

NPC1L1 mediates dietary cholesterol uptake and is the target of the drug ezetimibe. Others have concluded that cholesterol uptake requires sterol-dependent internalization. The present work shows that endocytosis is not required for NPC1L1 function and the drug does not alter endocytosis. Thus NPC1L1 may deliver cholesterol directly into the bilayer.
Abstract
Human NPC1L1 protein mediates cholesterol absorption in the intestine and liver and is the target of the drug ezetimibe, which is used to treat hypercholesterolemia. Previous studies concluded that NPC1L1-GFP protein trafficking is regulated by cholesterol binding and that ezetimibe blocks NPC1L1-GFP function by inhibiting its endocytosis. We used cell surface biotinylation to monitor NPC1L1-GFP endocytosis and show that ezetimibe does not alter the rate of NPC1L1-GFP endocytosis in cultured rat hepatocytes grown under normal growth conditions. As expected, NPC1L1-GFP endocytosis depends in part on C-terminal, cytoplasmically oriented sequences, but endocytosis does not require cholesterol binding to NPC1L1’s N-terminal domain. In addition, two small- molecule inhibitors of general (and NPC1L1-GFP) endocytosis failed to inhibit the ezetimibe-sensitive uptake of [3H]cholesterol from taurocholate micelles. These experiments demonstrate that cholesterol uptake by NPC1L1 does not require endocytosis; moreover, ezetimibe interferes with NPC1L1’s cholesterol adsorption activity without blocking NPC1L1 internalization in RH7777 cells.
INTRODUCTION
NPC1L1 is responsible for ∼70% of cholesterol absorption at the surface of intestinal epithelial cells (Altmann et al., 2004; Weinglass et al., 2008a; Jia et al., 2011). It is the target of the drug ezetimibe (Zetia), a drug discovered based on its ability to lower plasma low-density lipoprotein cholesterol levels (Rosenblum et al., 1998; van Heek et al., 2000; Davis et al., 2001). Although in mice, NPC1L1 is expressed uniquely in the intestine, humans also express NPC1L1 in the liver (Yu et al., 2006; Temel et al., 2007). In the liver, NPC1L1 is believed to mediate biliary readsorption of cholesterol, a process that is also inhibited by ezetimibe administration (Temel et al., 2007). The precise mechanism by which NPC1L1 protein transfers cholesterol across the membrane is unknown.
The predominant splice form of NPC1L1 yields a protein of 1332 amino acids with 13 transmembrane domains and three relatively large extracellular domains (Wang et al., 2009), analogous to the related NPC1 protein. The first, N-terminal, extracellular domain binds cholesterol in both NPC1 (Infante et al., 2008) and NPC1L1 (Kwon et al., 2011; Zhang et al., 2011). NPC2 protein is believed to transfer cholesterol to the NPC1L1-related protein NPC1 at the limiting membrane of late endosomes and lysosomes. No such partner is known for NPC1L1: bile salts likely solubilize cholesterol in mixed micelles for NPC1L1 binding (Yamanashi et al., 2007; Haikal et al., 2008). The second extracellular domain of NPC1L1 contributes significantly to the binding site for ezetimibe (Weinglass et al., 2008a).
At steady state in normal growth medium, NPC1L1-GFP is localized primarily to perinuclear recycling endosomes in rat hepatocyte cell lines, colocalizing with transferrin receptor and Rab11 GTPase (Yu et al., 2006; Ge et al., 2008). Yu et al. (2006) were the first to show that NPC1L1-GFP cycles between the endocytic recycling compartment and the cell surface. Nevertheless, most NPC1L1 resides in recycling endosomes at steady state. McArdle RH7777/CRL-1601 hepatoma cells (Yu et al., 2006; Ge et al., 2008; Li et al., 2014) and CaCo-2 (Yamanashi et al., 2007) and MDCKII cell lines (Weinglass et al., 2008a, and references therein) have been used to study NPC1L1-dependent cholesterol uptake in cell culture.
Intracellular transport of NPC1L1 has been reported to be cholesterol regulated (Yu et al., 2006; Ge at al., 2008). When NPC1L1-expressing cells are treated with 1.5% cyclodextrin (plus compactin and mevalonate to block de novo cholesterol biosynthesis), GFP-NPC1L1 accumulates at the cell surface. This relocalization is reversed upon readdition of cholesterol to the growth medium. Although at first this seems very exciting for a cholesterol-binding protein, it has long been known that treatment of cells with cyclodextrin also inhibits clathrin-mediated endocytosis of transferrin and epidermal growth factor receptors (Rodal et al., 1999; Subtil et al., 1999). Thus, by the same criteria, transferrin and epidermal growth factor receptor transport is also regulated by cholesterol.
In this study, we used a direct assay for NPC1L1 endocytosis to reinvestigate whether cholesterol uptake by NPC1L1 requires endocytosis and whether ezetimibe blocks cholesterol uptake by interfering with NPC1L1 internalization. Our findings demonstrate that under normal growth conditions, ezetimibe blocks NPC1L1 function by an entirely different mechanism and show that endocytosis is not required for NPC1L1-mediated cholesterol uptake under normal cellular growth conditions.
RESULTS AND DISCUSSION
We used a quantitative assay to monitor the trafficking of NPC1L1-GFP protein in stably expressing RH7777/CRL-1601 cells that have been used in previous studies of NPC1L1–green fluorescent protein (GFP) trafficking (Yu et al., 2006; Ge et al., 2008; Li et al., 2104). The presence of the C-terminal GFP moiety did not alter the fraction of NPC1L1 that acquires complex oligosaccharides, consistent with proper folding and transport through the Golgi complex (Yu et al., 2006). Although we cannot rule out the possibility that the presence of the GFP may influence the efficiency of endocytosis, it has not interfered with the ability of the tagged protein to enhance cholesterol uptake in a manner that is sensitive to ezetimibe inhibition (Yu et al., 2006; Ge et al., 2008; this study).
Schmid and coworkers pioneered the use of membrane-impermeable reducing agents as sensitive tools with which to monitor endocytosis of disulfide-linked biotin molecules (Schmid and Carter, 1990; Schmid and Smythe, 1991). After labeling the surface with a disulfide-linked biotin or biotinylated ligand, cells are warmed for various times and endocytosis protects internalized constituents from subsequent reduction upon addition of membrane-impermeant β-mercaptoethane sulfonate.
To measure NPC1L1-GFP endocytosis, we transferred RH7777 hepatoma cells stably expressing human NPC1L1-GFP to ice and incubated them with a thiol-cleavable, biotinylation reagent to label all cell surface proteins. Cells were then warmed for various times to permit endocytosis, then chilled and treated with β-mercaptoethane sulfonate to remove biotin from remaining, cell surface–localized molecules. Endocytosed NPC1L1-GFP, still biotinylated, was collected on NeutrAvidin agarose and detected by immunoblot analysis. Total NPC1L1-GFP, as well as the NPC1L1-GFP that was not biotinylated and flowed through the streptavidin agarose column, was monitored in parallel by immunoblot.
As shown in Figure 1A, NPC1L1-GFP was biotinylated at the surface of RH7777 hepatoma cells, as detected by immunoblot of streptavidin agarose–eluted proteins. On incubation of cells for 20 min at 37°C, ∼45% of total cell surface NPC1L1 was endocytosed, as determined by its protection from reducing agent action (Figure 1A). Protection from reducing agent required 37°C incubation to permit internalization, as NPC1L1 was no longer detected on the surface of cells held at 0°C (see later discussion) or in which endocytosis was inhibited (see later discussion). The abundant, control cytosolic protein GDP dissociation inhibitor (GDI) (Figure 1B) was easily detected in input samples of whole-cell lysates (7.5% analyzed) but not after binding to NeutrAvidin resin (40% analyzed). This shows that biotinylation was specific for surface localized proteins. Capture of biotinylated proteins on NeutrAvidin resin was essentially 100% as determined by monitoring the capture of bulk, unidentified, biotinylated cell surface proteins (Figure 1C). These results indicate that this assay monitors cell surface–localized NPC1L1-GFP molecules.
FIGURE 1:
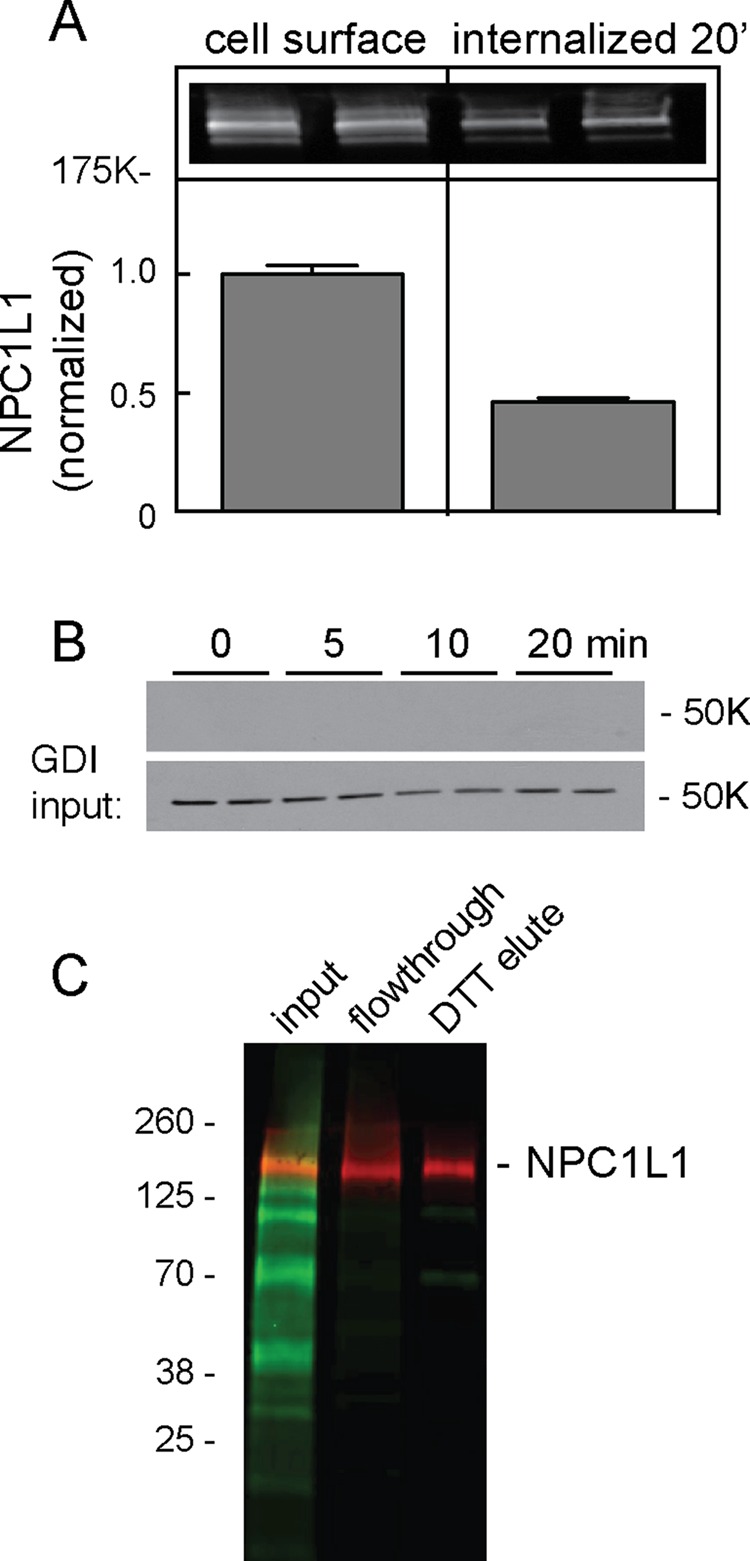
NPC1L1 biotinylation and internalization. (A) RH7777 cells stably expressing NPC1L1-GFP were surface labeled with sulfo-NHS-biotin-EZ link on ice for 30 min. Cells were then kept on ice (cell surface) or warmed for 20 min to permit endocytosis. The endocytosis plate was treated with 100 mM membrane-impermeant reducing agent so that only endocytosed proteins would remain biotinylated. Biotinylated proteins were collected on streptavidin beads. Inset, LI-COR image of resulting immunoblot using anti-GFP antibody; bar graph, quantitation of LI-COR data. Error bars represent SEM; N = 6. (B) Anti-GDI immunoblot of surface- biotinylated cells after indicated times at 37°C as in A; bottom, input of total extract (7.5%) to compare loading; top, (40%) NeutrAvidin-captured proteins. (C) LI-COR image of total biotinylated proteins (green) detected with IRDye 800CW NeutrAvidin or NPC1L1-GFP (red) detected with anti-GFP antibodies. Shown is a NeutrAvidin bead–binding experiment; little if any total biotinylated proteins were detected in the flowthrough fractions (5% input and flowthrough loaded). NPC1L1-GFP was eluted with DTT so that biotin remains on the resin and NPC1L1-GFP is eluted (25% loaded).
To verify that cell surface biotinylation is a reliable means to monitor NPC1L1 trafficking, we compared the efficiency of biotin labeling with direct antibody labeling. Antibodies to native NPC1L1 extracellular domain were not available; thus we introduced a 3xMyc tag into the third extracellular domain between residues S986 and L987 as described previously (Wang et al., 2009). Antibody labeling of transiently expressed, cell surface NPC1L1-3xMyc, as measured by either flow cytometry or immunoblot, detected ∼14% of this protein at the cell surface (Figure 2). Biotin labeling of cells transiently expressing either NPC1L1-3xMyc-GFP or non–Myc-tagged NPC1L1-GFP or stably expressing NPC1L1-GFP showed slightly higher labeling of surface molecules (16–18%) than antibody labeling (Figure 2C). These data confirm that biotin labeling is a highly efficient method for labeling NPC1L1 at the cell surface. In addition, introduction of a Myc tag did not alter significantly the fraction of NPC1L1 present at the cell surface at steady state (Figure 2). Note that all surface biotinylation was lost upon addition of membrane-impermeant reducing agent (Figure 3A, time zero; see later discussion of Figures 4, 5, and 7).
FIGURE 2:
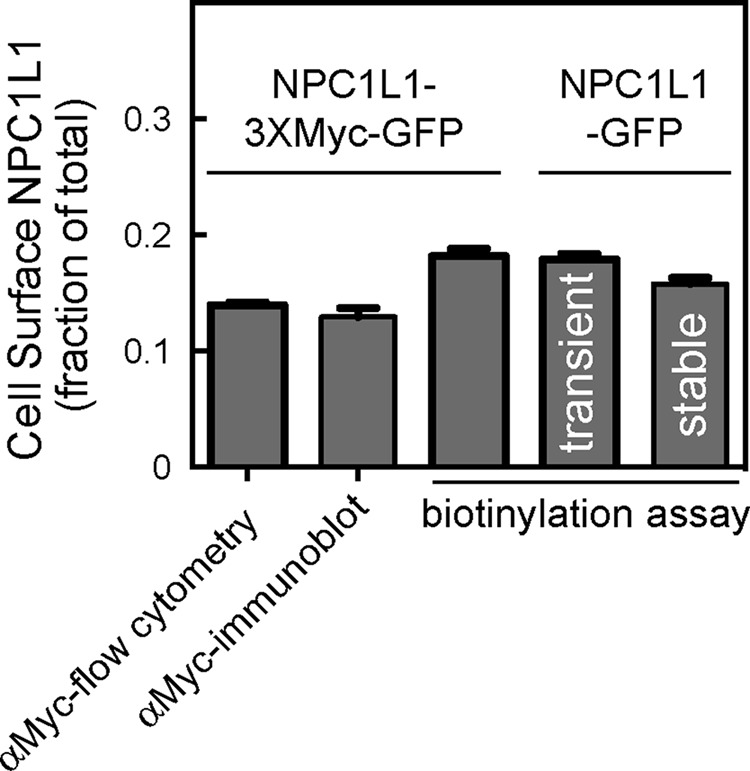
Comparison of methods to determine surface NPC1L1-3xMyc-GFP and NPC1L1-GFP. Cells transiently expressing NPC1L1-3xMyc-GFP were surface labeled with anti-Myc antibody and analyzed by (bar 1) flow cytometry (αMyc; >7000 cells counted in each of two experiments); bar 2, immunoblotting; bar 3, cell surface biotinylation as in Figure 1. Alternatively, cells expressing (non-Myc-tagged) NPC1L1-GFP transiently (bar 4) or stably (bar 5) were surface labeled with biotin as in Figure 1 and analyzed by immunoblotting. Flow cytometry and immunoblotting were also used to determine the total amount of NPC1L1; shown is the fraction of NPC1L1 detected at the surface. For flow cytometry totals, 5266 or 2430 cells were counted. Stably expressed NPC1L1-GFP was determined in five different experiments in triplicate or quadruplicate (N = 19). For other experiments, the average of triplicate determinations is shown; error bars represent SEM.
FIGURE 3:
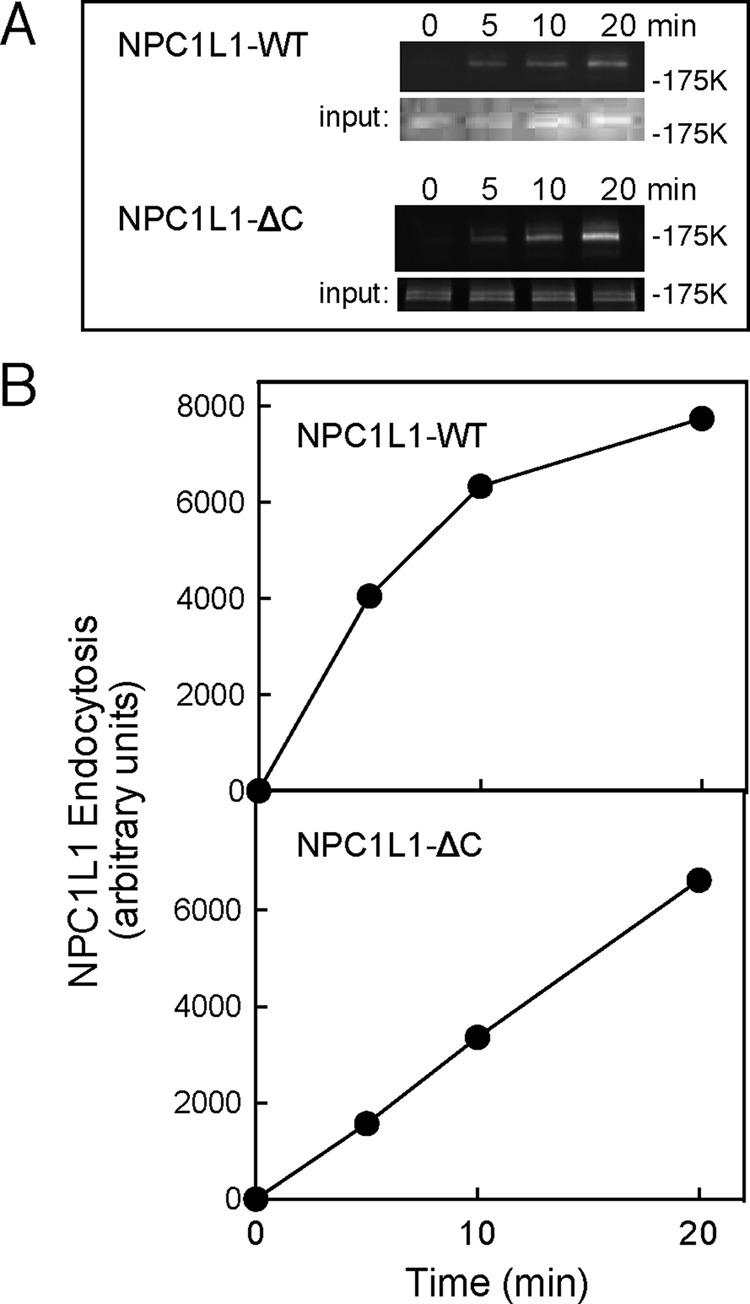
Cell surface biotinylation assays of NPC1L1 endocytosis. RH7777 cells expressing NPC1L1-GFP or NPC1L1ΔC-GFP were surface labeled and assayed for endocytosis as in Figure 1. (A) Anti-GFP blots: input, total cell extract (8%); top, streptavidin-captured, surface-biotinylated proteins (40%). For NPC1L1-GFP, a representative experiment from six experiments carried out in duplicate is shown; the combined data were used for the normalized, control panels of Figures 4B and 5, B and C. Note that in this and the following experiments, most of the input represents internal NPC1L1; it provides a loading control for each sample, but the absolute signals cannot be compared with the endocytosis data, as they were obtained from separate gels. (B) Quantitation of data in A.
FIGURE 4:
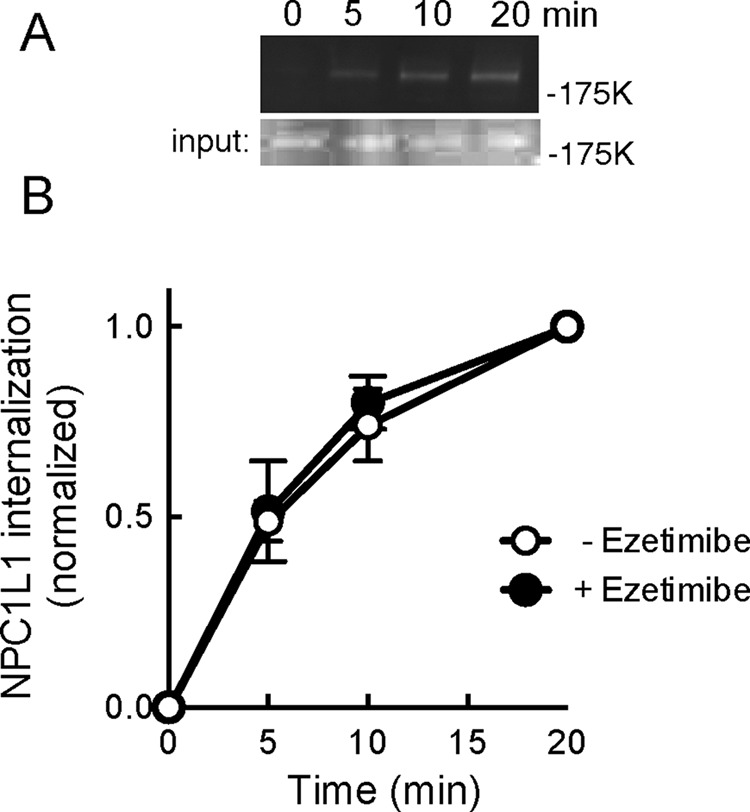
Effect of ezetimibe addition on NPC1L1 endocytosis. Cells were pretreated for 60 min with 30 μM ezetimibe; endocytosis was carried out in the continued presence of ezetimibe. (A) Representative gel in the presence of ezetimibe as in Figure 2B. (B) Combined results of three experiments with each point assayed in duplicate. Control values are from the experiments presented in Figure 3. Error bars represent SEM.
FIGURE 5:
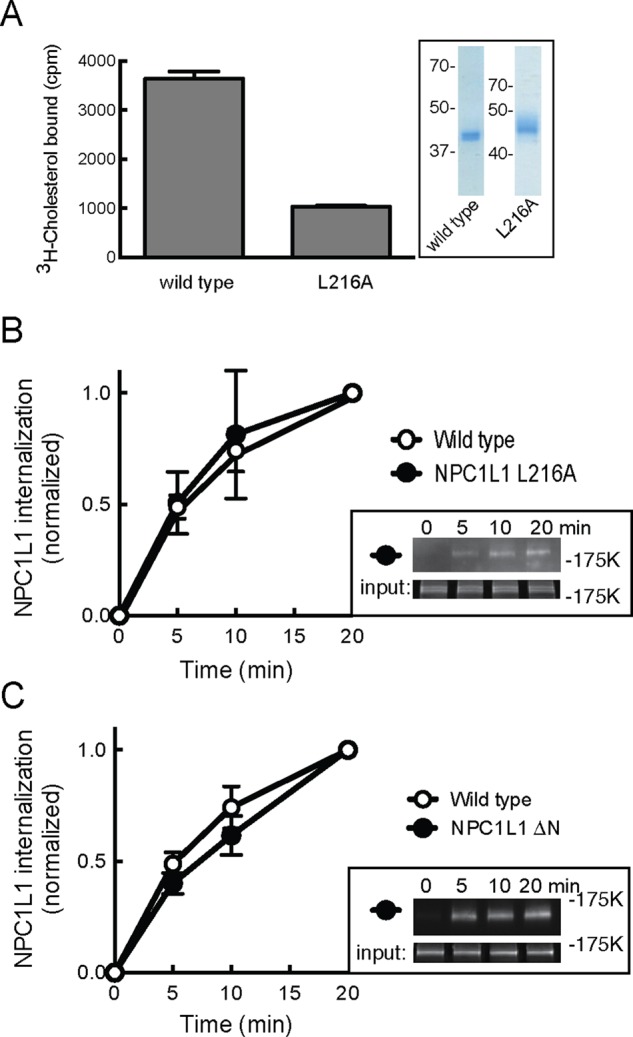
NPC1L1-GFP endocytosis does not require cholesterol binding to the N-terminal domain’s cholesterol-binding site. (A) The [3H]cholesterol binding to purified, soluble NPC1L1 N-terminal domain for wild-type and L216A mutant proteins. Inset, SDS–PAGE of soluble domains used; numbers represent molecular mass in kilodaltons. (B, C) Kinetics of internalization of wild-type or NPC1L1 L216A protein (B; four separate experiments in duplicate) or NPC1L1ΔN (C; one experiment in quadruplicate) as described and determined in Figure 3 except that 7% input is shown in the insets. Error bars represent SEM.
FIGURE 7:

Dyngo-4a and Pitstop 2 block NPC1L1 endocytosis but not cholesterol uptake. (A) Immunoblot (anti-GFP) showing endocytosis of NPC1L1-WT-GFP (as described in Figure 2) ± 30 min pretreatment with 20 μM Dyngo-4a. (B) Graph of NPC1L1 endocytosis measured in the absence (open circles) or presence (filled circles) of 20 μM Dyngo-4a. Values represent means of duplicates from a representative experiment. (C) The [3H]cholesterol uptake (20 min) from sodium taurocholate micelles ± Dyngo-4a; 18.5 μg of each sample was counted. Values represent the mean of quadruplicates from a representative experiment. The p values for pair comparisons ± NPC1L1 are 0.0096 and 0.003 (t test). (D) Immunoblot (anti-GFP) showing endocytosis (as described in Figure 2) of NPC1L1-WT-GFP ± 10 min pretreatment with 25 μM PitStop 2. Values represent means of duplicates from a representative experiment. (E) NPC1L1 endocytosis measured in the absence (open circles) or presence (filled circles) of 25 μM Pitstop 2. (F) The [3H]cholesterol uptake (20 min) from sodium taurocholate micelles ± ezetimibe and/or Pitstop 2 as indicated; 29 μg of each sample was counted. Values represent the mean of quadruplicates from a representative experiment. Error bars represent SEM. The p values for pairwise comparisons ± ezetimibe are ****p < 0.0001 and **p < 0.0027 (unpaired t test).
Figure 3 shows the kinetics of internalization of cell surface NPC1L1-GFP protein, which was roughly linear for the first 10 min and then began to plateau. An NPC1L1 mutant protein missing the C-terminal 27 amino acid residues showed a somewhat slower initial internalization rate that was linear for the first 20 min. Thus, as expected, deletion of this C-terminal portion of NPC1L1, previously shown to influence endocytosis (Li et al., 2014), yielded a protein with a roughly twofold slower initial rate of cell surface internalization.
Because each experiment was carried out multiple times in quadruplicate and involved fluorescence quantitation of separate immunoblots, the easiest way for us to consolidate the many data points from replicate samples and multiple experiments was to normalize all graphs to the amount of endocytosis detected after 20 min at 37°C. Thus all subsequent endocytosis graphs will be presented in this manner, with an example of actual gel data included.
Ezetimibe does not alter NPC1L1 endocytosis rate in RH7777 cells
Previous reports evaluated the effect of ezetimibe by monitoring the internalization of NPC1L1 in cells pretreated with cyclodextrin (Ge et al., 2008; Li et al., 2014). Under those conditions, internalization of NPC1L1 protein is cholesterol dependent. In contrast, when the rate of NPC1L1 internalization was measured in normal medium, it was completely unchanged after addition of 30 μM ezetimibe, a concentration that is far above the KD for drug binding (Figure 4, A and B). It is important to note that the preparations of ezetimibe used in these experiments were fully capable of blocking NPC1L1-dependent uptake of cholesterol from bile salts (see later discussion). These data show that in RH7777 cells, NPC1L1 internalization from the cell surface relies at least in part on cytoplasmic domain sequences (as expected) but is insensitive to ezetimibe addition.
The N-terminal extracellular domain of NPC1L1 protein binds cholesterol (Zhang et al., 2011), and mutations in this domain have been reported to both block cholesterol binding (Zhang et al., 2011) and influence NPC1L1 transport (Ge et al., 2008). To investigate the importance of cholesterol binding directly to NPC1L1 for its endocytosis, we tested an NPC1L1 mutant protein (L216A) that was shown previously to display impaired cholesterol binding (Zhang et al., 2011). A purified, soluble version of NPC1L1’s N-terminal domain (residues 22–284) carrying this mutation (from insect cells) showed a significant decrease in its capacity to bind [3H]cholesterol in vitro compared with the wild-type NPC1L1 protein (from 293F cells; Figure 5A). Thus, although our proteins were obtained from different cell lines, these findings are entirely consistent with previous reports (Zhang et al., 2011).
As shown in Figure 5B, despite impaired cholesterol binding via N-terminal domain sequences, full-length NPC1L1-GFP L216A protein was endocytosed at a rate that was indistinguishable from that of wild type NPC1L1-GFP. To verify further the importance of N-terminal domain cholesterol binding on NPC1L1 endocytosis, we obtained a construct that lacks the entire N-terminal, cholesterol-binding domain (Zhang et al., 2011) and monitored its uptake in RH7777 hepatocytes. Figure 5C shows that the complete absence of an N-terminal cholesterol-binding site had little, if any, effect on the initial rate of NPC1L1 internalization. These data show that under normal culture conditions, endocytosis of NPC1L1 protein is independent of cholesterol binding to its N-terminal, cholesterol-binding domain.
NPC1L1 function does not require endocytosis
NPC1L1 picks up cholesterol from bile salts in the intestine. To explore further the mechanism of ezetimibe action, we used an established assay (Reboul et al., 2011) to monitor ezetimibe-sensitive cholesterol uptake from bile salt micelles, using cultured RH7777 hepatoma cells. Although some labs have monitored NPC1L1 function using cyclodextrin-cholesterol complexes as substrate donor, we were not able to detect NPC1L1-dependent cholesterol uptake after 20 min using that donor substrate in our cell culture system (unpublished data); this may be because others used longer incubation times of 1–2 h. Nevertheless, we succeeded in monitoring NPC1L1-dependent cholesterol uptake when cholesterol was provided to cells in bile salt micelles (Figure 6).
FIGURE 6:

(A) Ezetimibe-sensitive cholesterol uptake in cells expressing NPC1L1 protein. Parental RH7777 cells (right) or RH7777 cells expressing human NPC1L1-GFP (left) were monitored for [3H]cholesterol uptake from sodium taurocholate micelles ± 30 μM ezetimibe for 20 min as described in Materials and Methods; 39 μg of each sample was counted. The average of two experiments carried out in quadruplicate. ****p < 0.0001 for ± ezetimibe for NPC1L1 cells (unpaired t test). (B) Cholesterol uptake in RH7777 cells expressing NPC1L1-WT or NPC1L1-ΔC proteins; 19 μg of each sample was counted. A representative experiment carried out in quadruplicate. Error bars represent SEM.
Briefly, cultures were incubated with [3H]cholesterol taurocholate micelles; NPC1L1-mediated uptake was determined by comparing cholesterol uptake with that detected for the parental hepatoma cell line, which does not express NPC1L1 protein. Although prolonged incubation of cultured cells with bile salts can lead to issues of toxicity, this was mitigated by monitoring cholesterol uptake and endocytosis within a 20-min time frame. Presumably, the extensive glycocalyx of the intact intestine protects cells from toxicity, and well-polarized monolayers of CaCo2 cells may also be less sensitive over longer incubations (Yamanashi et al., 2007). In CaCo2 cells, cholesterol uptake from bile salt micelles continues linearly for several hours (Yamanashi et al., 2007).
As shown in Figure 6A, NPC1L1-expressing cells displayed about twice the amount of total cholesterol uptake from bile salt micelles compared with the control, RH7777 parental cell line, entirely comparable to previous reports (Yamanashi et al., 2007; Weinglass et al., 2008b; Reboul et al., 2011). As expected, all NPC1L1-dependent cholesterol uptake was lost if cells were pretreated with 30 μM ezetimibe for 60 min with continued treatment during cholesterol uptake. This confirms that the additional cholesterol uptake seen in RH7777 cells expressing NPC1L1-GFP is mediated by NPC1L1 protein.
NPC1L1ΔC showed a decreased endocytosis rate (Figure 3, B and C). Nevertheless, cells expressing the NPC1L1ΔC protein showed wild-type levels of cholesterol adsorption when monitored after a 20 min of incubation with bile salt micelles (Figure 5B). This suggested that endocytosis might not be needed for cholesterol uptake by NPC1L1; moreover, the data demonstrate that the C-terminal sequences are not needed for cholesterol uptake from bile salt micelles. To test more directly the importance of endocytosis for NPC1L1-mediated cholesterol uptake, we also used small-molecule endocytosis inhibitors.
We first tested Dyngo-4a, a drug that inhibits endocytosis by blocking the GTPase activity of Dynamin1 (Robertson et al., 2014) and may have additional targets (Park et al., 2013). Figure 7, A and B, shows that Dyngo-4a blocked NPC1L1 endocytosis by >90%, as monitored by cell surface biotinylation. Nevertheless, Dyngo-4a had no effect on NPC1L1-dependent (or -independent) cholesterol uptake (Figure 7C).
We also tested a second inhibitor of endocytosis, Pitstop 2, which was designed to block the binding of partner proteins to the N-terminal domain of clathrin; it is a potent inhibitor of endocytosis but may function in cells by a less specific mechanism (von Kleist et al., 2011; Willox et al., 2014). Figure 7, D and E, shows that Pitstop 2 blocked NPC1L1 endocytosis completely. Consistent with our findings with Dyngo-4a, Pitstop 2 had no effect on total cholesterol uptake or on ezetimibe-sensitive, NPC1L1-mediated cholesterol uptake (Figure 7F). Pitstop 2 showed some toxicity when cells were incubated for times >20 min. Nevertheless, cholesterol uptake proceeded normally in 20-min assays (Figure 7, E and F). These data demonstrate that NPC1L1-dependent cholesterol uptake from bile salt micelles does not require endocytosis.
In summary, the mechanism by which human NPC1L1 protein catalyzes cholesterol transport in the intestine and liver is not yet known. Others have proposed that the protein acts as an endocytic transporter of cholesterol, shuttling it to intracellular compartments, perhaps one or a few molecules at a time (Ge et al., 2008; Li et al., 2014). In this study, we showed that ezetimibe-sensitive, NPC1L1-mediated cholesterol import occurs normally in cells in which endocytosis of NPC1L1 (and other cell surface proteins) has been completely blocked. Furthermore, using a cell surface biotinylation assay to monitor NPC1L1 endocytosis quantitatively, we showed that ezetimibe does not change the rate with which NPC1L1 protein is internalized from the cell surface and delivered to intracellular compartments. These experiments suggest that NPC1L1 is capable of transferring bound cholesterol from the extracellular space to intracellular stores. Precisely how the protein accomplishes this transfer and how ezetimibe binding blocks cholesterol import will be important issues for future investigation.
MATERIALS AND METHODS
Materials
McArdle RH7777/CRL-1601 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in DMEM containing 12% fetal bovine serum plus 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Stable cell lines expressing NPC1L1-GFP protein were established after transfection and G418 selection; cells were maintained in 0.75 mg/ml G418. The NPC1L1ΔN mutant lacks residues 18–260 (deleted from the signal sequence junction); NPC1L1ΔC lacks residues 1306–1332 at the end of the C-terminus. Chicken anti-GFP antibodies were from Life Technologies (Grand Island, NY); goat anti-chicken Dylight488 antibodies were from Abcam (Cambridge, MA); anti–chicken-horseradish peroxidase conjugate was from Promega (Sunnyvale, CA); and rabbit anti-bovine GDI was raised in the lab. All were used at 1:1000 for immunoblots. Goat anti-mouse Alexa Fluor 647 and mouse anti-Myc were from Invitrogen (Waltham, MA) and used at 1:500 and 10 μg/ml, respectively. Mouse anti-Myc monoclonal antibody was produced in the lab. IRDye 680RD donkey anti-chicken and IRDye 800CW streptavidin were from LI-COR (Lincoln, NE) and used at 1:10,000. EZ-link Sulfo-NHS-SS-Biotin was from Life Technologies. The [3H]cholesterol was from American Radiolabeled Chemicals (St. Louis, MO); cholesterol, sodium taurocholate, mono-olein, oleic acid, phosphatidylcholine (2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine), sodium 2-mercaptoethanesulfonate, iodoacetamide, paraformaldehyde, and protease inhibitor cocktail were from Sigma Aldrich (St. Louis, MO); ezetimibe and lysophosphatidylcholine (1-palmitoyl-sn-glycero-3-phosphocholine) were from Santa Cruz Biotechnology (Santa Cruz, CA); Pitstop 2 and Dyngo-4a were from Abcam; Ni-nitriloacetic acid (NTA) resin was from Qiagen (Valencia, CA); Lipofectamine 2000, Protein G agarose resin, and NeutrAvidin agarose were from Life Technologies (Carlsbad, CA).
Cell surface biotinylation
Endocytosis was measured as described (Weixel and Bradbury, 2002): RH7777 cells stably expressing NPC1L1-GFP (wild type or mutants) were plated on collagen-coated plates in DMEM containing 12% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. At 24 h after plating, cells were pretreated for 1 h in DMEM plus 12% FBS and 30 μM ezetimibe or 0.3% ethanol at 37°C. Medium was removed, and cells were placed on ice and washed three times with ice-cold phosphate-buffered saline (PBS). Cell proteins were surface labeled with 1 mg/ml sulfo-NHS-biotin-EZ link on ice for 30 min. Excess biotin reagent was removed by washing three times with 1% bovine serum albumin (BSA) in PBS, pH 7.4, on ice. Ice-cold (zero time point) or warm DMEM containing ezetimibe or ethanol was then placed on cells. Cells were kept on ice (zero time point) or warmed for indicated time points to permit endocytosis. After returning cells to ice, cell surface biotin on all plates was removed by treating cells with 100 mM β-mercaptoethane sulfonate. Unreacted reducing agent was quenched with three 5-min washes in 5 mg/ml iodoacetamide in PBS, pH 7.4, on ice. Cells were lysed with buffer containing 1.25% Triton X-100, 0.25% SDS, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 5 mg/ml iodoacetamide, and 1× protease inhibitor cocktail. Cleared lysate was incubated overnight with 25 μl of NeutrAvidin resin. After overnight incubation, resin was spun 700 × g for 3 min and loaded into a 1-ml syringe fitted with a frit. Resin was washed five times with 0.5 ml of cold wash buffer (0.5% Triton X-100, 0.1% SDS, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA). Syringes were spun at 1000 rpm for 5 s to remove wash buffer; resin was then transferred to a 1.5-ml tube containing 100 μl of SDS–PAGE sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 100 mM dithiothreitol [DTT], 10% glycerol, and 0.02% bromophenol blue [BPB]) and heated for 20 min at 37°C. Duplicate 40-μl portions of each sample were analyzed on Bio-Rad (Hercules, CA) Mini-PROTEAN TGX 4–20% gradient gels. After transfer to polyvinylidene fluoride membrane and antibody incubation, immunoblots were visualized using a VersaDoc Imager (Bio-Rad) and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). For endocytosis experiments with Pitstop2 and Dyngo-4a, cells were grown overnight in DMEM only and then incubated for 1 h with DMEM containing 5% lipoprotein-deficient serum (LPDS; Kalen Biomedical, Montgomery Village, MD) as in cholesterol uptake assay experiments (see later discussion). Cells were pretreated for 10 min with 25 μM Pitstop 2 or 30 min 20 μM Dyngo-4a. Endocytosis was carried out in the presence of sodium taurocholate micelles to confirm that inhibitors were still functional under these conditions. Immunoblots were visualized using the LI-COR Odyssey Imaging System and analyzed using ImageJ software. For assays with endocytosis inhibitors, all bands (± inhibitor) were normalized to the 20-min time point with no inhibitor present.
Cholesterol uptake
Uptake (20 min) was monitored using micelles as described (Reboul et al., 2011): RH7777 cells stably expressing NPC1L1-WT-GFP, or RH7777 cells as control, were plated in 12-well collagen-coated plates at 2.5 × 105 cells/well, four wells per condition. After allowing several hours for cells to adhere, the growth medium was removed and replaced with DMEM containing 2 mM l-glutamine. The next day, cells were incubated 1 h with DMEM containing 5% LPDS plus 2 mM l-glutamine. Ezetimibe stock was prepared in ethanol and used at a final concentration of 30 μM, final ethanol concentration 0.3%, and added during the incubation with the 5% LPDS medium. Pitstop 2 stock was prepared in dimethyl sulfoxide (DMSO) and used at a final concentration of 25 μM, final DMSO concentration 0.5%. Dyngo-4a stock was prepared in DMSO and used at a final concentration of 20 μM, final DMSO concentration 0.1%. Cells were treated for 10 min with PitStop 2 or 30 min with Dyngo-4a in serum-deficient DMEM containing 2 mM l-glutamine before cholesterol incubation. Drugs were also included during cholesterol uptake. Micelle lipids were first dissolved in ethanol and combined in a glass vial, and solvents were evaporated under N2. The dried lipids were incorporated into DMEM containing 24 mM sodium taurocholate by vortexing for 5 min. The solution was then diluted using DMEM containing 5% LPDS to final concentrations of 5 mM sodium taurocholate, 0.5 mM oleic acid, 0.035 mM phosphatidylcholine, 0.08 mM lysophosphatidylcholine, 0.3 mM mono-olein, 0.005 mM cholesterol, and 4 nM radiolabeled cholesterol. After 20 min at 37°C, the micelle mixture was removed and cells washed four times with ice-cold 5 mM sodium taurocholate in PBS. Cells were lysed using 100 μl of lysis buffer containing 1% Triton X-100, 50 mM Tris, pH 8, 150 mM NaCl, and 5 mM EDTA. Equal amounts of cleared lysate, measured by protein concentration, were analyzed by scintillation counting to determine cholesterol uptake.
Protein purification
pFastBac NPC1L1-WT-N-terminal domain plasmid was obtained from AddGene (Cambridge, MA) and used to make virus for infection of Sf9 insect cells. At 72 h after infection, Sf9 culture was spun down and ammonium sulfate added to the supernatant to achieve 75% saturation. The resulting precipitate was resuspended in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, and 150 mM NaCl and incubated with Ni-NTA resin overnight at 4°C. After elution with 20 mM HEPES, pH 7.4, 150 mM NaCl, and 400 mM imidazole, the protein was further purified using Q-Sepharose (GE Healthcare Life Sciences, Pittsburgh, PA). GFP-NPC1L1-N-terminal domain-L216A mutant protein was expressed in HEK293F cells. After 72 h the culture was spun down, and the supernatant was incubated with Ni-NTA resin overnight, eluted as described, and dialyzed into 50 mM HEPES, pH 7.4, and 150 mM NaCl.
NPC1L1 cholesterol binding
The assay was based on Infante et al. (2008): 80-μl solutions containing histidine-tagged NPC1L1-N-terminal domain wild-type and L216A cholesterol-binding mutant purified proteins (31.2 nM) were incubated with cholesterol (20 nM, 1:3 cold:hot) in 50 mM Tris, pH 7.4, 150 mM NaCl, 0.004% NP-40, and BSA (192 nM) overnight at 4°C. Solutions were loaded onto 1-ml syringes fitted with a frit and 20 μl of Ni-NTA resin. After 10 min, the resin was washed six times with 1 ml of wash buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.004% NP-40) and eluted with 1 ml of elution buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.004% NP-40, 400 mM imidazole). Eluted samples were mixed with BioSafeII (Research Products International, Mt. Prospect, IL), and radioactivity was determined using a scintillation counter.
Labeling with antibody for flow cytometry
RH7777 cells were transfected with NPC1L1-GFP containing a 3xMyc tag between residues S986 and L987 (Wang et al., 2009). At 48 h after transfection, the cells were incubated for 5 s with 0.05% trypsin and 10 mM EDTA in PBS. The trypsin was removed and immediately quenched with 10% FBS and 10 mM EDTA in PBS. Cells were spun at 1000 rpm for 5 min, washed once with PBS, and then divided into two portions. For surface labeling, one portion was gently resuspended in mouse anti-Myc supernatant and rotated for 1 h at 4°C. Both portions were fixed with 3.5% paraformaldehyde in PBS for 15 min at room temperature. For total NPC1L1 labeling, the other portion was permeabilized with 0.1% Triton X-100 and 1% BSA in PBS for 5 min at room temperature. After permeabilization, cells were resuspended in mouse anti-Myc supernatant and rotated for 1 h at 4°C. After being washed three times with PBS plus 1% BSA, portions were incubated with 1:500 goat anti-mouse Alexa Fluor 647 with 1% BSA in PBS for 1 h at room temperature. Cells were analyzed by flow cytometry, gating for single-cell GFP-positive cells, monitoring fluorescence intensity at 647 nm.
Labeling with antibody followed by immunoblot analysis
RH7777 cells transiently expressing NPC1L1-GFP containing a 3xMyc tag were washed three times with ice-cold PBS and then incubated on ice for 1 h with gentle rotation in 10 μg/ml mouse anti-Myc in PBS with 1% BSA. Unbound antibody was removed by washing three times with ice-cold PBS before lysis with 0.5% Triton X-100, 50 mM Tris, pH 8, 150 mM NaCl, and 5 mM EDTA. For total NPC1L1 labeling, cells were lysed, and the cleared lysate was incubated with 10 μg/ml mouse anti-Myc for 1 h at 4°C. Antibody-labeled protein was incubated with protein G resin overnight at 4°C. Protein was eluted in sample buffer (50 mM Tris-HCl, pH 6.8, 4% SDS, 100 mM DTT, 10% glycerol, 0.02% BPB) at 60°C for 20 min. Samples were loaded onto a 4–20% gradient gel and immunoblotted as described.
Other methods
Protein was determined by bicinchoninic acid protein assay (Life Technologies) using BSA as standard; for the cholesterol uptake experiments, protein was determined using Advanced Protein Assay reagent (Cytoskeleton, Denver, CO).
Acknowledgments
We thank B. L. Song for providing the NPC1L1 ΔN construct and Liang Ge for comments on the manuscript. This research was supported in part by funds from the Investigator Initiated Studies Program of Merck Sharp and Dohme Corp. and the National Institutes of Health (DK37332 to S.R.P. and T90DK070090 to T.A.J.). The opinions expressed in this article are those of the authors and do not necessarily represent those of Merck Sharp and Dohme Corp.
Abbreviations used:
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- GDI
GDP dissociation inhibitor
- GFP
green fluorescent protein
- LPDS
lipoprotein-deficient serum
- NPC1L1
Niemann-Pick C1-Like 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0154) on April 13, 2016.
REFERENCES
- Altmann SW, Davis HR, Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, et al. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- Davis HR, Jr, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2001;21:2032–2038. doi: 10.1161/hq1201.100260. [DOI] [PubMed] [Google Scholar]
- Ge L, Wang J, Qi W, Miao HH, Cao J, Qu YX, Li BL, Song BL. The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab. 2008;7:508–519. doi: 10.1016/j.cmet.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Haikal Z, Play B, Landrier JF, Giraud A, Ghiringhelli O, Lairon D, Jourdheuil-Rahmani D. NPC1L1 and SR-BI are involved in intestinal cholesterol absorption from small-size lipid donors. Lipids. 2008;43:401–408. doi: 10.1007/s11745-008-3172-7. [DOI] [PubMed] [Google Scholar]
- Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, Brown MS. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem. 2008;283:1064–1075. doi: 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. doi: 10.1146/annurev-physiol-012110-142233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HJ, Palnitkar M, Deisenhofer J. The structure of the NPC1L1 N-terminal domain in a closed conformation. PLoS One. 2011;6:e18722. doi: 10.1371/journal.pone.0018722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PS, Fu ZY, Zhang YY, Zhang JH, Xu CQ, Ma YT, Li BL, Song BL. The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L1. Nat Med. 2014;20:80–86. doi: 10.1038/nm.3417. [DOI] [PubMed] [Google Scholar]
- Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci. 2013;126:5305–5312. doi: 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul E, Soayfane Z, Goncalves A, Cantiello M, Bott R, Nauze M, Tercé F, Collet X, Coméra C. Respective contributions of intestinal Niemann-Pick C1-like 1 and scavenger receptor class B type 1 cholesterol tocopherol uptake: in vivo v. in vitro studies. Br J Nutr. 2011;107:1296–1304. doi: 10.1017/S0007114511004405. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, Deane FM, Robinson PJ, McCluskey A. Synthesis of Dynole 34-2, Dynole 2-24 and Dyngo 4a for investigating dynamin GTPase. Nat Protoc. 2014;9:851–870. doi: 10.1038/nprot.2014.046. [DOI] [PubMed] [Google Scholar]
- Rodal SK, Skretting G, Garred O, Vilhardt F, van Deurs B, Sandvig K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell. 1999;10:961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum SB, Huynh T, Afonso A, Davis HR, Jr, Yumibe N, Clader JW, Burnett DA. Discovery of 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4 -hydroxyphenyl)-2-azetidinone (SCH 58235): a designed, potent, orally active inhibitor of cholesterol absorption. J Med Chem. 1998;41:973–980. doi: 10.1021/jm970701f. [DOI] [PubMed] [Google Scholar]
- Schmid SL, Carter LL. ATP is required for receptor-mediated endocytosis in intact cells. J Cell Biol. 1990;111:2307–2318. doi: 10.1083/jcb.111.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SL, Smythe E. Stage-specific assays for coated pit formation and coated vesicle budding in vitro. J Cell Biol. 1991;114:869–880. doi: 10.1083/jcb.114.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil A, Gaidarov I, Kobylarz K, Lampson MA, Keen JH, McGraw TE. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc Natl Acad Sci USA. 1999;96:6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. 2007;117:1968–1978. doi: 10.1172/JCI30060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heek M, Farley C, Compton DS, Hoos L, Alton KB, Sybertz EJ, Davis HR., Jr Comparison of the activity and disposition of the novel cholesterol absorption inhibitor, SCH58235, and its glucuronide, SCH60663. Br J Pharmacol. 2000;129:1748–1754. doi: 10.1038/sj.bjp.0703235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang J, Chu BB, Ge L, Li BL, Yan Y, Song BL. Membrane topology of human NPC1L1, a key protein in enterohepatic cholesterol absorption. J Lipid Res. 2009;50:1653–1662. doi: 10.1194/jlr.M800669-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinglass AB, Kohler M, Schulte U, Liu J, Nketiah EO, Thomas A, Schmalhofer W, Williams B, Bildl W, McMasters DR, et al. Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc Natl Acad Sci USA. 2008a;105:11140–11145. doi: 10.1073/pnas.0800936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinglass AB, Köhler MG, Nketiah EO, Liu J, Schmalhofer W, Thomas A, Williams B, Beers L, Smith L, Hafey M, et al. Madin-Darby canine kidney II cells: a pharmacologically validated system for NPC1L1-mediated cholesterol uptake. Mol Pharmacol. 2008b;73:1072–1084. doi: 10.1124/mol.107.043844. [DOI] [PubMed] [Google Scholar]
- Weixel K, Bradbury N. Analysis of CFTR endocytosis by cell surface biotinylation. Methods Mol Med. 2002;70:323–340. doi: 10.1385/1-59259-187-6:323. [DOI] [PubMed] [Google Scholar]
- Willox AK, Sahraoui YM, Royle SJ. Non-specificity of Pitstop 2 in clathrin-mediated endocytosis. Biol Open. 2014;3:326–331. doi: 10.1242/bio.20147955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanashi Y, Takada T, Suzuki H. Niemann-Pick C1-like 1 overexpression facilitates ezetimibe-sensitive cholesterol and beta-sitosterol uptake in CaCo-2 cells. J Pharmacol Exp Ther. 2007;320:559–64. doi: 10.1124/jpet.106.114181. [DOI] [PubMed] [Google Scholar]
- Yu L, Bharadwaj S, Brown JM, Ma Y, Du W, Davis MA, Michaely P, Liu P, Willingham MC, Rudel LL. Cholesterol-regulated translocation of NPC1L1 to the cell surface facilitates free cholesterol uptake. J Biol Chem. 2006;281:6616–6624. doi: 10.1074/jbc.M511123200. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Ge L, Qi W, Zhang L, Miao HH, Li BL, Yang M, Song BL. The N-terminal domain of NPC1L1 protein binds cholesterol and plays essential roles in cholesterol uptake. J Biol Chem. 2011;286:25088–25097. doi: 10.1074/jbc.M111.244475. [DOI] [PMC free article] [PubMed] [Google Scholar]