

Abstract
Affibody molecules specific for H-Ras and Raf-1 were evaluated for their ability to inhibit synovial cell function. Affibody molecules targeting H-Ras (Zras122, Zras220, and Zras521) or Raf-1 (Zraf322) were introduced into the MH7A synovial cell line using two delivery methods: transfection with plasmids encoding the affibody molecules or direct introduction of affibody protein using a cell-penetrating peptide reagent. Interleukin-6 (IL-6) and prostaglandin E2 (PGE2) production by MH7A cells were analyzed by enzyme-linked immunosorbent assay after stimulation with tumor necrosis factor-alpha (TNF-α). Cell proliferation was also analyzed. Phosphorylation of extracellular signal-regulated kinase (ERK) was analyzed by western blot. All affibody molecules could inhibit IL-6 and PGE2 production in TNF-α-stimulated MH7A cells. The inhibitory effect was stronger when affibody molecules were delivered as proteins via a cell-penetrating peptide reagent than when plasmid-DNA encoding the affibody moelcules was transfected into the cells. Plasmid-expressed Zras220 inhibited phosphorylation of ERK in TNF-α-stimulated MH7A cells. Protein-introduced Zraf322 inhibited the production of IL-6 and PGE2 and inhibited cell proliferation in MH7A cells. These findings suggest that affibody molecules specific for H-Ras and Raf-1 can affect intracellular signal transduction through the MAP kinase pathway to inhibit cell proliferation and production of inflammatory mediators by synovial cells.
Electronic supplementary material
The online version of this article (doi:10.1186/s13568-014-0082-3) contains supplementary material, which is available to authorized users.
Keywords: Affibody molecules, Inflammation, Interleukin-6, Intracellular signal transduction, Prostaglandin E2, Rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is an inflammatory disease characterized by the progressive destruction of joints. In RA, hyperplasia of synovial lining cells is observed along with angiogenesis and inflammatory mononuclear cell infiltration (Kitano et al. [2006]). A wide variety of molecules produced by both synovial cells and infiltrating inflammatory mononuclear cells is thought to be involved in RA pathology. Specifically, proinflammatory cytokines such as interleukin-6 (IL-6), IL-17 (Schlegel et al. [2013]) and tumor necrosis factor alpha (TNF-α) play a key role in RA pathogenesis, including the activation of synovial fibroblasts, osteoclasts, and inflammatory mononuclear cells, which results in irreversible damage to the soft tissues and bones (Olsen and Stein [2004]).
One of the conventional disease-modifying antirheumatic drugs (DMARDs), methotrexate, is the standard for RA pharmacotherapy (Upchurch and Kay [2012]). However, its treatment outcomes are not satisfactory and often cause high levels of toxicity and adverse effects for patients (Smolen and Steiner [2003]). In recent years, various biological agents have been developed and used clinically as important options for RA treatment (Saag et al. [2008]). These biological agents include an anti-CD20 antibody (rituximab) (Mélet et al. [2013]), TNF antagonists (e.g., adalimumab, etanercept, golimumab, and infliximab), an IL-6 receptor antagonist (tocilizumab) (Smolen and Aletaha [2011]), and a recombinant fusion protein consisting of the extracellular domain of human CTLA-4 (abatacept) (Gómez-Reino [2012]). Although these biological agents provide favorable treatment outcomes for RA patients, in view of medical economics, biological therapies are more costly than traditional DMARDs. Thus, a more cost-effective treatment option is desirable because healthcare payers and providers are conscious of cost in light of the current period of budget constraints (Furneri et al. [2012]).
Several biologically active, non-immunoglobulin-based proteins have been created as candidates of novel biological agents. For instance, a monobody is a target-binding protein that is generated from the fibronectin type III domain (Koide et al. [2012]). Synthetic ankyrin repeat proteins have a binding scaffold and are potential diagnostic and therapeutic tools (Baumann et al. [2007]). In this study, we investigated another class of non-immunoglobulin ligands, referred to as affibody molecules. They are engineered binding proteins based on a scaffold of the three-α-helix bundle Z-domain (Friedman and Ståhl [2009]; Nilsson and Tolmachev [2007]; Nygren [2008]). The 6-kDa Z-domain is derived from the B-domain, one of the five domains in Staphylococcus aureus protein A (Gunneriusson et al. [1996]).
Affibody molecules associated with signal transduction or transcription in tumor development or immune responses have been screened and applied. For example, affibody molecules against c-Jun, an oncogenic transcription factor, have been selected and are able to detect c-Jun in a melanoma cell line (Lundberg et al. [2009]). Furthermore, other affibody molecules against human epidermal growth factor receptor (HER), specifically HER2 (Lindberg et al. [2012]) and HER3 (Malm et al. [2013]), have been investigated.
We have previously reported the selection of H-Ras-binding or Raf-1-binding affibody molecules (Grimm et al. [2010]). Affibody clones Zras122, Zras220, and Zraf322 were isolated by phage display selection from a library, and the specificity and affinity for H-Ras or Raf-1 proteins were determined in that study. Each molecule was demonstrated to selectively bind Ras or Raf-1 proteins, but not other human control proteins. Zras122, Zras220, and Zraf322 had binding affinities in the high nanomolar to low micromolar range, as demonstrated by a real-time biospecific interaction analysis. As an alternative or complement to current RA treatment regimens, these affibody molecules might have the potential to inhibit signal transduction mediated by Ras and Raf, since this signaling is thought to be important for efficient synthesis of several inflammatory mediators (Yamamoto et al. [2003]).
In this study, we investigated the effect of introducing H-Ras-binding and Raf-1-binding affibody molecules in the MH7A synovial cell line (Tsunemi et al. [2010]). First, we subcloned DNA encoding affibody molecules against H-Ras or Raf-1 into a plasmid DNA vector for mammalian expression, and the vectors were introduced into MH7A cells. Then, affibody proteins were produced using an Escherichia coli expression system and were introduced into MH7A cells. We investigated inhibition caused by H-Ras- or Raf-1-targeting affibody molecules on the production of the inflammatory mediators IL-6 and prostaglandin E2 (PGE2). Furthermore, we examined proliferation of MH7A cells.
Materials and methods
Cell line
The human MH7A synovial cell line (Riken, Saitama, Japan), which originated from intra-articular soft tissue of the knee joints of an RA patient, was established by transfection with the SV40 T antigen (Miyazawa et al. [1998]). MH7A cells were cultured in RPMI 1640 (Sigma, St. Louis, MO, USA) containing 10% heat-inactivated fetal bovine serum (Whittaker, Walkersville, MD, USA), 100 units/mL of penicillin, and 100 μg/mL of streptomycin (Invitrogen, CA, USA) at 37°C in an atmosphere of 5% CO2 in air.
Plasmid construction for affibodies and introduction into MH7A cells
The plasmids that were used to produce affibodies in mammalian cells were constructed by first amplifying DNA encoding anti-H-Ras and anti-Raf-1 affibodies (Zras122, Zras220, Zras521, and Zraf322) by PCR using primers 5'-AAGGGGATCCACCATGGGCAGCAGCCATCATCATCA-3' and 5'-AGGGGTTATGCTAGTTATTGCTCAGCGCGGAATTCTTA-3' (Grimm et al. [2010]). DNA sequences of affibody molecules are listed the additional file (Additional file 1). Each fragment was inserted into the multiple cloning site of pcDNA3.1(+) (Invitrogen). The nucleotide sequences of these constructs were confirmed by sequencing with an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Constructs were introduced into MH7A cells for transient expression using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Cells used for transfection were seeded at 1.7 × 105 cells/well in a 24-well plate (BD, Franklin Lakes, NJ, USA) and cultured for 1 d before transfection.
Western blot analysis of affibody expression
MH7A cells transfected with affibody-encoding plasmids (1 × 107) were plated in a 24-well plate. After 48 or 72 h of incubation, cells were lysed in Mammalian Lysis Buffer from the Qproteome Mammalian Protein Prep Kit (Qiagen, Hilden, Germany), and protein content was determined using the Bio-Rad Protein Assay Reagent (Bio-Rad, Hercules, CA, USA) with bovine serum albumin as the standard. Each sample was resolved by SDS-PAGE on 20% polyacrylamide gels and then transferred to 0.45-μm nitrocellulose membranes. After blocking overnight at 4°C with Blocking One Solution (Nacalai Tesque, Kyoto, Japan), membranes were incubated with goat polyclonal antibody specific for the affibody (Abcam, Cambridge, UK) as the primary antibody (1:400 dilution in phosphate-buffered saline; PBS) overnight at 4°C. After washing the membranes with Tris-buffered saline-0.05% Tween 20 (washing buffer), target proteins were detected with alkaline phosphatase (AP)-labeled ReserveAP Rabbit anti-goat immunoglobulin G (IgG) (KPL, Gaithersburg, MD, USA; 1:1,000 dilution in 1% PBS containing 0.05% Tween 20). NBT/BCIP (Roche, Mannheim, Germany) was used as the AP colorimetric substrate for visualization.
Measurement of IL-6 or PGE2 levels
MH7A cells (2 × 104) that were transfected with an affibody-encoding plasmid were plated in a 24-well plate and cultured in the presence or absence of 100 ng/mL TNF-α. After 24 or 48 h of incubation, IL-6 and PGE2 levels were determined by assaying supernatants with an Enzyme-Linked Immunosorbent Assay (ELISA) Kit for IL-6 (R&D Systems, Minneapolis, MN, USA) or PGE2 (ENZO Life Sciences, Farmingdale, NY, USA), respectively, according to the instructions.
Cell proliferation studies
Cell viability was determined using a Cell Counting Kit (CCK)-8 (Dojindo, Kumamoto, Japan), in which 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt was used as a substrate (Ohuchida et al. [2004]). Absorbance was measured using a SpectraMax M2 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm (Shibasaki et al. [2012]).
Western blot analysis for phosphorylated extracellular signal-regulated kinase (p-ERK)
Western blot analysis was performed as described above. Briefly, MH7A cells transfected with affibody-encoding plasmids (1 × 107) were plated in a 24-well plate, incubated for 24 h, and then stimulated with 100 ng/mL TNF-α (Sigma). After an additional incubation of 16 or 24 h, the soluble fraction of the cells was analyzed by western blot analysis using IgG against p-ERK (Santa Cruz Biotechnology, Dallas, TX, USA) as a primary antibody. Goat polyclonal IgG against actin (Santa Cruz) was used as a control.
Affibody protein production in E. coli
Expression of the pAffi-Zras122, -Zras220, -Zras521, and -Zraf322 plasmids in E. coli BL21(DE3) produced H-Ras and Raf-1 affibody molecules with an N-terminal 6 × His tag. The E. coli cells were inoculated in 2 mL of Luria-Bertani (LB) broth containing 100 μg/L ampicillin and were incubated in a shaking incubator overnight at 37°C. The 2-mL overnight cultures were used to inoculate fresh LB medium containing 100 μg/L ampicillin (125 mL), and cultures were grown at 37°C to an OD600 of 0.6-0.7. Gene expression was then induced by the addition of isopropyl β-D-thiogalactoside (Wako Pure Chemical, Osaka, Japan) to a final concentration of 1 mM. After 3-4 h of incubation at 37°C, the cells were harvested by centrifugation (4,000× g, 15 min, 4°C). The cell pellets were subsequently resuspended in 10 mL of B-PER Bacterial Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, MA, USA) and shaken gently for 10 min at room temperature (25°C). Cell debris was pelleted by centrifugation at 27,000 × g for 20 min. Supernatants containing soluble proteins were purified by recovering the 6 × His-Zras122, -Zras220, -Zras521, and -Zraf322 fusion proteins after passage through nickel-chelated agarose columns (Thermo Fisher Scientific). The columns were equilibrated with 10 mL of B-PER Bacterial Protein Extraction Reagent before application of the supernatant. After washing the column with B-PER wash buffer, the bound proteins were released with elution buffer (50 mM Tris, 300 mM NaCl, 200 mM imidazole, 10% [v/v] glycerol), according to a previous protocol (Shibasaki et al. [2013]). The concentration of each protein was determined by the BCA assay (REF), and the molecular size was confirmed by SDS-PAGE.
Affibody protein introduction into MH7A cells
MH7A cells were seeded at 1.7 × 105 cells/well in a 24-well plate and cultured for 1 d before the introduction of the purified proteins. Each affibody protein (5 μg) was mixed with Xfect Protein Buffer (Takara Bio, Shiga, Japan) to a total volume to 20 μL and then mixed with Xfect Protein Transfection Reagent. As a negative control, PBS was used in the place of the affibody molecule. After substitution of culture medium with serum-free medium for the cells, a diluted protein mixture was added to cultured MH7A cells and incubated at 37°C, according to a previous protocol (Kato et al. [2013]).
Statistical analysis
Results are expressed as the mean ± SD. The significance of the difference between the experimental results and control values was determined by Student's t-test. P values less than 0.05 were considered significant.
Results
Expression of affibody molecules in MH7A cells
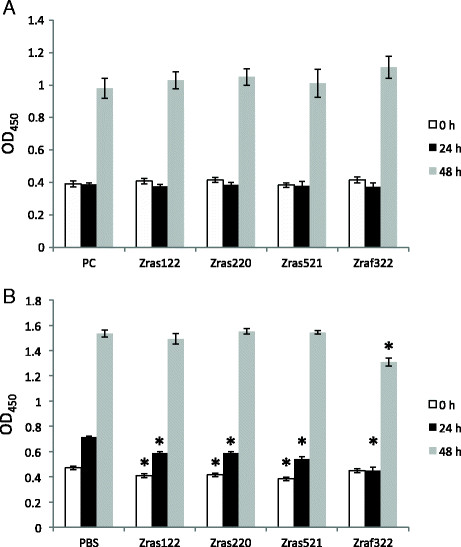
Each pcDNA3.1-based plasmid encoding Zras122, Zras220, or Zras521 was designed to yield head-to-tail homodimers of the molecules. The plasmid encoding Zraf322 was designed to yield a monomeric molecule. Each plasmid was introduced into MH7A cells, and protein expression was confirmed by western blot analysis. The dimeric affibody molecules, Zras122, Zras220, and Zras521, appeared to be approximately 12 kDa, and the monomeric affibody molecule, Zraf322, appeared to be approximately 6 kDa. The Zras122 affibody protein was expressed in a greater quantity than the other affibody proteins at 48 and 72 h after transfection (Figure 1).
Figure 1.
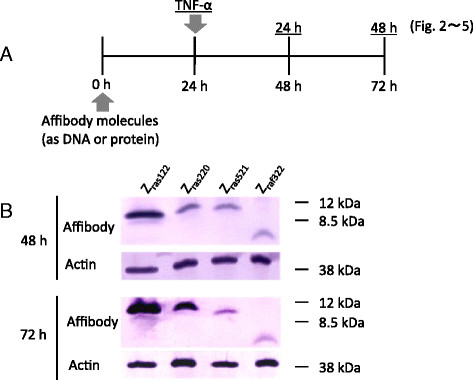
Experimental protocol (A) and western blot analysis for confirmation of cytosolic synthesis of affibody molecules (B). (A) Affibody molecules were introduced into MH7A cells encoded by DNA or as proteins. After 24-h culture, the cells were stimulated with TNF-α (100 ng/mL) and cultured for an additional 24-48 h. (B) Western blot analysis of each soluble fraction from MH7A cells harboring plasmid-encoded affibody protein molecules was analyzed at 48 and 72 h after the introduction of the plasmid into MH7A cells. Actin was analyzed as a control.
Inhibition of IL-6 and PGE2 production by plasmid-expressed affibody proteins
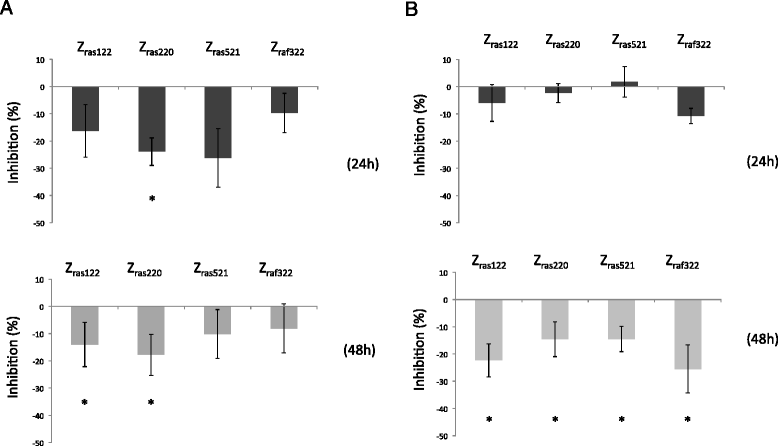
Each plasmid expressing the affibody molecules was introduced into MH7A cells to observe any inhibition of IL-6 production. At 24 h after transfection, TNF-α was added to induce synthesis of IL-6 and PGE2 (Figure 1A). After an additional 24-48 h of culture, the supernatant was collected, and IL-6 production was evaluated by ELISA. Zras220 reduced IL-6 production by approximately 20% relative to the empty plasmid (pcDNA3.1) at 24 and 48 h after TNF-α stimulation; in contrast, expression of Zraf322 reduced IL-6 production by approximately 10% (Figure 2A). The inhibition of IL-6 production by each plasmid-expressed affibody was greater at 24 h than at 48 h after TNF-α stimulation (Figure 2A). Possible inhibition of PGE2 production in MH7A cells was also analyzed upon expression of the affibody molecules. Expression of Zraf322 reduced PGE2 production by approximately 10% relative to the empty plasmid at 24 h after TNF-α stimulation, while expression of Zraf122. Zras220, and Zras521 did not significantly affect PGE2 expression at 24 h (Figure 2B). Zras122 and Zraf322 reduced PGE2 production by approximately 20% relative to the control plasmid at 48 h after TNF-α stimulation; in contrast to IL-6 production, the inhibition of PGE2 production by each plasmid-expressed affibody was greater at 48 h than at 24 h after TNF-α stimulation (Figure 2B).
Figure 2.
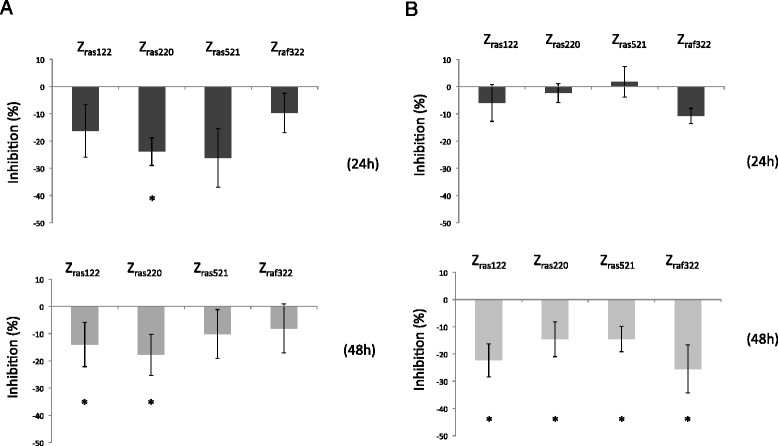
IL-6 (A) and PGE2 (B) production by MH7A cells with plasmid-expressed affibodies. MH7A cells were transfected with plasmids encoding affibodies. After 24 h of culture, the cells were stimulated with TNF-α (100 ng/mL) and cultured for an additional 24-48 h. Data represent the percent inhibition of IL-6 or PGE2 production by MH7A cells. Percent inhibition was calculated as follows: ([(IL-6 or PGE2 levels in MH7A cells transfected with affibody DNA) - (IL-6 or PGE2 levels in MH7A cells transfected with pcDNA3.1)]/[IL-6 or PGE2 levels in MH7A cells transfected with pcDNA3.1]) × 100. Data represent the means × SD of three independent experiments. *p <0.05.
Phosphorylation of ERK
ERK1/2 is situated downstream of Ras/Raf in the MAP kinase signal transduction cascade. To determine if affibody molecules affected signaling through this pathway, phosphorylation of ERK1/2 was evaluated in MH7A cells at 16 and 24 h after TNF-α stimulation. Western blot analysis of each cell lysate was performed, using an anti-p-ERK antibody with an anti-actin antibody serving as a control. The expression of Zras220 inhibited phosphorylation of ERK1/2 at 16 h after TNF-α stimulation (Figure 3). This result indicated that Zras220, but not the other affibody proteins, was able to block ERK1/2 phosphorylation through binding to Ras.
Figure 3.
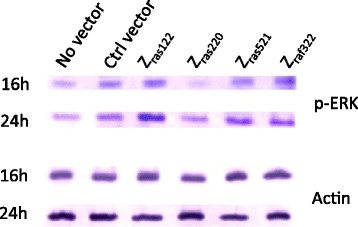
Western blot analysis for the confirmation of ERK1/2 phosphorylation. MH7A cells were transfected with plasmids encoding affibodies. After 24 h of culture, the cells were stimulated with TNF-α (100 ng/mL) and cultured for an additional 24-48 h. Each soluble fraction of MH7A cells harboring plasmid-encoded affibody molecules was analyzed via western blot for the expression of p-ERK and actin at 16 and 24 h after TNF-α stimulation. Representative data are shown for p-ERK and actin expression by MH7A cells.
Inhibition of IL-6 and PGE2 production by affibody molecules as proteins in MH7A cells
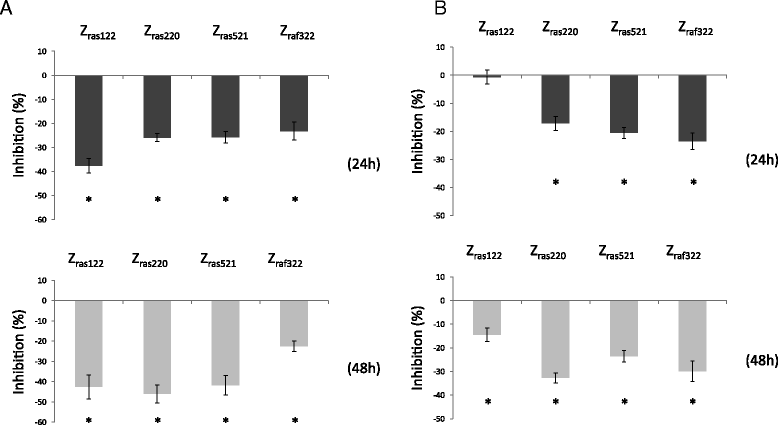
The affibody molecules were expressed in Escherichia coli and purified to >95% purity. SDS-PAGE analysis of the fractions from the last chromatographic step revealed essentially pure proteins of the expected molecular weight (data not shown). Individually, Zras122, Zras220, Zras521, and Zraf322 proteins were introduced into MH7A cells by incubation with the cell-penetrating peptide reagent Xfect. Although the inhibition of IL-6 production by Zras122 was stronger than the inhibition by the other proteins at 24 h, Zras220 protein strongly inhibited IL-6 production by approximately 46% relative to the control penetrating peptide alone at 48 h after TNF-α stimulation (Figure 4A). The Zras220 protein also strongly inhibited PGE2 production by 32% relative to the control penetrating peptide alone at 48 h after TNF-α stimulation (Figure 4B).
Figure 4.
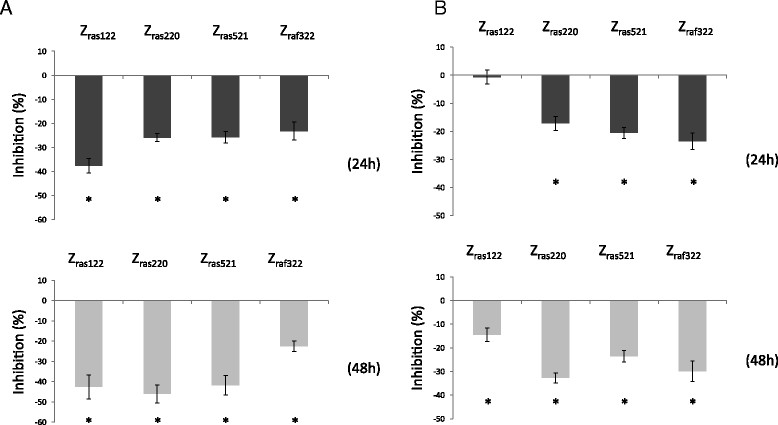
IL-6 (A) and PGE2 (B) production by affibody protein-introduced MH7A cells. MH7A cells were transfected with affibody proteins. After 24 h of culture, the cells were stimulated with TNF-α (100 ng/mL) and cultured for an additional 24-48 h. IL-6 and PGE2 levels were analyzed, and percent inhibition of IL-6 or PGE2 production was determined. Percent inhibition was calculated as follows: ([(IL-6 or PGE2 levels in affibody protein-introduced MH7A cells) - (IL-6 or PGE2 levels in MH7A cells introduced with standard peptides)]/([IL-6 or PGE2 levels in MH7A cells introduced with standard peptides]) × 100. Data represent the means × SD of three independent experiments. *p <0.05.
Effect of affibody molecules on cell proliferation
The proliferation of MH7A cells was also investigated upon expression of the affibody molecules by plasmid transfection. No change in proliferation could be observed at 24 or 48 h after TNF-α stimulation for any of the constructs, compared to the cells harboring the empty plasmid (Figure 5A). We next investigated a possible effect on MH7A cell proliferation after introduction of affibody proteins. MH7A cells transfected with a control peptide proliferated 151% from 0-24 h, while those transfected with Zraf322 proteins proliferated almost 0% from 0-24 h (Figure 5B). Furthermore, during 0-48 h, control cells and Zraf322 proteins-transfected cells proliferated 327% and 291%, respectively. These results indicate an inhibitory effect of Zraf322 protein introduction on proliferation.
Figure 5.
Cell proliferation assay for MH7A cells transfected with plasmid-expressed affibody molecules (A) and MH7A cells introduced affibody molecules as protein (B). After 24 h of culture, the cells were stimulated with TNF-α (100 ng/mL) and cultured for an additional 24-48 h. Cell proliferation was analyzed with a CCK assay. Data represent the means × SD of three independent experiments. *p <0.05.
Discussion
In this study, we investigated whether affibody molecules targeting H-Ras or Raf-1 can inhibit cell proliferation and the production of inflammatory mediators in vitro. To date, it has been demonstrated that intracellularly expressed affibody molecules can bind to target molecules and inhibit their functions. For example, an affibody molecule that has an affinity for epidermal growth factor receptor (EGFR) retains EGFR in the secretory compartment. This constraint led to decreased phosphorylation of EGFR and a reduced proliferation rate (Vernet et al. [2009]). An affibody molecule targeting HER-3 inhibits proliferation of a cancer cell line (Göstring et al. [2012]). However, we demonstrated for the first time that an affibody targeting intracellular signal transduction molecules could inhibit cell proliferation and the production of inflammatory mediators. We demonstrated that H-Ras- (Zras122, Zras220, Zras521) or Raf-1- (Zraf322) targeting affibody molecules inhibited the production of IL-6 and PGE2 in a synovial cell line. These affibody molecules were introduced into the cells via DNA-encoding plasmids or as purified proteins. In both cases, inhibition of IL-6 and PGE2 production was observed for almost all affibody molecules.
Several previous reports indicated suppression of synovial cell function by Ras inhibition. For instance, gene transfer of a dominant negative mutant of the Ras gene (Yamamoto et al. [2003]) or gene silencing using a locked nucleic acid against Ras (de Launay et al. [2010]) in synovial cells inhibited the production of inflammatory mediators, such as IL-6. Consistent with these reports, we observed that affibody molecules binding to H-Ras or Raf-1 could inhibit the production of IL-6. PGE2 production by RA synovial cells is regulated by Ras (Na et al. [2004]). Therefore, we analyzed PGE2 levels and demonstrated that these affibody molecules also inhibited PGE2 production. Matrix metalloproteinase (MMP)-3 is a protease that is produced by synovial cells upon TNF-α stimulation, and its production is regulated by Ras (Abreu et al. [2009]). We analyzed MMP-3 levels and demonstrated that these affibody molecules also inhibited MMP-3 production (data not shown). These results indicate that affibody molecules binding to H-Ras or Raf-1 can inhibit inflammatory mediators or proteases, such as IL-6, PGE2, and MMP-3, which are crucial to RA pathogenesis.
Regarding the expression of affibody molecules in MH7A cells, we observed stronger expression of Zras122 than other affibody proteins. In addition, Zras122 protein levels increased, while Zraf322 and Zras521 protein levels decreased between 24 and 48 h (Figure 1). These results might be related to the stability of the Zras122 affibody protein afforded by the plasmid construct. Supporting this notion, we observed that Zras220 and Zras521 inhibited IL-6 production by 23.9% and 26.2% relative to the control vector treatment at 24 h, while these inhibitory effects were decreased to 17.8% and 10.1% at 48 h after TNF-α stimulation, respectively. In contrast, inhibition by Zras122 was maintained up to 48 h after TNF-α stimulation (Figure 2A), suggesting increased stability of the affibody. With regard to PGE2 production, inhibition by all affibodies at 48 h increased compared with that at 24 h after TNF-α stimulation. Of interest, the timing of maximal inhibition was different for IL-6 and PGE2, but the underlying mechanism is still unknown. The difference could be attributed to the difference in the signal transduction cascade of these inflammatory molecules.
To determine whether affibody molecules inhibit signal transduction via ERK, we examined the phosphorylation of ERK1/2, a downstream molecule of Ras/Raf signaling. Zras220 inhibited ERK1/2 phosphorylation in the early stage (16 h) after stimulation by TNF-α (Figure 3), although we did not observe distinct inhibition by other affibodies. This transient inhibition of ERK1/2 phosphorylation might be caused relatively by Zras220 via interruption of the interaction between Raf-1 and MEK1/2 (Chen et al. [2001]). However, this inhibition on ERK1/2 phosphorylation could not be kept after 16-24 h, so that differences of phosphorylation among all affibody molecules here should be investigate more precisely in further study.
We utilized two different methods to deliver affibody molecules into synovial cells: 1) transfection of plasmids encoding the affibody molecules and 2) introduction of affibody proteins using a cell-penetrating peptide reagent. Protein-introduced affibody molecules exhibited stronger inhibition of IL-6 and PGE2 production than plasmid-expressed affibody molecules (Figures 2 and 4). Moreover, the inhibition resulting from the introduced proteins was less variable than that resulting from plasmid transfection. When cell proliferation was examined using plasmid-encoded affibody molecules, distinctive differences were not observed (Figure 5A). On the contrary, introduction of Zraf322 proteins partially inhibited cell proliferation (Figure 5B), suggesting that a plasmid-based expression system did not generate a sufficient concentration of affibody in MH7A cells to inhibit signal transduction. In the experiments using plasmids, cell proliferation was not observed at 24 (Figure 5A), while in the experiments using proteins, cell proliferation was observed at the same period (Figure 5B). This might be based on higher cytotoxicity of liposome compared with peptides used as a carrier (Suhorutsenko et al. [2011]). These results indicate that protein-introduced affibody molecules more effectively inhibited cytokine production and cell proliferation in synovial cells than plasmid-expressed affibody molecules. Although the effect on synovial cell proliferation was not strong, our results demonstrate that it is possible to inhibit synovial cell proliferation by selecting useful affibody clones and by improving transduction efficiency of affibody protein molecules.
Inhibition of IL-6 and PGE2 production caused by the protein-introduced affibodies varied. For example, at 48 h after TNF-α stimulation, inhibition of IL-6 production was greater than 40% for three affibodies (Zras122, Zras220, and Zras521), whereas that of PGE2 was approximately 30% for two affibodies (Zras220 and Zraf322). These results could be attributed to the fact that the PGE2 receptor, which couples with G proteins, is integrated into a positive feedback loop for PGE2 production (Nishimura et al. [2013]); thus, the effect of an affibody molecule on PGE2 production might be weaker than on IL-6. The differential effectiveness of each affibody molecule on synovial cell function might also be explained by differences in the amino acid sequences of the affibodies themselves. Each clone was selected using a phage display library of Z variants, and the amino acid sequences encoded by each clone are different.
In conclusion, we demonstrated that affibody molecules could inhibit cell proliferation and the production of inflammatory mediators by blocking the Ras/Raf signal transduction cascade. Although inhibition of cell function could be enhanced, and delivery methods should be examined further, we propose that these molecules are candidates to be investigated as a molecular-targeting therapy for RA. Studies are in progress to select useful affibody clones, to improve delivery methods of affibody protein molecules, and to verify the efficiency of affibodies in an RA model.
Additional file
Electronic supplementary material
Additional file 1: DNA sequence of affibody molecules in this study. (PDF 32 KB)
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was partially supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, with a Grant-in-Aid for Young Scientists (B).
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors' contributions
SS, TG and PN carried out the genetic construction of affibody molecules. MK carried out Western blot analysis and ELISA. SS and MK carried out cell proliferation assays and productions of affibody molecules using E. coli. HS and TI participated in the design, coordination of the study. SS, TG, PN and TI contributed in the drafting of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Seiji Shibasaki, Email: seiji@huhs.ac.jp.
Miki Karasaki, Email: m.karasaki@huhs.ac.jp.
Torbjörn Gräslund, Email: torbjorn@kth.se.
Per-Åke Nygren, Email: perake@biotech.kth.se.
Hajime Sano, Email: hsano@hyo-med.ac.jp.
Tsuyoshi Iwasaki, Email: tsuyo-i@huhs.ac.jp.
References
- Abreu JR, de Launay D, Sanders ME, Grabiec AM, van de Sande MG, Tak PP, Reedquist KA. The Ras guanine nucleotide exchange factor RasGRF1 promotes matrix metalloproteinase-3 production in rheumatoid arthritis synovial tissue. Arthritis Res Ther. 2009;11:R121. doi: 10.1186/ar2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MJ, Stadler BM, Vogel M. Potential applications of designed ankyrin repeat proteins in diagnostics and therapeutics. Exp Opin Med Diagn. 2007;1:409–421. doi: 10.1517/17530059.1.3.409. [DOI] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci U S A. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Launay D, Vreijling J, Hartkamp LM, Karpus ON, Abreu JR, van Maanen MA, Sanders ME, Grabiec AM, Hamann J, Ørum H, Vervoordeldonk MJ, Fluiter K, Tak PP, Reedquist KA. Silencing the expression of Ras family GTPase homologues decreases inflammation and joint destruction in experimental arthritis. Am J Pathol. 2010;177:3010–3024. doi: 10.2353/ajpath.2010.091053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M, Ståhl S. Engineered affinity proteins for tumour-targeting applications. Biotechnol Appl Biochem. 2009;53:1–29. doi: 10.1042/BA20080287. [DOI] [PubMed] [Google Scholar]
- Furneri G, Mantovani LG, Belisari A, Mosca M, Cristiani M, Bellelli S, Cortesi PA, Turchetti G. Systematic literature review on economic implications and pharmacoeconomic issues of rheumatoid arthritis. Clin Exp Rheumatol. 2012;30:S72–S84. [PubMed] [Google Scholar]
- Gómez-Reino J. Biologic monotherapy as initial treatment in patients with early rheumatoid arthritis. Rheumatology. 2012;51(Suppl 5):v31–v37. doi: 10.1093/rheumatology/kes116. [DOI] [PubMed] [Google Scholar]
- Göstring L, Malm M, Höidén-Guthenberg I, Frejd FY, Stähl S, Löfblom J, Gedda L. Cellular effects of HER3-specific affibody molecules. PLoS One. 2012;7:e40023. doi: 10.1371/journal.pone.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S, Lundberg E, Yu F, Shibasaki S, Vernet E, Skogs M, Nygren PÅ, Gräslund T. Selection and characterisation of affibody molecules inhibiting the interaction between Ras and Raf in vitro. N Biotechnol. 2010;27:766–773. doi: 10.1016/j.nbt.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Gunneriusson E, Samuelson P, Uhlen M, Nygren PA, Stähl S. Surface display of a functional single-chain Fv antibody on staphylococci. J Bacteriol. 1996;178:1341–1346. doi: 10.1128/jb.178.5.1341-1346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Tanaka M, Oba M. Protein transfection study using multicellular tumor spheroids of human hepatoma Huh-7 cells. PLoS One. 2013;8:e82876. doi: 10.1371/journal.pone.0082876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H, Saba JD, Tam YY. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 2006;54:742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J Mol Biol. 2012;415:393–405. doi: 10.1016/j.jmb.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg H, Hofström C, Altai M, Honorvar H, Wållberg H, Orlova A, Stähl S, Gräslund T, Tolmachev V. Evaluation of a HER2-targeting affibody molecule combining an N-terminal HEHEHE-tag with a GGGC chelator for 99mTc-labelling at the C terminus. Tumour Biol. 2012;33:641–651. doi: 10.1007/s13277-011-0305-z. [DOI] [PubMed] [Google Scholar]
- Lundberg E, Brismar H, Gräslund T. Selection and characterization of Affibody ligands to the transcription factor c-Jun. Biotechnol Appl Biochem. 2009;52:17–27. doi: 10.1042/BA20070178. [DOI] [PubMed] [Google Scholar]
- Malm M, Kronqvist N, Lindberg H, Gudmundsdotter L, Bass T, Frejd FY, Höidén-Guthenberg I, Varasteh Z, Orlova A, Tolmachev V, Stähl S, Löfblom J. Inhibiting HER3-mediated tumor cell growth with affibody molecules engineered to low picomolar affinity by position-directed error-prone PCR-like diversification. PLoS One. 2013;8:e62791. doi: 10.1371/journal.pone.0062791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mélet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab-induced T cell depletion in patients with rheumatoid arthritis: association with clinical response. Arthritis Rheum. 2013;65:2783–2790. doi: 10.1002/art.38107. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Okudaira H. Establishment and characterization of a novel human rheumatoid fibroblast-like synoviocyte line, MH7A, immortalized with SV40 T antigen. J Biochem. 1998;124:1153–1162. doi: 10.1093/oxfordjournals.jbchem.a022233. [DOI] [PubMed] [Google Scholar]
- Na HJ, Lee SJ, Kang YC, Cho YL, Nam WD, Kim PK, Ha KS, Chung HT, Lee H, Kwon YG, Koh JS, Kim YM. Inhibition of farnesyltransferase prevents collagen-induced arthritis by down-regulation of inflammatory gene expression through suppression of p21(ras)-dependent NF-kappaB activation. J Immunol. 2004;173:1276–1283. doi: 10.4049/jimmunol.173.2.1276. [DOI] [PubMed] [Google Scholar]
- Nilsson FY, Tolmachev V. Affibody molecules: new protein domains for molecular imaging and targeted tumor therapy. Curr Opin Drug Discov Devel. 2007;10:167–175. [PubMed] [Google Scholar]
- Nishimura T, Zhao X, Gan H, Koyasu S, Remold HG. The prostaglandin E2 receptor EP4 is integral to a positive feedback loop for prostaglandin E2 production in human macrophages infected with Mycobacterium tuberculosis. FASEB J. 2013;27:3827–3836. doi: 10.1096/fj.13-228858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren PA. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008;275:2668–2676. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- Ohuchida T, Okamoto K, Akahane K, Higure A, Todoroki H, Abe Y, Kikuchi M, Ikematsu S, Muramatsu T, Itoh H. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer. 2004;100:2430–2436. doi: 10.1002/cncr.20266. [DOI] [PubMed] [Google Scholar]
- Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N Engl J Med. 2004;350:2167–2179. doi: 10.1056/NEJMra032906. [DOI] [PubMed] [Google Scholar]
- Saag KG, Teng GG, Patkar NM, Anuntiyo J, Finney C, Curtis JR, Paulus HE, Mudano A, Pisu M, Elkins-Melton M, Outman R, Allison JJ, Suarez Almazor M, Bridges SL, Jr, Chatham WW, Hochberg M, MacLean C, Mikuls T, Moreland LW, O'Dell J, Turkiewicz AM, Furst DE. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum. 2008;59:762–784. doi: 10.1002/art.23721. [DOI] [PubMed] [Google Scholar]
- Schlegel PM, Steiert I, Kötter I, Müller CA. B cells contribute to heterogeneity of IL-17 producing cells in rheumatoid arthritis and healthy controls. PLoS One. 2013;8:e82580. doi: 10.1371/journal.pone.0082580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki S, Fujita A, Usui C, Watanabe S, Kitano S, Sano H, Iwasaki T. Effect of transient expression of fluorescent protein probes in synovial and myoblast cell lines. Springer Plus. 2012;1:36. doi: 10.1186/2193-1801-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki S, Aoki W, Nomura T, Miyoshi A, Tafuku S, Sewaki T, Ueda M. An oral vaccine against candidiasis generated by a yeast molecular display system. Pathog Dis. 2013;69:262–268. doi: 10.1111/2049-632X.12068. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Aletaha D. Interleukin-6 receptor inhibition with tocilizumab and attainment of disease remission in rheumatoid arthritis: the role of acute-phase reactants. Arthritis Rheum. 2011;63:43–52. doi: 10.1002/art.27740. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2:473–488. doi: 10.1038/nrd1109. [DOI] [PubMed] [Google Scholar]
- Suhorutsenko J, Oskolkov N, Arukuusk P, Kurrikoff K, Eriste E, Copolovici DM, Langel U. PepFects, show no evidence of toxicity and immunogenicity in vitro and in vivo. Bioconjug Chem. 2011;22:2255–2262. doi: 10.1021/bc200293d. [DOI] [PubMed] [Google Scholar]
- Tsunemi S, Iwasaki T, Kitano S, Imado T, Miyazawa K, Sano H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin Immunol. 2010;136:197–204. doi: 10.1016/j.clim.2010.03.428. [DOI] [PubMed] [Google Scholar]
- Upchurch KS, Kay J. Evolution of treatment for rheumatoid arthritis. Rheumatology. 2012;51(Suppl 6):vi28–vi36. doi: 10.1093/rheumatology/kes278. [DOI] [PubMed] [Google Scholar]
- Vernet E, Lundberg E, Friedman M, Rigamonti N, Klausing S, Nygren PA, Gräslund T. Affibody-mediated retention of the epidermal growth factor receptor in the secretory compartments leads to inhibition of phosphorylation in the kinase domain. N Biotechnol. 2009;25:417–423. doi: 10.1016/j.nbt.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Fukuda A, Seto H, Miyazaki T, Kadono Y, Sawada Y, Nakamura I, Katagiri H, Asano T, Tanaka Y, Oda H, Nakamura K, Tanaka S. Suppression of arthritic bone destruction by adenovirus-mediated dominant-negative Ras gene transfer to synoviocytes and osteoclasts. Arthritis Rheum. 2003;48:2682–2692. doi: 10.1002/art.11214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: DNA sequence of affibody molecules in this study. (PDF 32 KB)