyl-2-carbonitrile ((rac)-25)
Under an argon atmosphere 1 (0.05 g, 0.11 mmol, 1.00 eq) was dissolved in 8 mL of DCM. The solution was cooled to 0°C and mCPBA (0.03 g, 0.11 mmol, 1.00 eq) was added. The reaction mixture was allowed to warm to rt and was stirred for 2 d. After confirming the completion of the reaction with HPLC, saturated sodium sulphite solution was added to the mixture. The organic layer was washed with saturated NaHCO3 solution and dried over MgSO4. Purification was performed by column chromatography (cyclohexane/EtOAc 1:1) to give (0.04 g, 79%) of compound (rac)-25 as a colorless solid. HPLC: 99%, tR = 7.71 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.31 – 4.33 (m, 2H), 4.35 – 4.36 (m, 2H), 4.92 (q, J = 12.57 Hz, 2H), 7.09 (d, J = 8.56 Hz, 1H), 7.48 – 7.50 (m, 3H), 7.53 (dd, J = 2.11 Hz, J = 8.41 Hz, 1H), 7.56 – 7.57 (m, 1H), 7.57 – 7.58 (m, 1H), 7.62 (dd, J = 5.40 Hz, J = 8.56 Hz, 1H), 7.68 (td, J = 2.67 Hz, J = 8.56 Hz, 1H), 7.96 (dd, J = 2.67 Hz, J = 8.56 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 58.2, 64.2, 64.7, 111.8, 115.3, 115.9, 117.4, 118.5, 120.6 (d, JC-F = 24 Hz), 120.9, 121.2 (d, JC-F = 23 Hz), 129.2 (2C), 130.1, 131.0 (2C), 132.6 (d, JC-F = 8 Hz), 137.2, 140.6, 144.1, 147.6, 165.0 (d, JC-F = 243 Hz), 165.5. EI-MS: m/z (%): 461 (100, [M+]), 445 (43, [M+ - O], 462 (25, [M+ + H], 463 (6, [M+ + 2H]).
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-sulfonyl)methyl)-4-fluorbiphenyl-2-carbonitrile (26)
Under argon atmosphere 2 (0.03 g, 0.07 mmol, 1.00 eq) was dissolved in 6 mL of DCM. The solution was cooled to 0°C and mCPBA (0.04 g, 0.20 mmol, 3.00 eq) was added. The reaction mixture warmed to 30°C and was stirred for 3 d. In the case of full implementation the reaction was stopped by adding saturated NaSO3 solution. The organic layer was washed with saturated NaHCO3 solution and dried over MgSO4. Purification was performed by column chromatography (cyclohexane/EtOAc 1:1) to give (0.02 g, 68%) of compound 26 as a colorless solid. HPLC: 97%, tR = 8.07 min. 1H-NMR (DMSO, 500 MHz, 300 K): δ (ppm) = 4.34 (ddd, J = 2.7 Hz, 5.9 Hz, 7:8 Hz, 4H), 5.33 (s, 2H), 7.09 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 2.1 Hz, 1H), 7.50 (dd, J = 2.1 Hz, J = 8.5 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.63 (m, 3H), 7.69 (td, J = 2.7 Hz, J = 8.5 Hz, 1H), 7.98 (dd, J = 2.7 Hz, J = 8.7 Hz, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 60.2, 64.1, 64.6, 111.7 (d, JC-F = 10 Hz), 114.6, 116.0, 117.2, 118.4, 120.5 (d, JC-F = 25 Hz), 121.1, 121.1 (d, JC-F = 23 Hz), 126.8, 129.1 (2C), 131.7 (2C), 132.4 (d, JC-F = 8 Hz), 137.7, 140.4, 143.9, 147.9, 160.7, 160.9 (d, JC-F = 227 Hz), 165.9. EI-MS: m/z (%): 477 (100, [M+]), 478 (28, [M+ + H]), 479 (8, [M+ + 2H]).
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorbiphenyl-2-carboxamide (27)
Under an argon atmosphere 2 (0.05 g, 0.11 mmol, 1.00 Äq.) was added to a dried vessel. Trifluoroacetic acid (0.8 mL) and concentrated sulphoric acid (0.2 mL) were added and the red mixture was stirred 6h at 70°C. Subsequently the reaction is poured into a water/ice-mixture (50 mL), extracted with CH2Cl2, washed with saturated NaHCO3 solution and dried over MgSO4. Purification by column chromatography (cyclohexane/EtOAc 1:2) gave (0.04 g, 78%) of compound 27 as a colorless solid. HPLC: 96%, tR = 6.93 min. 1H-NMR (DMSO-d6, 500 MHz, 300 K): δ (ppm) = 4.32 (td, J = 5.2 Hz, J = 3.7 Hz, 4H), 4.60 (s, 2H), 7.05 (d, J = 8.4 Hz, 1H), 7.25 (dd, J = 9.1 Hz, J = 2.7 Hz, 1H), 7.31 (td, J = 8.6 Hz, J = 2.8 Hz, 1H), 7.38 (m, 3H), 7.41 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.73 (s, 1H). 13C-NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 114.3 (d, JC-F = 21 Hz), 115.0, 115.9 (d, JC-F = 21 Hz), 115.9, 118.1, 120.0, 128.6 (2C), 128.8 (2C), 132.0 (d, JC-F = 8 Hz), 134.8, 135.5, 138.9, 139.0 (d, JC-F = 7 Hz), 143.8, 146.7, 160.9 (d, JC-F = 247 Hz), 162.7, 164.9, 169.5. EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
Supplementary Material
Acknowledgments
The authors thank the students Martin Brodrecht, Anne Schieferdecker and David Fiebig (Technische Universität Darmstadt), who supported this work. The authors thank Dennis Bensinger (Technische Universität Darmstadt) for help with scripting. We thank the Hans und Ilse Breuer-Stiftung for financial support. K.S. is a Leukemia and Lymphoma Society Scholar, and this work was supported in part by R01 CA140292.
Footnotes
Molecular docking, determination of aqueous solubility, in vitro activity in GSK-3α (h) and GSK-3β (h), in vitro activity in AML cell lines, in vivo on golden zebrafish embryo. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Theresa Neumann, Email: theresa_neumann@web.de.
Boris Schmidt, Email: schmidt_boris@t-online.de.
References and notes
- 1.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2(10):769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 2.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359(1):1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal Structure of Glycogen Synthase Kinase 3β: Structural Basis for Phosphate-Primed Substrate Specificity and Autoinhibition. Cell. 2001;105(6):721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 4.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29(2):95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Lei P, Ayton S, Bush AI, Adlard PA. GSK-3 in Neurodegenerative Diseases. Int J Alzheimers Dis. 2011;2011:189246. doi: 10.4061/2011/189246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko Z, Cocco L, Martelli AM, Cervello M. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5(10):2881–2911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhat RV, Budd Haeberlein SL, Lindquist JM. Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors. John Wiley & Sons, Inc; 2006. Inhibition of GSK-3 as Therapeutic Strategy in Disease: Efficacy of AR-A014418; pp. 243–255. [Google Scholar]
- 8.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo Monte F, Kramer T, Gu J, Anumala UR, Marinelli L, La Pietra V, Novellino E, Franco B, Demedts D, Van Leuven F, Fuertes A, Dominguez JM, Plotkin B, Eldar-Finkelman H, Schmidt B. Identification of Glycogen Synthase Kinase-3 Inhibitors with a Selective Sting for Glycogen Synthase Kinase-3α. J Med Chem. 2012;55(9):4407–4424. doi: 10.1021/jm300309a. [DOI] [PubMed] [Google Scholar]
- 10.Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, Kakoza RM, Chow KT, Ross L, Alexe G, Tolliday N, Inguilizian H, Galinsky I, Stone RM, DeAngelo DJ, Roti G, Aster JC, Hahn WC, Kung AL, Stegmaier K. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J Clin Invest. 2012;122(3):935–947. doi: 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:1–40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen Synthase Kinase 3α-Specific Regulation of Murine Hepatic Glycogen Metabolism. Cell Metab. 2007;6(4):329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Kaidanovich-Beilin O, Lipina T, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers J, Fletcher P, MacAulay K, Doble B, Henkelman M, Miyakawa T, Roder J, Woodgett J. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2(1):1–23. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoeflich KP, Luo J, Rubie EA, Tsao M-S, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3[beta] in cell survival and. Nature. 2000;406(6791):86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 15.Brown CMS, Larsen SR, Iland HJ, Joshua DE, Gibson J. Leukaemias into the 21st century: part 1: the acute leukaemias. Intern Med J. 2012;42(11):1179–1186. doi: 10.1111/j.1445-5994.2012.02938.x. [DOI] [PubMed] [Google Scholar]
- 16.Elert E. Living with leukaemia. Nature. 2013;498(7455):S2–S3. doi: 10.1038/498S2a. [DOI] [PubMed] [Google Scholar]
- 17.Molecular Operating Environment (MOE) 2014.09. Chemical Computing Group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2014. [Google Scholar]
- 18.Neudert G, Klebe G. DSX: A Knowledge-Based Scoring Function for the Assessment of Protein–Ligand Complexes. J Chem Inf Model. 2011;51(10):2731–2745. doi: 10.1021/ci200274q. [DOI] [PubMed] [Google Scholar]
- 19.Cerep-2. [accessed 26.03.2013]; http://www.cerep.fr/cerep/users/pages/catalog/Affiche_CondExp_Test.asp?test=2842.
- 20.Cerep. [accessed 26.03.2013]; http://www.cerep.fr/cerep/users/pages/catalog/Affiche_CondExp_Test.asp?test=2879.
- 21.Soft C. Chem Bio Draw Ultra. 2013. [Google Scholar]
- 22.Meijer L, Skaltsounis A-L, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-Selective Inhibitors Derived from Tyrian Purple Indirubins. Chemistry & Biology. 2003;10(12):1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Henderson JL, Kormos BL, Hayward MM, Coffman KJ, Jasti J, Kurumbail RG, Wager TT, Verhoest PR, Noell GS, Chen Y, Needle E, Berger Z, Steyn SJ, Houle C, Hirst WD, Galatsis P. Discovery and Preclinical Profiling of 3-[4-(Morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a Highly Potent, Selective, Brain Penetrant, and in Vivo Active LRRK2 Kinase Inhibitor. J Med Chem. 2015;58(1):419–432. doi: 10.1021/jm5014055. [DOI] [PubMed] [Google Scholar]
- 24.Test Guideline 107. OECD; Paris: 1981. [Google Scholar]
- 25.Nishiya N, Oku Y, Kumagai Y, Sato Y, Yamaguchi E, Sasaki A, Shoji M, Ohnishi Y, Okamoto H, Uehara Y. A Zebrafish Chemical Suppressor Screening Identifies Small Molecule Inhibitors of the Wnt/β-catenin Pathway. Chem Biol (Oxford, U K) 2014;21(4):530–540. doi: 10.1016/j.chembiol.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Hassan FG, Mohaimen NA, Al-Maliki JG. The Wnt signal pathways. Int J Adv Res. 2014;2(8):816822. [Google Scholar]
- 27.Paquet D, Bhat R, Sydow A, Mandelkow E-M, Berg S, Hellberg S, Fälting J, Distel M, Köster RW, Schmid B, Haass C. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119(5):1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.