

Abstract
Autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis, multiple sclerosis, autoimmune hepatitis, and inflammatory bowel disease have complex pathogeneses and the courses of events leading to these diseases are not well understood. The immune surveillance is a delicate balance between self and foreign as well as between tolerance and immune response. Exposure to certain environmental factors may impair this equilibrium, leading to autoimmune diseases, cancer, and the so-called “lifestyle diseases” such as atherosclerosis, heart attack, stroke, and obesity, among others. These external stimuli may also alter the epigenetic status quo and may trigger autoimmune diseases such as SLE in genetically susceptible individuals. This review aims to highlight the role of epigenetic (dys-)regulation in the pathogenesis of SLE.
Electronic supplementary material
The online version of this article (doi:10.1007/s40744-015-0014-y) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Gene imprinting, Histone modifications, Methylation, Non-coding RNA, Nucleosome remodeling, Systemic lupus erythematosus (SLE)
Introduction
One interesting hypothesis concerning autoimmune diseases is that environmental effects on immune responses could be mediated by alterations in the epigenetic profile. Indeed, there is evidence that environmental factors may be the reason for the high discordance rate for autoimmune diseases in identical twins [1–4]. Advances in molecular genetics have illustrated that genomes are not a static entity for the deposition of genetic information. These findings imply dynamic response to external stimuli and a high genomic plasticity that is affected by epigenetic gene regulation. This kind of gene regulation relies on inducible and/or heritable patterns of gene expression, which are not based on changes of the genomic DNA sequence. Major mechanisms include DNA methylation, histone modification, non-coding RNA expression, gene imprinting and chromatin remodeling. Although in neither of these cases is gene expression modified by changes in the base sequence, these mechanisms interact with each other in a complex manner to regulate the expression and silencing of genes.
As epigenetic gene regulation is a new and highly promising research field (in autoimmune diseases), this article reviews the role of epigenetic regulation in systemic lupus erythematosus (SLE). This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
What is Epigenetics?
Although known before, the word “epigenetics” was introduced in modern science in 1942 by Conrad Hal Waddington, a British developmental biologist [5]. The concept of epigenetics is defined as the study of regulatory mechanisms that account for (potentially) heritable and reversible patterns in gene expression without affecting the nucleotide sequence of the genome. As the Greek prefix “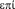 “ (epi) means “upon, over, on top of”, the “epigenome” is thought to be an additional, secondary informational level on top of the genetic code. A classic example of epigenetic regulation in mammals is the dosage compensation by silencing of one X chromosome in females. A condensed chromatin configuration prevents expression of genes on the silenced X chromosome, while the other X chromosome in the same nucleus is actively transcribed [6, 7]. It has been shown in humans that the silencing of individual gene loci by imprinting (in combination with micro deletions and mutations) leads to developmental abnormalities, known as Beckwith–Wiedemann, Angelman and Prader–Willi syndromes [8–10].
“ (epi) means “upon, over, on top of”, the “epigenome” is thought to be an additional, secondary informational level on top of the genetic code. A classic example of epigenetic regulation in mammals is the dosage compensation by silencing of one X chromosome in females. A condensed chromatin configuration prevents expression of genes on the silenced X chromosome, while the other X chromosome in the same nucleus is actively transcribed [6, 7]. It has been shown in humans that the silencing of individual gene loci by imprinting (in combination with micro deletions and mutations) leads to developmental abnormalities, known as Beckwith–Wiedemann, Angelman and Prader–Willi syndromes [8–10].
As the molecular basis of inheritance was unknown at that time, the term was initially used in an unspecific sense. This in conjunction with the description of the DNA double-helix structure by Watson and Crick, which demonstrated its eminent role in inheritance [11], “have cast a shadow over this discipline for decades” [12]. The term was reintroduced no more than four decades later, as studies on chromatin structure had identified the molecular basis of epigenetics.
The year 1974 marked the “birth-year” of modern, molecular-based epigenetics. There, Kornberg and colleagues published that chromatin is “a repeating unit of histones and DNA” [13]. These repetitive units were then called “nucleosomes”. However, it took until 1996 before two studies provided the first clear connection between histone acetylation and transcriptional regulation [14, 15] and it took further 4 years before a functional link between histone methylation and chromatin structure could be established [16].
In transcription and DNA replication, changes in the chromatin structure are essential to overcome steric hindrances for DNA binding factors [17–19]. These fundamental alterations are called “chromatin remodeling”. Chromatin remodeling is, amongst other things, based on post-translational modifications of histones by histone-modifying enzymes, which induce a complex cascade of post-translational modifications that can either activate or repress transcription [20]. This has led to the notion that defined patterns of histone modifications alter the structure of higher order chromatin to recruit effector molecules [21]. The different combinations of these modifications are thought to constitute a code, which has led to the so-called “histone code” hypothesis [20].
General Epigenetic Mechanisms
Among the approximately 3 billion base pairs of the mammalian genome, there are 20,000–30,000 protein-coding genes [22–24], which need instructions for where and when to be expressed or silenced. Only the accurate interplay of these genes results in functioning cells, organs and organisms. Like cancer and other complex diseases, autoimmune diseases seem to be the result of multistep processes in which genetic predisposition and epigenetic alterations interact and contribute to the pathological changes of this susceptible interplay.
Two major groups of cellular compounds are affected by epigenetic changes: the genome and the histones. Major mechanisms of epigenetic gene regulation include DNA methylation, histone modification, non-coding RNA expression, gene imprinting, and chromatin remodeling (Fig. 1). Mainly, three types of epigenetic modifications of chromatin transition are known: histone hypoacetylation, methylation of lysine 9 of histone H3 [Me(Lys9)H3], and DNA methylation at CpG dinucleotides. However, Me(Lys9)H3 is typically not found on regulated genes. It is characteristic for so-called constitutive heterochromatin, whereas genes that are shut off are typically localized in ‘facultative’ heterochromatin, which is marked by tri-methylated lysine 27 on histone H3 (H3K27me3).
Fig. 1.

Epigenetics and autoimmune disease. Five different epigenetic mechanisms have been identified so far: histone modification, non-coding RNA expression, DNA methylation, gene imprinting, and chromatin remodeling. These mechanisms interact with each other in a complex manner to regulate the expression and silencing of genes. Environmental factors may be the reason for the (high) discordance rate for autoimmune diseases in identical twins. These environmental factors may alter the epigenetic status quo and may trigger autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS), inflammatory bowel disease, as well as autoimmune diabetes, thyroid disease and hepatitis
Histones are subject to diverse post-translational modifications including acetylation, methylation, phosphorylation, and ubiquitylation [21, 25, 26]. In 1996, Taunton et al. [15] identified the first histone deacetylase (HDAC), a human homolog of the yeast transcriptional regulator Rpd3p. Since that time, large and ancient families of HDACs have been identified in yeast as well as in mammals [27–29]. It is believed that HDACs reverse the regulatory acetylation of histone proteins and silence genes by stabilizing a transcription-incompetent condition of nucleosomes [27]. It has been demonstrated in animal models that HDAC inhibitors are therapeutic for several inflammatory diseases [30], suggesting a potential therapeutic use of these inhibitors in autoimmune diseases.
Methylation of lysine 9 of histone H3 is catalyzed by the mammalian homolog of the fruit fly suppressor of variegation 3–9 [Su(var)3–9], which is a histone lysine methyltransferase that selectively methylates histone H3 at this site [16]. This modification generates a binding site for HP1 proteins, a family of heterochromatic transcriptional repressors that establish a repressed chromatin state [31–33], which is also called “gene silencing”.
DNA methylation patterns are established and maintained by methyltransferases [34]. Any deletion of these enzymes is lethal during embryogenesis [35–37]. Methylated cytosines within CpG dinucleotides are recognized by methyl-CpG binding proteins that recruit HDACs and thereby induce an inhibitory chromatin configuration [38]. Histone and DNA modifications are highly dynamic, a property that is crucial for the regulation and control of cellular proliferation, differentiation and survival [39]. Moreover, they are substantial constituents of the so-called “epigenetic code” [40].
In the last three decades, major advances have been made in understanding the interaction between DNA methylation, histone modification, and gene expression. This fundamental research demonstrated that the interplay between the individual components is highly complex and opened the new field of epigenetics. In the last few years, it became evident that epigenetic changes are not only involved in cancer and developmental processes but also play a significant role in the etiopathology of autoimmune diseases.
Epigenetic Changes in Autoimmune Diseases
The mechanisms underlying epigenetic changes are of great importance to human autoimmune diseases. However, they are poorly understood (Fig. 2). Over the years, increasing evidence has demonstrated the important role for aberrant epigenetics in the pathogenesis of SLE [41].
Fig. 2.

Possible roles of epigenetic alterations involved in SLE pathogenesis. Genetic disposition in combination with environmental factors can alter epigenetic marks, such as DNA (de-)methylation, histone modifications and transcriptional regulation by non-coding RNA. These epigenetic modifications may lead to aberrant gene expression profiles in (autoreactive) T cells that activate proinflammatory and repress anti-inflammatory genes. Aberrant and continuous expression of chemokines and cytokines mobilizes autoreactive B cells, which may trigger and aggravate SLE. SLE systemic lupus erythematosus
A number of genes have been claimed to be associated with susceptibility to anti-self responses. Because of their considerable heterogeneity, the immunoglobulin genes, the T cell receptor genes and the major histocompatibility complex (MHC) genes have soon been suspected of playing a distinct role in the pathology of SLE and other autoimmune diseases. In the past few years, progress has been made in identifying SLE susceptibility genes in mice [42]. However, these models only try to decipher the complex genetic background of SLE, but not the comparably complex environmental background of the disease. In general, mice are housed under (standardized) specific pathogen-free conditions and, thus, do not fully resemble the human disease. Interestingly, the data on several autoimmune diseases (including SLE) disclose increasing disease rates over the past few decades [43]. As genetic determinants are unlikely to alter disease rates within such short intervals, any rapid change indicates an environmental influence [43].
Since Holliday and Pugh proposed that “cytoplasmic components can have a powerful or overriding influence on genomic activity” in 1975 [44], many investigations broadened our understanding of epigenetic alterations in the pathogenesis of various complex disorders, including cancer and autoimmunity. One important epigenetic mechanism is the cytosine methylation/demethylation “switch” of regulatory DNA sequences. Highly simplified, methylation inactivates transcription, while hypomethylation associated with the activation of genes [45–47]. CpG dinucleotides are found at a lower frequency in the genome than would be expected due to random distribution, a phenomenon called “CG suppression”. This loss of CpGs has been explained by a spontaneous deamination of methylated cytosine residues [48]. In contrast, regions with a higher CpG content are found in approximately 40% of mammalian promoters [49]. These accumulations are called “CpG islands” [38, 50] and in most instances the CpG sites of these CpG islands are unmethylated if the genes are expressed. Environmentally induced hypomethylation of proinflammatory genes on the one hand and hypermethylation of anti-inflammatory genes on the other hand may have the effect that genetically susceptible individuals come down with an autoimmune disease.
How is SLE Influenced by Epigenetic Changes?
T cells from patients with active SLE have a 17% decrease in genomic deoxymethylcytosine content [51] and the inhibition of T cell DNA methylation causes autoreactivity in vitro and a SLE-like disease in vivo [51]. In murine models of drug-induced lupus erythematosus, it has been shown that mice receiving CD4+ T cells treated with demethylating agents, including procainamide and hydralazine, develop a SLE-like disease [52]. These drugs also inhibit T cell DNA methylation and induce autoreactivity in cloned T cell lines [53, 54]. CD4+ T cells treated with DNA methylation inhibitor 5-azacytidine become autoreactive and respond to self-class II MHC without the addition of exogenous antigen [55]. In lupus-prone MRL/lpr mice, defective DNA methylation and CD70 overexpression in CD4+ T cells could be detected [56].
It is well known that an exposure to ultraviolet light can trigger lupus flares [57, 58]. Moreover, brief exposures of T cells to ultraviolet light induce DNA hypomethylation and T cell autoreactivity [59], which supports an association between T cell DNA hypomethylation and autoimmunity [60]. Lymphocyte function-associated antigen-1 (LFA1) is a heterodimer consisting of the integrin alpha L (ITGAL) and the beta 2 chain (ITGB2), which is expressed on all leukocytes. In T cells from SLE, patients’ sequences flanking the ITGAL gene promoter region were demethylated, suggesting a mechanism for LFA-1 overexpression on an autoreactive subset of T cells [51]. Indeed, overexpression of LFA-1 [60, 61] and CD70 (TNFSF7) [62, 63], which, in turn, induces autoantibody synthesis in B cells [64], is thought to be involved in T cell autoreactivity in SLE. Additional studies have confirmed that DNA hypomethylation and histone hyperacetylation of CD11a and CD70 promoter regions contribute to their overexpression in SLE CD4+ T cells [51, 65, 66]. Hypermethylation of MHC class II transactivator (MHC2TA) and downregulation of human leukocyte antigen (HLA)-DR and MHC2TA could also be observed [67]. Different blood cell populations of SLE patients are characterized by a global loss of DNA methylation [68]. For instance, persistent hypomethylation of interferon genes and interferon-regulated genes can be found in CD4+ T cells [69], CD19+ B cells, CD14+ monocytes [70], and neutrophils [71] of patients with SLE. This process is associated with defects in extracellular-signal-regulated kinases (ERK) pathway signaling and consequent downregulation of the methyltransferase DNMT 1 [72]. These studies indicate that T cells hypomethylated by treatment with DNA methyltransferase inhibitors or ERK pathway inhibitors are sufficient to induce a SLE-like disease [73]. Recently, it could be shown that female but not male mice with an inducible ERK defect developed SLE-like symptoms in a transgenic mouse model, demonstrating ERK-dependent female predisposition for SLE [74]. Gorelik et al. [75] traced the SLE ERK pathway defect to impaired protein kinase C delta (PKCδ) phosphorylation. Additionally identified demethylation targets in SLE are genes involved in inflammation (CD40LG) [74, 76], cytokine pathway (IL-4 [77, 78], IL-6 [77, 79], IL-10 [80], IL-13 [80]) and IL1R2 [81], respectively) and cell lysis (perforin [82, 83]), which all can increase inflammation by stimulating the immune system.
Another example concerns the overexpression of the transcription regulatory factor cAMP-responsive element modulator alpha (CREMα) in T cells of patients with SLE and lupus-prone MRL/lpr mice. It binds to the CRE site in the promoter region of genes and contributes to epigenetic remodeling through the recruitment of DNA methyltransferase DNMT3A [84]. Then, DNMT3A mediates CpG hypomethylation, remodeling the CD8 cluster [85] and silencing of IL2 and IL17A [84]. On the other hand, it has recently been shown, that an increased histone H3 lysine 27 trimethylation enrichment at the hematopoietic progenitor kinase 1 (HPK1) promoter of SLE CD4+ T cells (relative to controls) inhibits the HPK1 expression and contributes to autoimmunity in SLE [86]. All these reports support a role for epigenetic DNA alterations in the pathogenesis of SLE.
However, DNA methylation is not a process whose effects are restricted to the DNA. As the methylation of DNA maintains chromatin in a condensed and hence, more inactive configuration, it acts synergistically or antagonistically on the diverse modifications of histone proteins [87]. For instance, hypomethylated CpG island chromatin is enriched in hyperacetylated histones and deficient in linker histones [88]. DNA methylation may also protect individuals from autoimmune diseases, such as SLE: as the estrogen receptor becomes hypermethylated during aging [89], this change may reduce the risk for women to come down with SLE or other sex-related autoimmune diseases. The identification of further genes that are deregulated by DNA (de-)methylation will successively account for a better understanding of the aberrant physiological pathways of SLE.
CD4+ lymphocytes undergo global histone H3 and H4 deacetylation and consequent skewed gene expression [68]. However, currently little is known about the relationship between alterations of histone modification patterns and SLE. However, histone H3 and H4 hypoacetylation as well as a lysine 9 on H3 (H3K9) hypomethylation have been described for SLE-derived CD4+ cells [90]. The same group observed increased histone H3 acetylation and dimethylated H3 lysine 4 (H3K4me2) levels in the TNFSF7 (CD70) gene promoter region in SLE CD4+ T cells, which correlated positively with disease activity [91]. Histone acetylation is regulated by histone acetyltransferases (HATs) and HDACs [92, 93]. Previous studies have shown that HDAC2 and HDAC7 levels are downregulated in active lupus erythematosus CD4+ T cells [90] and HDAC7 is decreased in MRL-lpr/lpr mice [94]. Moreover, it could be shown that the specific class I and II HDAC inhibitor ITF2357 was able to ameliorate SLE-like symptoms in NZB/W mice through regulation of T cell profiles [95].
Further evidence for a substantial role of histone modification in SLE pathogenesis comes from Lu and coworkers. They found that the transcription factor regulatory factor X-box 1 (RFX1) regulates CD70 and CD11a expression in T cells of patients with SLE by recruiting histone methyltransferase SUV39H1 [96]. The downregulation of RFX1 contributes to DNA hypomethylation and histone H3 hyperacetylation at the CD11a and CD70 promoters in CD4+ T cells of patients with SLE, which trigger immune responses [66].
A recent study detected a correlation between the 5-hydroxymethylcytosine (5-hmC) level in the peripheral blood and SLE [97]. The oxidation of 5-methylcytosine (5-mC) to 5-hmC is an epigenetic mechanism which is present in the DNA of mammalian cells. First seen in bacteriophages in 1952 [98], it was then found in high levels in neurons of the central nervous system in human and mouse as well as in embryonic stem cells [99, 100]. The exact function of this sixth DNA base is not fully understood, but it is thought to regulate gene expression and prompt DNA demethylation. This hypothesis is supported by the observation that hydroxylation of 5-mC to 5-hmC by TET1 actively promotes DNA demethylation [101]. Reduction of hmC levels in DNA is also a hallmark of cancers and, contrary to DNA methylation, which occurs immediately during replication, hmC forms slowly during the first 30 h following DNA synthesis [102].
Role of Micro RNA in SLE Pathogenesis
Micro RNA (miRNA) was initially discovered in 1993 [103] but little attention was given to these small RNAs until 2001 [104–108]. miRNAs are an important class of endogenous regulatory small RNAs [109, 110] which (amongst others) regulate the expression of genes involved in immune activation [111]. For cancer, it has been demonstrated that the miR-29 family induces DNA hypomethylation by directly targeting DNA methyltransferases thereby leading to a re-expression of hypermethylated silenced tumor suppressor genes [112, 113]. These studies have shown that miRNAs are involved in disease pathogenesis by targeting DNA methylation. miRNAs are also implicated in the pathology of SLE [114, 115]. One of these miRNAs, miR-146a, is a negative regulator of the IFN pathway. Underexpression of miR-146a contributes to alterations in the type I IFN pathway in lupus patients by targeting the key signaling proteins [114]. Dai et al. [116] identified several miRNAs that are differentially expressed in the peripheral blood mononuclear cells of SLE patients whose expression profiling may provide a useful clue for the etiology of SLE. Expression of miR-21 and miR-148a is highly upregulated in CD4+ T cells from both patients with SLE and MRL/lpr mice [117]. These two miRNAs promote CD4+ T cell hypomethylation by repressing DNA (cytosine-5)-methyltransferase 1 (DNMT1) expression and inducing the expression of autoimmune-associated methylation-sensitive genes [117]. Another miRNA that is upregulated in CD4+ T cells from SLE patients is miR-126 [118]. The overexpression of miR-126 in CD4+ T cells from healthy donors caused demethylation and upregulation of the genes ITGAL (encoding CD11a) and CD70, thereby causing T cell and B cell hyperactivity. It could also be shown that the expression of the mir-126 host gene EGFL7 was upregulated in CD4+ T cells from patients with SLE and that the degree of overexpression is associated with the hypomethylation of its promoter [119]. CCL5 (RANTES) is a chemokine expressed by circulating T cells which recruits leukocytes to sites of inflammation. It could be shown that serum levels of RANTES were significantly elevated in patients with SLE when compared with normal controls [120]. miR-125a negatively regulates RANTES expression by targeting KLF13 in activated T cells [121]. Thus, underexpression of miR-125a contributes to the elevated expression of RANTES in SLE. Tissue factor (TF) is the main initiator of the blood coagulation cascade and it could be shown that monocytes of patients with SLE are characterized by a high TF expression and low miR-19b and -20a levels [122]. Reporter assays demonstrated that miR-20a binds to TF mRNA [123]. Thus, downregulation of miR-19b and miR-20a could contribute to increased TF expression provoking the hypercoagulable state characteristic of patients with SLE. Further miRNAs involved in SLE are miRNA-3148 [124], miRNA-1246 [125], and miRNA-let7A [126], respectively. Excessive activation of the innate immune system involving toll-like receptor 7 (TLR7) has been recognized as an important pathogenic mechanism in SLE [127] and miR-3148, with a predicted binding site at the 3′-untranslated region (3′-UTR) of TLR7 mRNA, modulates the allelic expression of this gene. Individuals carrying the G allele of the single nucleotide polymorphism (SNP) rs3853839 in the 3′-UTR of the TLR7 gene exhibited increased TLR7 expression at both the mRNA and protein level and decreased transcript degradation [124]. In contrast, in people bearing the non-risk C allele of this SNP, miR-3148 perfectly matches the 3′-UTR of the TLR7-mRNA, leading to a faster and more effective degradation of non-risk allele containing TLR7 transcripts. For miR-1246, it was shown that its expression was significantly decreased in B cells from SLE patients and that it specifically targeted the 3′-UTR of the early B cell factor 1 (EBF1) mRNA [125]. These findings provide a causal role of miR-1246 in the pathogenesis of SLE: EBF1 contributes to the development, activation, and proliferation of B cells through activation of the AKT signaling pathway. A downregulation of the miR-1246 expression may decrease the degradation rate of the EBF1 mRNA, leading to a B cell overactivation in patients with SLE. Let-7a is implicated in SLE pathogenesis due to its responsiveness to immune stimulation and its reported inflammatory targets [128, 129]. It could be shown that its overexpression may contribute to hyperplasia and a proinflammatory response, including inflammatory mediator production. Recent studies have shown that a significant fraction of miRNAs themselves is regulated by epigenetic mechanisms [130–132], demonstrating the entire complexity of eukaryotic gene regulation.
We now know that changes in DNA methylation, mRNA, and miRNA expression are characteristic for SLE and correlate with the phenotype of this severe disease [133]. However, more studies are required to consolidate the role of miRNAs in SLE pathology. Until then, the question of if these non-coding RNAs are “hope or hype” [134] remains unanswered for SLE.
Conclusion
Deciphering the contribution of epigenetic alterations to the pathogenesis of SLE will provide promising insights in this complex autoimmune disease. Epigenetic alterations are (potentially) reversible and hence candidates for the development of new therapeutics. However, to attain this goal, many questions remain to be answered in the promising field of epigenetics.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors wish to thank Prof. René Ketting for his valuable comments and suggestions to improve the manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval of the version to be published. This work was supported by a grant of the ‘‘Rheinland-Pfalz Stiftung fur Innovation’’ (Grant 1032).
Conflict of interest
Manfred Relle, Bernd Föhr, and Andreas Schwarting declare that they have no conflict of interest.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.Block SR, et al. Studies of twins with systemic lupus erythematosus. A review of the literature and presentation of 12 additional sets. Am J Med. 1975;59(4):533–552. doi: 10.1016/0002-9343(75)90261-2. [DOI] [PubMed] [Google Scholar]
- 2.Block SR, et al. Proceedings: twin studies in systemic lupus erythematosus (SLE) Arthritis Rheum. 1975;18(3):285. [PubMed] [Google Scholar]
- 3.Gregersen PK. Discordance for autoimmunity in monozygotic twins. Are “identical” twins really identical? Arthritis Rheum. 1993;36(9):1185–1192. doi: 10.1002/art.1780360902. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan D. The onset of disease in twins and siblings with systemic lupus erythematosus. J Rheumatol. 1984;11(5):648–652. [PubMed] [Google Scholar]
- 5.Waddington CH. The epigenotype. Endeavour. 1942;1:18–20. [Google Scholar]
- 6.Valley CM, Willard HF. Genomic and epigenomic approaches to the study of X chromosome inactivation. Curr Opin Genet Dev. 2006;16(3):240–245. doi: 10.1016/j.gde.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14(1):9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 8.Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):365–376. doi: 10.1002/ajmg.c.30273. [DOI] [PubMed] [Google Scholar]
- 9.Henry I, et al. Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature. 1991;351(6328):665–667. doi: 10.1038/351665a0. [DOI] [PubMed] [Google Scholar]
- 10.Viljoen D, Ramesar R. Evidence for paternal imprinting in familial Beckwith-Wiedemann syndrome. J Med Genet. 1992;29(4):221–225. doi: 10.1136/jmg.29.4.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 12.Cavalli G. Chromatin and epigenetics in development: blending cellular memory with cell fate plasticity. Development. 2006;133(11):2089–2094. doi: 10.1242/dev.02402. [DOI] [PubMed] [Google Scholar]
- 13.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(139):868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 14.Brownell JE, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84(6):843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 15.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272(5260):408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 16.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 17.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Groth A, et al. Chromatin challenges during DNA replication and repair. Cell. 2007;128(4):721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 19.Kohn KW, et al. Chromatin challenges during DNA replication: a systems representation. Mol Biol Cell. 2008;19(1):1–7. doi: 10.1091/mbc.E07-06-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11(11):1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 21.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 22.Church DM, et al. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009;7(5):e1000112. doi: 10.1371/journal.pbio.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 24.Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 25.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13(9):373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner BM. Histone acetylation and an epigenetic code. BioEssays. 2000;22(9):836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262(2):75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 29.Leipe DD, Landsman D. Histone deacetylases, acetoin utilization proteins and acetylpolyamine amidohydrolases are members of an ancient protein superfamily. Nucleic Acids Res. 1997;25(18):3693–3697. doi: 10.1093/nar/25.18.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shakespear MR, et al. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32(7):335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Wallrath LL. Unfolding the mysteries of heterochromatin. Curr Opin Genet Dev. 1998;8(2):147–153. doi: 10.1016/s0959-437x(98)80135-4. [DOI] [PubMed] [Google Scholar]
- 32.Kwon SH, Workman JL. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cells. 2008;26(3):217–227. [PubMed] [Google Scholar]
- 33.Lomberk G, et al. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol. 2006;8(4):407–415. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- 34.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 35.Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 36.Lei H, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122(10):3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 37.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 38.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 41.Ballestar E, Esteller M, Richardson BC. The epigenetic face of systemic lupus erythematosus. J Immunol. 2006;176(12):7143–7147. doi: 10.4049/jimmunol.176.12.7143. [DOI] [PubMed] [Google Scholar]
- 42.Wakeland EK, et al. Genetic dissection of systemic lupus erythematosus. Curr Opin Immunol. 1999;11(6):701–707. doi: 10.1016/s0952-7915(99)00039-4. [DOI] [PubMed] [Google Scholar]
- 43.Shapira Y, Agmon-Levin N, Shoenfeld Y. Geoepidemiology of autoimmune rheumatic diseases. Nat Rev Rheumatol. 2010;6(8):468–476. doi: 10.1038/nrrheum.2010.86. [DOI] [PubMed] [Google Scholar]
- 44.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–232. [PubMed] [Google Scholar]
- 45.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 46.Richardson B, Yung R. Role of DNA methylation in the regulation of cell function. J Lab Clin Med. 1999;134(4):333–340. doi: 10.1016/s0022-2143(99)90147-6. [DOI] [PubMed] [Google Scholar]
- 47.Nakao M. Epigenetics: interaction of DNA methylation and chromatin. Gene. 2001;278(1–2):25–31. doi: 10.1016/s0378-1119(01)00721-1. [DOI] [PubMed] [Google Scholar]
- 48.Cooper DN, Krawczak M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum Genet. 1989;83(2):181–188. doi: 10.1007/BF00286715. [DOI] [PubMed] [Google Scholar]
- 49.Fatemi M, et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33(20):e176. doi: 10.1093/nar/gni180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 51.Lu Q, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002;46(5):1282–1291. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 52.Richardson B, Ray D, Yung R. Murine models of lupus induced by hypomethylated T cells. Methods Mol Med. 2004;102:285–294. doi: 10.1385/1-59259-805-6:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornacchia E, et al. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140(7):2197–2200. [PubMed] [Google Scholar]
- 54.Scheinbart LS, et al. Procainamide inhibits DNA methyltransferase in a human T cell line. J Rheumatol. 1991;18(4):530–534. [PubMed] [Google Scholar]
- 55.Richardson B. Effect of an inhibitor of DNA methylation on T cells. II. 5-Azacytidine induces self-reactivity in antigen-specific T4+ cells. Hum Immunol. 1986;17(4):456–470. doi: 10.1016/0198-8859(86)90304-6. [DOI] [PubMed] [Google Scholar]
- 56.Sawalha AH, Jeffries M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. Eur J Immunol. 2007;37(5):1407–1413. doi: 10.1002/eji.200636872. [DOI] [PubMed] [Google Scholar]
- 57.Caricchio R, McPhie L, Cohen PL. Ultraviolet B radiation-induced cell death: critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J Immunol. 2003;171(11):5778–5786. doi: 10.4049/jimmunol.171.11.5778. [DOI] [PubMed] [Google Scholar]
- 58.Sanders CJ, et al. Photosensitivity in patients with lupus erythematosus: a clinical and photobiological study of 100 patients using a prolonged phototest protocol. Br J Dermatol. 2003;149(1):131–137. doi: 10.1046/j.1365-2133.2003.05379.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhu XH, et al. Effects of ultraviolet B exposure on DNA methylation in patients with systemic lupus erythematosus. Exp Ther Med. 2013;5(4):1219–1225. doi: 10.3892/etm.2013.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richardson B, et al. Lymphocyte function-associated antigen 1 overexpression and T cell autoreactivity. Arthritis Rheum. 1994;37(9):1363–1372. doi: 10.1002/art.1780370915. [DOI] [PubMed] [Google Scholar]
- 61.Yung R, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97(12):2866–2871. doi: 10.1172/JCI118743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo Y, Zhao M, Lu Q. Demethylation of promoter regulatory elements contributes to CD70 overexpression in CD4+ T cells from patients with subacute cutaneous lupus erythematosus. Clin Exp Dermatol. 2010;35(4):425–430. doi: 10.1111/j.1365-2230.2009.03611.x. [DOI] [PubMed] [Google Scholar]
- 63.Balada E, et al. Associations between the expression of epigenetically regulated genes and the expression of DNMTs and MBDs in systemic lupus erythematosus. Plos One. 2012;7(9):e45356. [DOI] [PMC free article] [PubMed]
- 64.Kobata T, et al. CD27-CD70 interactions regulate B-cell activation by T cells. Proc Natl Acad Sci. 1995;92(24):11249–11253. doi: 10.1073/pnas.92.24.11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005;174(10):6212–6219. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 66.Zhao M, et al. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J Autoimmun. 2010;35(1):58–69. doi: 10.1016/j.jaut.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 67.Rupasree Y, et al. Epigenetic modulation of RFC1, MHC2TA and HLA-DR in systemic lupus erythematosus: association with serological markers and six functional polymorphisms of one-carbon metabolic pathway. Gene. 2014;536(1):45–52. doi: 10.1016/j.gene.2013.11.094. [DOI] [PubMed] [Google Scholar]
- 68.Javierre BM, Richardson B. A new epigenetic challenge: systemic lupus erythematosus. Adv Exp Med Biol. 2011;711:117–136. doi: 10.1007/978-1-4419-8216-2_9. [DOI] [PubMed] [Google Scholar]
- 69.Coit P, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Absher DM, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. Plos Genet. 2013;9(8):e1003678. [DOI] [PMC free article] [PubMed]
- 71.Coit P, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. 2015;58:59–66. doi: 10.1016/j.jaut.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sawalha AH, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9(4):368–378. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gorelik G, Richardson B. Key role of ERK pathway signaling in lupus. Autoimmunity. 2010;43(1):17–22. doi: 10.3109/08916930903374832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strickland FM, et al. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J Autoimmun. 2011;38(2–3):J135–43. [DOI] [PMC free article] [PubMed]
- 75.Gorelik GJ, Yarlagadda S, Richardson BC. Protein kinase C delta oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum. 2012;64(9):2964–2974. doi: 10.1002/art.34503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou Y, et al. T cell CD40LG gene expression and the production of IgG by autologous B cells in systemic lupus erythematosus. Clin Immunol. 2009;132(3):362–370. doi: 10.1016/j.clim.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mi XB, Zeng FQ. Hypomethylation of interleukin-4 and -6 promoters in T cells from systemic lupus erythematosus patients. Acta Pharmacol Sin. 2008;29(1):105–112. doi: 10.1111/j.1745-7254.2008.00739.x. [DOI] [PubMed] [Google Scholar]
- 78.Richardson B. DNA methylation and autoimmune disease. Clin Immunol. 2003;109(1):72–79. doi: 10.1016/s1521-6616(03)00206-7. [DOI] [PubMed] [Google Scholar]
- 79.Garaud S, et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol. 2009;182(9):5623–5632. doi: 10.4049/jimmunol.0802412. [DOI] [PubMed] [Google Scholar]
- 80.Zhao M, et al. Hypomethylation of IL10 and IL13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:931018. doi: 10.1155/2010/931018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin SY, et al. A whole genome methylation analysis of systemic lupus erythematosus: hypomethylation of the IL10 and IL1R2 promoters is associated with disease activity. Genes Immun. 2012;13(3):214–220. doi: 10.1038/gene.2011.74. [DOI] [PubMed] [Google Scholar]
- 82.Kaplan MJ, et al. Demethylation of promoter regulatory elements contributes to perforin overexpression in CD4+ lupus T cells. J Immunol. 2004;172(6):3652–3661. doi: 10.4049/jimmunol.172.6.3652. [DOI] [PubMed] [Google Scholar]
- 83.Luo Y, et al. DNA demethylation of the perforin promoter in CD4(+) T cells from patients with subacute cutaneous lupus erythematosus. J Dermatol Sci. 2009;56(1):33–36. doi: 10.1016/j.jdermsci.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 84.Hedrich CM, et al. cAMP response element modulator alpha controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci. 2012;109(41):16606–16611. doi: 10.1073/pnas.1210129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hedrich CM, et al. cAMP responsive element modulator (CREM) alpha mediates chromatin remodeling of CD8 during the generation of CD3(+)CD4(−)CD8(−) T Cells. J Biol Chem. 2014;289(4):2361–2370. doi: 10.1074/jbc.M113.523605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Q, et al. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J Autoimmun. 2011;37(3):180–189. doi: 10.1016/j.jaut.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 87.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10(5):295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 88.Bird AP, Wolffe AP. Methylation-induced repression–belts, braces, and chromatin. Cell. 1999;99(5):451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 89.Issa JP, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 90.Hu N, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol. 2008;35(5):804–810. [PubMed] [Google Scholar]
- 91.Zhou Y, et al. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus. 2011;20(13):1365–1371. doi: 10.1177/0961203311413412. [DOI] [PubMed] [Google Scholar]
- 92.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20(8):615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 93.Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011;2011:371832. doi: 10.1155/2011/371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Long H, et al. Abnormal expression pattern of histone demethylases in CD4(+) T cells of MRL/lpr lupus-like mice. Lupus. 2009;18(14):1327–1328. doi: 10.1177/0961203309104869. [DOI] [PubMed] [Google Scholar]
- 95.Regna NL, et al. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin Immunol. 2014;151(1):29–42. doi: 10.1016/j.clim.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao M, et al. RFX1 regulates CD70 and CD11a expression in lupus T cells by recruiting the histone methyltransferase SUV39H1. Arthritis Res Ther. 2011;12(6):R227. doi: 10.1186/ar3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sui W, et al. Genome-wide analysis of 5-hmC in the peripheral blood of systemic lupus erythematosus patients using an hMeDIP-chip. Int J Mol Med. 2015;35(5):1467–1479. doi: 10.3892/ijmm.2015.2149. [DOI] [PubMed] [Google Scholar]
- 98.Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952;170(4338):1072–1073. doi: 10.1038/1701072a0. [DOI] [PubMed] [Google Scholar]
- 99.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science. 2009;324(5929):929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo JU, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bachman M, et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6(12):1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 104.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 105.Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004;116(2 Suppl):S89–92 (1 p following S96). [DOI] [PubMed]
- 106.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15(2):188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lagos-Quintana M, et al. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 108.Lau NC, et al. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 109.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 110.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stagakis E, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70(8):1496–1506. doi: 10.1136/ard.2010.139857. [DOI] [PubMed] [Google Scholar]
- 112.Garzon R, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fabbri M, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci. 2007;104(40):15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- 115.Te JL, et al. Identification of unique microRNA signature associated with lupus nephritis. PLoS One. 2010;5(5):e10344. doi: 10.1371/journal.pone.0010344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dai Y, et al. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007;16(12):939–946. doi: 10.1177/0961203307084158. [DOI] [PubMed] [Google Scholar]
- 117.Pan W, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184(12):6773–6781. doi: 10.4049/jimmunol.0904060. [DOI] [PubMed] [Google Scholar]
- 118.Zhao S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011;63(5):1376–86. [DOI] [PubMed]
- 119.Liang Y, et al. DNA methylation status of miR-126 and its host gene EGFL7 in CD4+ T cells from patients with systemic lupus erythematosus. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2013;38(8):793–797. doi: 10.3969/j.issn.1672-7347.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 120.Lu MM, et al. Increased serum RANTES in patients with systemic lupus erythematosus. Rheumatol Int. 2012;32(5):1231–1233. doi: 10.1007/s00296-010-1761-2. [DOI] [PubMed] [Google Scholar]
- 121.Zhao X, et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010;62(11):3425–3435. doi: 10.1002/art.27632. [DOI] [PubMed] [Google Scholar]
- 122.Nojima J, et al. Tissue factor expression on monocytes induced by anti-phospholipid antibodies as a strong risk factor for thromboembolic complications in SLE patients. Biochem Biophys Res Commun. 2008;365(1):195–200. doi: 10.1016/j.bbrc.2007.10.173. [DOI] [PubMed] [Google Scholar]
- 123.Teruel R, et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J Thromb Haemost. 2011;9(10):1985–1992. doi: 10.1111/j.1538-7836.2011.04451.x. [DOI] [PubMed] [Google Scholar]
- 124.Deng Y, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. Plos Genet. 2013;9(2):e1003336. [DOI] [PMC free article] [PubMed]
- 125.Luo SY, et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clinical: Epigenetics; 2015. p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chafin CB, et al. MicroRNA-let-7a promotes E2F-mediated cell proliferation and NF kappa B activation in vitro. Cell Mol Immunol. 2014;11(1):79–93. doi: 10.1038/cmi.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kontaki E, Boumpas DT. Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun. 2010;35(3):206–211. doi: 10.1016/j.jaut.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 128.Dai Y, et al. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. 2009;29(7):749–754. doi: 10.1007/s00296-008-0758-6. [DOI] [PubMed] [Google Scholar]
- 129.Chafin CB, et al. MicroRNA-let-7a expression is increased in the mesangial cells of NZB/W mice and increases IL-6 production in vitro. Autoimmunity. 2013;46(6):351–362. doi: 10.3109/08916934.2013.773976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Han L, et al. DNA methylation regulates MicroRNA expression. Cancer Biol Ther. 2007;6(8):1284–1288. doi: 10.4161/cbt.6.8.4486. [DOI] [PubMed] [Google Scholar]
- 131.Lujambio A, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci. 2008;105(36):13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tang JT, et al. MicroRNA 345, a methylation-sensitive microRNA is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis. 2011;32(8):1207–1215. doi: 10.1093/carcin/bgr114. [DOI] [PubMed] [Google Scholar]
- 133.Zhao M, et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun. 2014;54:127–136. doi: 10.1016/j.jaut.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 134.Huttenhofer A, Schattner P, Polacek N. Non-coding RNAs: hope or hype? Trends Genet. 2005;21(5):289–297. doi: 10.1016/j.tig.2005.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.