

Abstract
Stroke is the leading cause of adult disability. The past decade has seen advances in basic science research of neural repair in stroke. The brain forms new connections after stroke, which have a causal role in recovery of function. Brain progenitors, including neuronal and glial progenitors, respond to stroke and initiate a partial formation of new neurons and glial cells. The molecular systems that underlie axonal sprouting, neurogenesis, and gliogenesis after stroke have recently been identified. Importantly, tractable drug targets exist within these molecular systems that might stimulate tissue repair. These basic science advances have taken the field to its first scientific milestone; the elemental principles of neural repair in stroke have been identified. The next stages in this field involve understanding how these elemental principles of recovery interact in the dynamic cellular systems of the repairing brain. Emergent principles arise out of the interaction of the fundamental or elemental principles in a system. In neural repair, the elemental principles of brain reorganization after stroke interact to generate higher order and distinct concepts of regenerative brain niches in cellular repair, neuronal networks in synaptic plasticity, and the distinction of molecular systems of neuroregeneration. Many of these emergent principles directly guide the development of new therapies, such as the necessity for spatial and temporal control in neural repair therapy delivery and the overlap of cancer and neural repair mechanisms. This review discusses the emergent principles of neural repair in stroke as they relate to scientific and therapeutic concepts in this field. Ann Neurol 2016;79:895–906
Emergent properties arise out of more fundamental properties in a given context and are distinct or irreducible. As individual fish form a school, the synchronous and coordinated movements of the group are distinct from the properties of the single fish.1 A walk on a leafy street in the summertime Midwest brings the sound of the cicada. As cicadas sing in a group, the song takes on oscillating waves that emerge as properties with unique harmonics and function, distinct from the screech of the single male.2 In these cases, the properties of the group do not directly relate to properties of the single member, but emerge as elements uniquely related to the single members when they aggregate.
Closer to home, synaptic plasticity is an emergent property of the molecular connections and transmitter release in a single synapse, which reflects the activity patterns of that synapse together with its interactions from adjacent synapses.3 The migration of groups of cells in a 3‐dimensional environment takes on properties that are not present in isolated cell migration. Migrating cells signal through group interactions that relay distant cues across the whole population,4 something metaphorically similar to a school of fish.
Tissue reorganization and repair after stroke show emergent properties that stem from interactions of individual elements in reorganizing brain tissue. The basic or elemental properties of neural repair include axonal sprouting, neurogenesis, gliogenesis, and changes in neuronal excitability in peri‐infarct tissue. Each of these cellular events is a distinct and definable property of the brain tissue response to stroke, and has irreducible elements in its response. For example, reactive astrocytes downregulate the uptake of the inhibitory transmitter γ‐aminobutyric acid (GABA), and this causes an elevation in tonic GABA signaling and hypoexcitability of pyramidal neurons adjacent to stroke.5 In poststroke neurogenesis, angiogenic vessels release chemokines and growth factors that simulate immature neurons to migrate to areas of brain injury adjacent to the stroke core.6, 7, 8 These individual neural repair events interact in the aggregate and evolve over time to produce properties that are different, larger, and more important than the properties of the single reactive astrocyte or migrating neuroblast. In a definitional sense, the formation of new neurons,6, 7, 8 new oligodendrocytes,9, 10 or new connections11, 12, 13 has been interchangeably described as “repair” or “regeneration.” Both terms connote a process of renewal and growth of tissues after injury, and are literally true in a limited fashion in the central nervous system (CNS) after injury.12 This review will discuss emergent properties of neural repair from the epidemiology of stroke to the synapse, highlighting areas of clinical translation opportunity. These emergent properties are reflected in the following sections: Stroke Is Not a Killer but a Chronic and Progressive Disabling Disease, Behavioral Activity Shapes Tissue Regeneration, The Suffered is the Learned, Plasticity Is a Risk for Neuroprotection, The Brain Forms Regenerative Cellular Niches during Repair and Recovery, Engaging CNS Tissue Regeneration Is Like Activating a Cancer, Neural Repair Therapies Require Directed Effect, and Regeneration Does Not Recapitulate Development.
Stroke Is Not a Killer but a Chronic and Progressive Disabling Disease
Stroke mortality is declining. Two years ago, stroke slipped from the third leading cause of death to the fourth,14 and this year it fell to the fifth leading cause of death.15 This decline in mortality is welcome and stems from implementation of stroke guidelines and improved care in the acute setting. Recent epidemiological studies indicate a decline in stroke incidence.16 However, even with this decline in stroke incidence, mortality is declining faster than the reduction in incidence.16 The prevalence of stroke is thus increasing. An emergent property of stroke epidemiology is that stroke is ever more a chronically disabling disease; stroke victims survive their stroke but not their disability. This stroke disability is substantial. Up to 80% of stroke patients may ultimately recover the ability to walk short distances, but most do not achieve the ability for community ambulation.17 Initially, 70 to 80% of people who sustain a stroke have upper‐extremity impairment.18, 19 As with gait, most of these patients will recover, but many do not regain functional use of the paretic arm. In terms of personal function, 6 months after stroke a substantial proportion (25−53%) of people remain dependent in at least one activities of daily living task.20, 21
What makes matters worse is that not only is stroke disabling as reflected in these statistics, but stroke victims themselves show declines over time after their initial gains from neurorehabilitation.22, 23, 24, 25 Most of this decline may be due to inactivity and lack of task‐specific practice.26 This means that stroke disability progresses. The decline of stroke patients in initially recovered neurological function over time after stroke, through disuse or nonuse of the affected function, is a major target of outpatient neurorehabilitative therapies, such as occupational, physical, and speech therapy, but to only limited success. Overall in the epidemiology of stroke, the elemental facts of declining mortality, increasing prevalence, and progressing disability mean that the emergent epidemiology of stroke is that it is now a chronically disabling and progressive disease. With no medical therapies that address neurological recovery after stroke, developing treatments for this chronically disabling and progressive disease becomes a research priority.
Behavioral Activity Shapes Tissue Regeneration
Stroke alters behavioral activity by directly changing neurological function, such as producing a hemiparesis, incoordination, or aphasia. Clinicians in turn alter behavioral activity after stroke through neurorehabilitation, in which patients are placed in regimens of repetitive and task‐specific overuse of their affected function. The behavioral and neuronal activity of the brain regions that undergo repair alter the cellular events that are occurring during tissue repair of these brain regions. An emergent property in stroke neural repair is that behavioral activity interacts with elements of cellular repair after stroke to create properties that are unique and unexpected.
An elemental process of repair is that axonal sprouting and the formation of new connections after stroke occur in brain regions that are damaged or partially deafferented from the stroke. Stroke triggers new connections to form in motor, somatosensory, and premotor cortex adjacent to the stroke site, and in projections from cortex contralateral to the stroke site into distant connections in the striatum, midbrain, and spinal cord.11, 12, 13, 27, 28, 29, 30, 31, 32 In both the cortex contralateral to the stroke, and ipsilateral or peri‐infarct cortex, animal models indicate that axonal sprouting establishes new patterns of connections that are causally linked to recovery. In peri‐infarct cortex, when new connections are stimulated from motor to premotor cortex motor recovery is enhanced, and when these same connections are blocked from forming motor recovery is reduced.13, 33 In contralateral cortex, when new connections form in the cervical spinal cord, reaching into the portion of the cervical spinal cord that has lost its original projection from the stroke site, this process mediates part of the motor recovery in a rat stroke model.29 It appears that axonal sprouting is a widespread process after stroke, and that axonal sprouting in different functional areas can control recovery in distinct neuronal systems, such as corticospinal or premotor connections. These studies come from several distinct models of stroke in rats, mice, and primates. Axonal sprouting cannot be definitively identified yet in human imaging studies, but the most extensive cortical remapping after stroke occurs in the same functional systems and relative areas of peri‐infarct cortex that undergo axonal sprouting in primates, rats, and mice.12 Also, the paradigmatic axonal sprouting marker GAP43 was first studied in its association with human stroke, where is induced in peri‐infarct tissue.34
Behavioral activity is also highly likely to modulate the structure of axonal fiber tracts after stroke in addition to the distal axonal connections of these fiber tracts in the spinal cord or peri‐infarct cortex. Human studies show that the structure of specific white matter tracts are associated with functional recovery after stroke, and can respond to behavioral activity.35, 36 The relationship of white matter tract structure and axonal sprouting has not been determined. Human studies image large white matter tracts and, through the diffusion of water, their myelination state and their directional organization.35, 37 Rodent studies image truly small axonal collaterals and fine synaptic terminations within the cervical spinal cord.27, 28, 29, 30, 31 One set of studies is looking at the cross‐country electrical power transmission lines and the other is looking at the electrical wiring of the end‐user's house. It is quite possible that a rehabilitation therapy may affect white matter structure and myelination, and not affect axonal terminal fields; or that axonal terminal fields may be affected and not gross structure of the myelin tract; or that both are affected by the same behavioral therapy after stroke. Because both myelinated fiber tracts and axonal terminal fields are sensitive to behavioral modulation,29, 36, 38 both are likely altered to some degree after stroke and through neurorehabilitative therapies. But at present, the role of behavioral activity in modulating white matter structure after stroke has not been determined in animal models and only larger white matter tracts can be studied in humans.39
The process of poststroke axonal sprouting has basic cellular and molecular properties. Axonal connections are confined by the expression of glial growth inhibitory molecules, such as NogoA, EphrinA5, and chondroitin sulfate proteoglycans.11, 12, 27, 28, 29, 30, 31, 32, 33 Opposed to these glial growth inhibitors, sprouting neurons activate a unique regenerative molecular program.11, 12, 13 Outside of stroke, behavioral activity in the normal adult brain, such as overuse or learning in a forelimb task, induces local changes in synaptic connections on a small scale within the corresponding motor cortex.33, 40, 41 However, when the behavioral activity patterns of neurorehabilitative therapy are added to the axonal sprouting response in stroke, emergent properties appear that are not present in either of these 2 parent conditions.
One example of the interaction of behavioral brain activity and axonal sprouting is seen in peri‐infarct cortex after stroke. Stroke induces axonal sprouting in motor, somatosensory, and premotor areas in peri‐infarct cortex. When glial growth inhibitors are blocked after stroke, this axonal sprouting response is locally enhanced in these cortical areas.11, 33 When glial growth inhibitors are blocked and the animal is forced to overuse the affected forelimb, a model for the clinical approach of constraint‐induced motor therapy, there is a magnitude of axonal sprouting that is remarkable in the adult brain. New projections are formed from motor cortex to widespread areas in prefrontal, orbitofrontal, temporal, and parietal cortical areas in a substantial remapping of the ipsilateral hemisphere.33 The magnitude and pattern of axonal sprouting that are seen when behavioral activity is modified under conditions of manipulation of glial growth inhibitors emerge as unique, widespread, and unpredictable from the 2 individual conditions that underlie this response: behavioral activity alone and glial growth inhibition blockade alone.40 This emergent property suggests that neurorehabilitative therapy will have great power to shape neuronal connections when these connections are released from the normal inhibition of the poststroke brain (Fig 1).
Other types of poststroke axonal sprouting are also responsive to behavioral activity patterns. Axonal sprouting from the cortex contralateral to large strokes occurs in the projection from sensorimotor regions to the cervical spinal cord. This axonal sprouting response is into the portion of the cervical spinal cord that lost its corticospinal projection from the stroke site.12, 27 Blocking the myelin growth inhibitory protein NogoA or stimulating axonal sprouting with inosine enhances this axonal sprouting response.27, 28, 29, 30, 31 Stimulating behavioral activity after stroke, such as with an enriched environment or repetitive forelimb tasks, also stimulates axonal sprouting from the cortex contralateral to the stroke into this denervated spinal cord.28, 31, 32 When increased behavioral activity and blockade of Nogo signaling are combined, there is a significant increase in spinal cord axonal sprouting in stroke. However, if the behavioral activity increase occurs at the time of the Nogo blockade, the axonal sprouting is exuberant and aberrant in the spinal cord and overall functional recovery is actually degraded—worse than in stroke alone. If Nogo blockade is induced first, followed by the rehabilitative activity, axonal sprouting is more targeted in the spinal cord and behavioral recovery is enhanced.29 These findings, like those noted above in peri‐infarct cortex, indicate that the combination of altered behavioral activity and blockade of growth inhibitors produces emergent properties that are not expected from the effects of either physical activity or growth‐inhibitory interventions alone.
The Suffered Is the Learned
The process of spontaneous recovery after stroke shares similarities with the process of normal motor learning. Both involve similar neuropsychological characteristics, such as learned nonuse, mass action, contextual interference, and distributed practice.42 Both occur with similar brain imaging changes in which an initially diffuse network of brain areas is funneled down with learning, training, or recovery into a core set of brain areas directly involved in the task. On a cellular level, processes of memory formation and network changes in the poststroke brain are both associated with long‐term potentiation–like phenomena and dendritic spine morphogenesis. On a molecular level, learning and memory paradigms, such as in the hippocampus, are associated with expression changes in stathmin, RB3, GAP43, and the Nogo signaling system, and these same molecular pathways are involved in recovery from stroke.12, 43 An emergent property in neural repair after stroke is that molecular systems that mediate the synaptic plasticity underlying learning and memory are coopted in brain injury to produce recovery of function.
There has been recent experimental testing of this idea. Two signaling processes that impact the synaptic signaling in learning and memory have been implicated in recovery after stroke. Tonic GABA receptors respond to extrasynaptic or ambient levels of GABA, are more sensitive than phasic (synaptic) GABAa receptors, and desensitize more slowly. These inhibitory receptors can thus mediate an inhibitory chloride current that controls the baseline firing threshold of pyramidal neurons.44 Tonic or extrasynaptic GABA signaling is a target in the learning and memory field, as blocking this current promotes neuronal excitability in the hippocampus and enhances learning and memory in many animal models.44, 45, 46 In stroke, tonic GABA signaling is increased in peri‐infarct cortex in a zone of cortex near the stroke site (0.2mm adjacent to the stroke) and produces a hypoexcitable state in pyramidal neurons in brain regions that normally mediate recovery of function. This can be pharmacologically reversed to enhance recovery in several rodent models of stroke at a considerable delay after the infarct.5, 47
The finding of increased tonic inhibition or effectiveness of agents that block tonic GABA inhibition in promoting behavioral recovery has been reported in 2 models of cortical stroke in 2 species (rat and mouse) by independent investigators.5, 47 This replication meets part of the goals of stroke drug development guidelines (STAIR and STEPS criteria48, 49) and is fairly rigorous in this field in terms of support. There remain additional studies that might enhance these findings, such as testing in other stroke models. Clinically, the literature on tonic GABA inhibition in humans after stroke is not strong because of the inability to measure tonic versus phasic GABA signaling in clinically available techniques. No technique in human studies (theta burst magnetic stimulation [TMS], transcranial direct current stimulation [tDC], magnetic resonance imaging [MRI] positron emission tomography) can specifically measure tonic and not phasic inhibition.50 Perhaps the closest ability to replicate the findings behind the mechanism of increased tonic GABA inhibition in a human study would be in measuring total extrasynaptic GABA levels, which in the mouse are elevated because of reduced astrocyte uptake of GABA after stroke.5 However, the only ability to measure GABA in humans is with [18]F‐flumazenil (which measures synaptic GABA receptor binding occupancy) and GABA magnetic resonance spectroscopy MRI (which measures total GABA levels in a 2 × 2 mm block of brain tissue). Neither of these approaches is sensitive to phasic (synaptic) versus tonic (extrasynaptic) GABA levels or activity.50, 51 Nonetheless, there are reports that good functional recovery is correlated with declining GABA levels after stroke52, 53 and that motor learning is associated with a reduced GABA level in motor cortex.54
These human and rodent findings on levels of inhibition after stroke imply generally that manipulation of cortical excitability may modulate recovery. tDC and TMS are methods to do this in humans. Unlike the pharmacological manipulation of tonic GABA in rodent models of stroke, which specifically manipulate excitatory neurons and preferentially those that have been impacted by stroke,5 tDC and TMS more grossly affect all of the brain tissue in the electrical or magnetic field, and induce many distinct types of cortical circuits.55 Nonetheless, small‐scale studies indicate that excitatory tDC or TMS over the ipsilesional hemisphere in stroke, or inhibitory tDC over the contralateral hemisphere, may improve recovery after stroke.55 Larger‐scale trials are under way for this application.56, 57
Synaptic activity mediated by the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) subtype of the glutamate receptor is also a key process in memory formation. Manipulations that enhance AMPA receptor signaling increase long‐term potentiation and produce increased performance in many models of learning and memory.58 Positive allosteric modulators of the AMPA receptor, which enhance AMPA receptor signaling only when glutamate is bound to the receptor, were originally developed as memory‐enhancing drugs.59 Termed AMPAkines, these drugs stimulate learning and memory in normal conditions and disease models.59, 60 In stroke, AMPAkines also promote recovery of function. Specifically, high‐impact AMPAkines, which induce downstream production of brain‐derived neurotrophic factor (BDNF), promote motor recovery. This action is via BDNF production, rather than through the increased excitability of AMPA receptor ion channel opening duration or amplitude. The action of AMPAkines after stroke occurs specifically in peri‐infarct cortex, further indicating that stroke specifically alters neuronal networks in regions of recovery in a way that parallels learning and memory function.61 The circuit‐specific and regional effect (peri‐infarct cortex) in modulating AMPA receptor signaling after stroke with pharmacological means contrasts with the more generalized action of tDC and TMS in inducing excitatory circuits in the brain.
These data in tonic GABA and AMPA receptor signaling are supported by other studies in which processes that affect molecular systems first described in learning and memory contexts also improve recovery after stroke.61 The ancient Greeks used the term “ta pathemeta mathmeta,” the suffered is the learned. An emergent property in neural repair after stroke is that the damage suffered from the infarct sets in place mechanisms of learning that can be manipulated to promote recovery.
Plasticity Is a Risk for Neuroprotection: Timing for a Neural Repair Therapy
The neuronal excitability in learning and memory systems after stroke that leads to enhanced recovery, and the increased behavioral activity in stroke rehabilitation that leads to enhanced recovery, also define a key emergent concept: mechanisms that enhance neuronal plasticity also destabilize a brain's ability to deal with stress. Put more simply, enhancing neuroplasticity at times in which the acute insult of the stroke is still present will make the stroke worse. In studies with blocking tonic GABA signaling or enhancing signaling through the AMPA glutamate receptor, when therapy was within 3 to 5 days from the stroke, GABA antagonists or AMPA drugs make the stroke worse by increasing infarct size. When these therapies are initiated after this 3‐ to 5‐day period, they do not change infarct size and enhance motor recovery.5, 60 Many neural repair therapies enhance endogenous neuronal plasticity in ways that activate neuronal excitability, such as with enhancing signaling of the transcription factor Creb,61 and might be expected to also follow this timeline. This general principle of a tradeoff between enhancing the plasticity of a cell versus ensuring its ability to withstand stress is seen in other neuronal systems. Manipulations of the growth cone protein GAP43 enhance the ability of a neuron to grow a new axon after injury but also increase the level of cell death from that very same injury.62, 63 Neuronal cell types with the greatest resistance to cell death after injury usually also have the lowest regenerative capacity, such as Purkinje cells, whereas neuronal cell types with the greatest regenerative capacity after injury also suffer the most cell death when injured, such as inferior olivary neurons.64 In neuronal sites as diverse as the optic nerve, dorsal root ganglion, and cortex, lesions closest to the cell body provoke the greatest amount of cell death, but also induce a greater regenerative response—compared to lesions distant from the cell body, in which there is little cell death and also little regeneration.64, 65, 66 Outside of these experimental lesion and molecular observations, increasing the activity of the motor system early after stroke may exacerbate the stroke insult itself in animal models even though this same activity will lead to improved recovery at a later period after stroke.65
Based on this transition from stroke damage to stroke repair, the timing of an activity or plasticity therapy in human stroke, based on animal model data, is not exactly clear. The early events of inflammatory cell infiltration and astrocytosis appear to follow similar time courses in rodents and humans. The recovery curve for rodents also has a similar shape and ceiling to that of humans, except that it is constricted to the first month after stroke in rodents and to the first 3 months after stroke in humans.67, 68 In humans, early and increased neurorehabilitative therapy after stroke may also worsen outcome compared to similar neurorehabilitative therapies introduced later.69 The early time period in this VECTORS trial was 9 days after stroke.69 Thus, it may be inferred that starting a neuroplasticity therapy earlier than the first week after stroke in humans positions this therapy within a window of risk for exacerbation of the initial stroke damage.
The opposite applies in this emergent principle as well; treatments that protect the brain in stroke may worsen behavioral performance if given during the recovery phase.60 Glutamate receptor antagonists and GABA receptor agonists reduce neuronal excitability and stroke size when given early after stroke, but degrade behavioral performance and recovery when given later after stroke.60, 70 The upshot of this emergent concept in stroke neural repair is that treatments that promote plasticity and recovery must clearly be distinguished from treatments that promote stability and protection, and a timeline must be developed in which each has its own window.
The Brain Forms Regenerative Cellular Niches during Repair and Recovery
Stroke triggers regenerative responses in neural stem cells and glial progenitors that mediate some aspects of repair. The term “regeneration” is far‐reaching in its implication but literally true in its application to the limited processes of neurogenesis,6, 7, 8 oligodendrocyte generation,9, 10 and formation of new connections after stroke12 in that these processes form new circuits and cells in the brain after destructive injury. In poststroke neurogenesis, the multipotent neural stem cells and transit amplifying cells in the subventricular zone respond to stroke and proliferate. Immature neurons produced from these progenitor cells migrate to areas of injury and can differentiate into mature neurons with local synaptic connections and long‐distance projections.6, 7, 8 Ablation of newly derived immature neurons after stroke causes reduced recovery.71 Immature neurons localize to angiogenic blood vessels in damaged tissue and are stimulated to migrate by growth factors or cytokines released by these vessels.6, 7, 8 However, despite a robust initial neurogenic response, most of these immature neurons die. Poststroke neurogenesis has been reported in human stroke, by utilizing tissue staining for protein markers of immature neurons in autopsy material.72, 73, 74 However, a lack of poststroke neurogenesis has been reported in human cortical stroke, using 14C labeling of newly born cells.75 Both techniques have limitations in specificity and sensitivity, and may also miss a transient neurogenic response after stroke that is limited in size and then stopped.76, 77 Because of the nature of human studies, a definitive finding of clinical poststroke neurogenesis remains lacking.
Stroke also stimulates oligodendrocyte progenitor cells (OPCs) to divide and partially differentiate adjacent to the lesion,9, 10 a form of poststroke gliogenesis. This has also been reported in human stroke.78 Further white matter remodeling occurs in humans with stroke and with therapies that promote recovery in human stroke.79, 80 OPCs carry the capacity to differentiate into mature oligodendrocytes and are in a position to mediate neural repair, as occurs in the initial stages of multiple sclerosis. However, glial progenitor cells after stroke do not appear to differentiate into oligodendrocytes as in multiple sclerosis and the damage produced to myelinated fiber tracts is even worse in aged animals after white matter stroke.81 This age effect on OPC and white matter responses appears to be mediated by greater local inflammation in the aged brain81 and by intrinsic differences in aged OPCs.82 In another aspect of gliogenesis, stroke induces proliferation of astrocytes adjacent to the lesion,83, 84 and a remarkable generation of new astrocytes from neural progenitor cells that migrate from the subventricular zone.85, 86 Astrocytes proliferate near human stroke,87 but the role of this generation of new astrocytes immediately adjacent to stroke in recovery remains to be defined.
Neurogenesis and gliogenesis involve elemental cellular programs of tropism, migration, and stimulation (neurogenesis) or inhibition (gliogenesis) by inflammation. However, these elemental cellular responses occur within the larger context of multicellular niches. Poststroke neurogenesis occurs within a neurovascular niche in which angiogenesis and neurogenesis are causally interconnected.6, 7, 8 Gliogenesis occurs in a zone of reactive astrocytes and damaged axons in both rodents and humans and appears limited by cues from these cellular compartments.81, 88 The emergent property of tissue regeneration after stroke is that neural progenitor responses occur in regenerative cellular niches within damaged tissues that are transient and unique to the injured brain. The concept of a progenitor niche in neural repair after stroke is informed by the concept of the stem cell niche. The original description of the stem cell niche explained the maintenance of bone marrow stem cells in aging and in response to chemotherapy.89 Scadden recently updated this concept: “Stem‐cell populations are established in 'niches'—specific anatomic locations that regulate how they participate in tissue generation, maintenance and repair…It constitutes a basic unit of tissue physiology, integrating signals that mediate the balanced response of stem cells to the needs of organisms.”90 Progenitor cells in stroke engage in tissue repair within transitory regenerative niches whose properties emerge from the constituent cells and from the forces that induce the initial progenitor response. These regenerative niches after stroke mediate cues from the surrounding environment, communicate signals that reflect age and likely comorbid diseases, and ultimately define the outcome of recovery. A promising area of future research will be to define the molecular signaling systems within these niches to enhance progenitor responses, maturation, and repair.
Engaging CNS Tissue Regeneration Is Like Activating a Cancer
Neural repair means in part activating a growth program in an adult neuron to form new connections or inducing progenitor responses to tissue injury signals.11, 12, 13, 28 In axonal sprouting, induction of oncogenes such as c‐myc 91 and RAS 92 promote the formation of new connections in stroke and spinal cord injury models. Blockade or inactivation of tumor suppressor proteins, such as PTEN and SOCS3, also promote axonal sprouting or functional recovery in stroke, spinal cord injury, optic nerve injury, and other types of CNS injury.93, 94 But PTEN is also mutated or inactivated in up to 70% of prostate cancers95, 96 and 40% of glioblastomas.97 SOCS3 is a tumor suppressor, and its function is lost in liver, lung, and squamous head and neck cancer.98 Molecular receptor systems, such as transforming growth factor β (TGFβ) receptors, induce a growth state and axonal sprouting in stroke13 and also are key molecules in metastatic transformation in cancer, such as the epithelial–mesenchymal transition.97, 99, 100 Recently described microRNAs that induce neural repair or axonal growth responses, such as miR‐9 and miR‐133,101, 102 are also closely linked to oncogenesis.103, 104, 105 Thus, there is a substantial molecular overlap between gene systems that promote tissue regeneration and recovery in brain and spinal cord injury, including stroke, and also induce initial formation of a cancerous state or promote tumor metastasis.
On a phenotypic level, there is similarity between neuronal and glial progenitor responses after stroke and cancer. Following a stroke, progenitor cells are induced into a growth program that involves cell division, migration to a tropic cue, and association with angiogenic vessels.6, 7, 8 This is a cellular response similar to tumor metastasis, in which primary tumor cells invade adjacent tissue, circulate, and localize to angiogenic vessels.97, 99 Like metastatic tumor cells,100 migrating progenitors after stroke secrete matrix metalloproteinases to digest their way to the target tissue.106 Thus, neurogenesis after stroke occurs in a neurovascular niche and tumor metastases home to and then create a similar vascular niche.
Each of the elemental properties of neural repair in stroke in neurogenesis and axonal sprouting, and in cancer in tumor initiation and metastasis, when taken into the larger context of biological responses in the body leads to this emergent property in CNS regeneration: molecular programs in cancer and in brain repair are shared. Outside of the CNS, similar pathways regulate tissue regeneration in other organ systems within molecular pathways that are also involved in cancer development.107 This emergent property of tissue regeneration is obviously problematic for the design of neural repair therapies. The emergent property of neural repair as a cancer program leads to the next emergent property of neural repair.
Neural Repair Therapies Require Directed Effect
Therapies that stimulate neural repair after stroke will need to be controlled in their effect in both time and space because of the overlap in molecular programs between oncogenesis and neural repair. Also, outside of this cancer overlap, many of the growth factors or cytokines that stimulate neural repair in preclinical models have widespread effects in other body tissues. Examples of these are erythropoietin, fibroblast growth factor, and granulocyte colony‐stimulating factor. In preclinical studies, these molecules stimulated axonal sprouting, neurogenesis, and other aspects of neural repair. However, in clinical trials their off‐CNS effect limited their use, with renal, hemodynamic, bone marrow, and thrombogenic complications.108, 109, 110 In addition to these past examples, recent discoveries in the neural repair field further make this point. The TGFβ family member GDF10 serves as a signal after stroke that activates a gene expression program in peri‐infarct tissue, which produces axonal sprouting and motor recovery.13 However, this molecule signals through TGFβRI and II, and this system also plays a role in tumor metastasis.100 A GDF10 therapeutic agent would need to be delivered locally to peri‐infarct tissue, or only for a brief period in the subacute phase after stroke when axonal sprouting is active, or both, so as to minimize potential oncogenic or non‐CNS complications. Similar therapeutic control has been proposed for PTEN inhibition,111 which would be another pathway to enhance axonal sprouting and recovery. One mechanism for spatial and temporal control of delivery of a neural repair drug is to use tissue bioengineering, with hydrogels that self‐assemble in brain and locally release a small molecule or biologic agent to the peri‐infarct tissue.11, 13, 33, 112
Regeneration Does Not Recapitulate Development
CNS tissue regeneration or repair has many similarities to neurodevelopment. In the cortex adjacent to the stroke core, neurons lose their perineuronal net, show altered intracortical inhibition, and become growth factor dependent.5, 12, 113, 114 These are hallmarks of neurons in the critical period of neuronal development, when cortex is uniquely plastic to environmental alterations in physiology and structure.115 In poststroke neurogenesis, multipotent neural stem cells give rise to immature neurons that migrate long distances and mature in small numbers into neurons with synaptic connections and long‐distance projections.6, 7, 8 This of course resembles both pyramidal neuron development in cortex and inhibitory neuron development in the forebrain.116 Such similarity in tissue repair in stroke to neurodevelopment has prompted suggestions that brain regeneration recapitulates development.117 Similar comparisons to regeneration and development have been made in other systems, such as kidney, bone, liver, and skin.118, 119, 120, 121
This comparison of regeneration to development takes its impetus from a similarity in phenotype: axonal extension and synaptogenesis, perineuronal net condition, growth factor dependency, neuroblast migration. However, what is critical for the biology of regeneration and for a possible regeneration therapeutic agent is whether the underlying molecular profile of CNS regeneration recapitulates the molecular profile of neurodevelopment. Initial transcriptional profiling of neurons that form new connections after stroke, retinal ganglion cells that regenerate an injured axon in the optic nerve, or dorsal root ganglion cells that regrow a connection after peripheral nerve injury suggest some overlap with genes that are active in the developing nervous system, but overall a distinct set of genes that is regulated in these injury responses.11, 122 However, direct comparison of the transcriptome of neurons exposed to a regenerating stimulus after stroke and the transcriptomes of neurons at several stages of neuronal development from many different laboratories clearly indicates that the molecular expression profile of regeneration is statistically and fairly dramatically distinct from the developmental transcriptome.13 Thus, an emergent property of neural repair is that on a molecular level regeneration does not recapitulate development. This principle is seen in other systems. The molecular control of regeneration is distinct from that in development in muscle repair.123 Even in highly regenerative animals, such as the newt or salamander in which a whole limb develops after injury, the process of regeneration is distinct from development.124
Conclusion
Recent studies have identified 7 emergent properties in neural repair after stroke. Stroke is not a killer but a chronic and progressively disabling disease. During the limited recovery that occurs after stroke, the brain activates growth programs in surviving neurons, which can form new connections in ways that are sensitive to behavioral activity or neurorehabilitative paradigms. These molecular programs are distinct to CNS regeneration or modified from their context and overall signaling partners from those in neurodevelopment. Tissue repair after stroke occurs within transient regenerative cellular niches that communicate cues to the regenerating cells that ultimately limit these events. The response of neurons to grow new connections or in neural progenitors to repair damaged tissue shares molecular features with malignant transformation of tissues and with metastasis. These emergent properties of neural repair present opportunities for future therapeutic development and offer challenges in the delivery of such therapies (see Fig 1). The field of neural repair has passed its first phase; the phenomenology of neural repair has been defined. Like walking down a summer path, the next steps in the field of neural repair are to appreciate the sounds of the cicadas, and more completely define how these individual neural repair phenomena interact, their emergent properties, so as to develop new approaches to enhance recovery.
Potential Conflicts of Interest
S.T.C. has received research grant support from Takeda Pharmaceuticals, Asterias Biotherapeutics, and Biotime. These entities have provided research funding for studies discussed in this review, but products or specific technologies in this funding are not discussed. S.T.C. receives no personal support from these entities.
Figure 1.
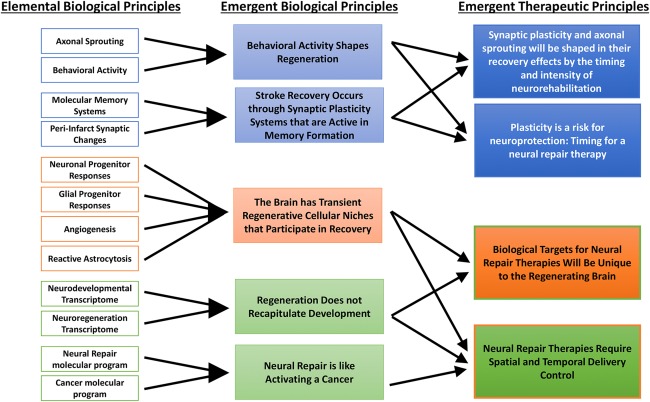
Evolution of scientific principles in the field of neural repair in stroke.
Acknowledgment
This work was supported by the Dr Miriam and Sheldon G. Adelson Medical Research Foundation, Richard Merkin Foundation for Neural Repair at UCLA, the American Stroke Association/Bugher Foundation, and the NIH (NS085019, NS081055, NS077521, NS071481).
References
- 1. Parrish JK, Viscido SV, Grunbaum, D. Self‐organized fish schools: an examination of emergent properties. Biol Bull 2002;202:296–305. [DOI] [PubMed] [Google Scholar]
- 2. Herbert‐Read JE, Nicolis S, Sumpter DJT. Collective waves of sound: call synchronisation in the Australian cicada, Henicopsaltria eydouxii Paper presented at: International Society for Behavioral Ecology Congress; July 31–August 5, 2014; New York, NY. [Google Scholar]
- 3. He Y, Kulasiri D, Samarasinghe S. Systems biology of synaptic plasticity: a review on N‐methyl‐D‐aspartate receptor mediated biochemical pathways and related mathematical models. Biosystems 2014;122:7–18. [DOI] [PubMed] [Google Scholar]
- 4. Londono C, Loureiro MJ, Slater B, et al. Nonautonomous contact guidance signaling during collective cell migration. Proc Natl Acad Sci U S A 2014;111:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clarkson AN, Huang BS, Macisaac SE, et al. Reducing excessive GABA‐mediated tonic inhibition promotes functional recovery after stroke. Nature 2010;468:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohab JJ, Carmichael ST. Poststroke neurogenesis: emerging principles of migration and localization of immature neurons. Neuroscientist 2008;14:369–380. [DOI] [PubMed] [Google Scholar]
- 7. Hermann DM, Chopp M. Promoting brain remodelling and plasticity for stroke recovery: therapeutic promise and potential pitfalls of clinical translation. Lancet Neurol 2012;11:369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kahle MP, Bix GJ. Neuronal restoration following ischemic stroke: influences, barriers, and therapeutic potential. Neurorehabil Neural Repair 2013;27:469–478. [DOI] [PubMed] [Google Scholar]
- 9. Sozmen EG, Kolekar A, Havton LA, Carmichael ST. A white matter stroke model in the mouse: axonal damage, progenitor responses and MRI correlates. J Neurosci Methods 2009;180:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miyamoto N, Maki T, Shindo A, et al. Astrocytes promote oligodendrogenesis after white matter damage via brain‐derived neurotrophic factor. J Neurosci 2015;35:14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li S, Overman JJ, Katsman D, et al. An age‐related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci 2010;13:1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmichael ST. The 3 Rs of stroke biology: radial, relayed, and regenerative. Neurotherapeutics 2016;13:348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li S, Nie EH, Yin Y, et al. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat Neurosci 2015;18:1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lackland DT, Roccella EJ, Deutsch AF, et al. Factors influencing the decline in stroke mortality: a statement from the American Heart Association/American Stroke Association. Stroke 2014;45:315–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Centers for Disease Control and Prevention. Stroke facts. 2015. Available at: http://www.cdc.gov/stroke/facts.htm. Accessed November 29, 2015.
- 16. Koton S, Schneider AL, Rosamond WD, et al. Stroke incidence and mortality trends in US communities, 1987 to 2011. JAMA 2014;312:259–268. [DOI] [PubMed] [Google Scholar]
- 17. Bogey R, Hornby GT. Gait training strategies utilized in poststroke rehabilitation: are we really making a difference? Top Stroke Rehabil 2007;14:1–8. [DOI] [PubMed] [Google Scholar]
- 18. Parker VM, Wade DT, Langton Hewer R. Loss of arm function after stroke: measurement, frequency, and recovery. Int Rehabil Med 1986;8:69–73. [DOI] [PubMed] [Google Scholar]
- 19. Nakayama H, Jørgensen HS, Raaschou HO, Olsen TS. Recovery of upper extremity function in stroke patients: the Copenhagen Stroke Study. Arch Phys Med Rehabil 1994;75:394–398. [DOI] [PubMed] [Google Scholar]
- 20. Gresham GE, Fitzpatrick TE, Wolf PA, et al. Residual disability in survivors of stroke–the Framingham study. N Engl J Med 1975;293:954–956. [DOI] [PubMed] [Google Scholar]
- 21. O'Mahony PG, Thomson RG, Dobson R, et al. The prevalence of stroke and associated disability. J Public Health Med 1999;21:166–171. [DOI] [PubMed] [Google Scholar]
- 22. Sonde L, Kalimo H, Fernaeus SE, Viitanen M. Low TENS treatment on poststroke paretic arm: a three‐year follow‐up. Clin Rehabil 2000;14:14–19 [DOI] [PubMed] [Google Scholar]
- 23. Kernan WN, Viscoli CM, Brass LM, et al. Decline in physical performance among women with a recent transient ischemic attack or ischemic stroke: opportunities for functional preservation a report of the Women's Estrogen Stroke Trial. Stroke 2005;36:630–634. [DOI] [PubMed] [Google Scholar]
- 24. Thorsén AM, Widén Holmqvist L, von Koch L. Early supported discharge and continued rehabilitation at home after stroke: 5‐year follow‐up of resource use. J Stroke Cerebrovasc Dis 2006;15:139–143. [DOI] [PubMed] [Google Scholar]
- 25. Dhamoon MS, Moon YP, Paik MC, et al. Trajectory of functional decline before and after ischemic stroke: the Northern Manhattan Study. Stroke 2012;43:2180–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dobkin B. The clinical science of neurologic rehabilitation. Oxford, UK: Oxford University Press, 2003. [Google Scholar]
- 27. Benowitz LI, Carmichael ST. Promoting axonal rewiring to improve outcome after stroke. Neurobiol Dis 2010;37:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zai L, Ferrari C, Subbaiah S, et al. Inosine alters gene expression and axonal projections in neurons contralateral to a cortical infarct and improves skilled use of the impaired limb. J Neurosci 2009;29:8187–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wahl AS, Omlor W, Rubio JC, et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science 2014;344:1250–1255. [DOI] [PubMed] [Google Scholar]
- 30. Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci 2004;24:6209–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lindau NT, Bänninger BJ, Gullo M, et al. Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti‐Nogo‐A therapy. Brain 2014;137:739–756. [DOI] [PubMed] [Google Scholar]
- 32. Lee KH, Kim JH, Choi DH, Lee J. Effect of task‐specific training on functional recovery and corticospinal tract plasticity after stroke. Restor Neurol Neurosci 2013;31:773–785. [DOI] [PubMed] [Google Scholar]
- 33. Overman JJ, Clarkson AN, Wanner IB, et al. A role for ephrin‐A5 in axonal sprouting, recovery, and activity‐dependent plasticity after stroke. Proc Natl Acad Sci U S A 2012;109:E2230–E2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng SC, de la Monte SM, Conboy GL, et al. Cloning of human GAP‐43: growth association and ischemic resurgence. Neuron 1988;1:133–139. [DOI] [PubMed] [Google Scholar]
- 35. Rüber T, Schlaug G, Lindenberg R. Compensatory role of the cortico‐rubro‐spinal tract in motor recovery after stroke. Neurology 2012;79:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rüber T, Lindenberg R, Schlaug G. Differential adaptation of descending motor tracts in musicians. Cereb Cortex 2015;25:1490–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raffin E, Dyrby TB. Diagnostic approach to functional recovery: diffusion‐weighted imaging and tractography. Front Neurol Neurosci 2013;32:26–35. [DOI] [PubMed] [Google Scholar]
- 38. Fields RD. A new mechanism of nervous system plasticity: activity‐dependent myelination. Nat Rev Neurosci 2015;16:756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lamar M, Zhou XJ, Charlton RA, et al. In vivo quantification of white matter microstructure for use in aging: a focus on two emerging techniques. Am J Geriatr Psychiatry 2014;22:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Overman JJ, Carmichael ST. Plasticity in the injured brain: more than molecules matter. Neuroscientist 2014;20:15–28. [DOI] [PubMed] [Google Scholar]
- 41. Kleim JA, Hogg TM, VandenBerg PM, et al. Cortical synaptogenesis and motor map reorganization occur during late, but not early, phase of motor skill learning. J Neurosci 2004;24:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krakauer JW. Motor learning: its relevance to stroke recovery and neurorehabilitation. Curr Opin Neurol 2006;19:84–90. [DOI] [PubMed] [Google Scholar]
- 43. Carmichael ST. Brain excitability in stroke: the yin and yang of stroke progression. Arch Neurol 2012;69:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 2007;56:763–770. [DOI] [PubMed] [Google Scholar]
- 45. Martin LJ, Zurek AA, MacDonald JF, et al. Alpha5GABAA receptor activity sets the threshold for long‐term potentiation and constrains hippocampus‐dependent memory. J Neurosci 2010;30:5269–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rudolph U, Möhler H. GABAA receptor subtypes: therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol 2014;54:483–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lake EM, Chaudhuri J, Thomason L, et al. The effects of delayed reduction of tonic inhibition on ischemic lesion and sensorimotor function. J Cereb Blood Flow Metab 2015;35:1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009;40:2244–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Savitz SI, Chopp M, Deans R, et al. Stem Cell Therapy as an Emerging Paradigm for Stroke (STEPS) II. Stroke 2011;42:825–829. [DOI] [PubMed] [Google Scholar]
- 50. Paulus W, Classen J, Cohen LG, et al. State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul 2008;1:151–163. [DOI] [PubMed] [Google Scholar]
- 51. Stagg CJ, Bachtiar V, Johansen‐Berg H. What are we measuring with GABA magnetic resonance spectroscopy? Commun Integr Biol 2011;4:573–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim YK, Yang EJ, Cho K, et al. Functional recovery after ischemic stroke is associated with reduced GABAergic inhibition in the cerebral cortex: a GABA PET study. Neurorehabil Neural Repair 2014;28:576–583. [DOI] [PubMed] [Google Scholar]
- 53. Blicher JU, Near J, Næss‐Schmidt E, et al. GABA levels are decreased after stroke and GABA changes during rehabilitation correlate with motor improvement. Neurorehabil Neural Repair 2015;29:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sampaio‐Baptista C, Filippini N, Stagg CJ, et al. Changes in functional connectivity and GABA levels with long‐term motor learning. Neuroimage 2015;1:106:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wessel MJ, Zimerman M, Hummel FC. Non‐invasive brain stimulation: an interventional tool for enhancing behavioral training after stroke. Front Hum Neurosci 2015;9:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. ClinicalTrials.gov. Search results for stroke TMS. Available at: https://www.clinicaltrials.gov/ct2/results?term=stroke+tms& recr=&rslt= &type=&cond =&intr=&titles=&outc=&spons= &lead=& id=&state1 =&cntry1=&state2=&cntry2=&state3=&cntry3=&locn=&gndr=&rcv_s=&rcv_e=&lup_s=&lup_e=. Accessed November 29, 2015.
- 57. ClinicalTrials.gov. Search results for stroke TDC. Available at: https://www.clinicaltrials.gov/ct2/results?term=stroke+tdc-recr=&rslt= &type=&cond=&intr=&titles=&outc=&spons=&lead=&id=&state 1=&cntry1=&state2=&cntry2=&state3=&cntry3=&locn=&gndr=& rcv_s=&rcv_e=&lup_s=&lup_e=. Accessed November 29, 2015.
- 58. Partin KM. AMPA receptor potentiators: from drug design to cognitive enhancement. Curr Opin Pharmacol 2015;20:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lynch G, Cox CD, Gall CM. Pharmacological enhancement of memory or cognition in normal subjects. Front Syst Neurosci 2014;8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clarkson AN, Overman JJ, Zhong S, et al. AMPA receptor‐induced local brain‐derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 2011;31:3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacDonald E, Van der Lee H, Pocock D, et al. A novel phosphodiesterase type 4 inhibitor, HT‐0712, enhances rehabilitation‐dependent motor recovery and cortical reorganization after focal cortical ischemia. Neurorehabil Neural Repair 2007;21:486–496. [DOI] [PubMed] [Google Scholar]
- 62. Harding DI, Greensmith L, Mason M, et al. Overexpression of GAP‐43 induces prolonged sprouting and causes death of adult motoneurons. Eur J Neurosci 1999;11:2237–2242. [DOI] [PubMed] [Google Scholar]
- 63. Huang C, Cen LP, Liu L, et al. Adeno‐associated virus‐mediated expression of growth‐associated protein‐43 aggravates retinal ganglion cell death in experimental chronic glaucomatous injury. Mol Vis 2013;19:1422–1432. [PMC free article] [PubMed] [Google Scholar]
- 64. Dusart I, Ghoumari A, Wehrle R, et al. Cell death and axon regeneration of Purkinje cells after axotomy: challenges of classical hypotheses of axon regeneration. Brain Res Brain Res Rev 2005;49:300–316. [DOI] [PubMed] [Google Scholar]
- 65. Richardson PM, Issa VM, Aguayo AJ. Regeneration of long spinal axons in the rat. J Neurocytol 1984;13:165–182. [DOI] [PubMed] [Google Scholar]
- 66. Mason MR, Lieberman AR, Anderson PN. Corticospinal neurons up‐regulate a range of growth‐associated genes following intracortical, but not spinal, axotomy. Eur J Neurosci 2003;18:789–802. [DOI] [PubMed] [Google Scholar]
- 67. Krakauer JW, Carmichael ST, Corbett D, Wittenberg GF. Getting neurorehabilitation right: what can be learned from animal models? Neurorehabil Neural Repair 2012;26:923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dobkin BH, Carmichael ST. The specific requirements of neural repair trials for stroke. Neurotherapeutics 2016;13:348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dromerick AW, Lang CE, Birkenmeier RL, et al. Very Early Constraint‐Induced Movement during Stroke Rehabilitation (VECTORS): a single‐center RCT. Neurology 2009;73:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lazar RM, Berman MF, Festa JR, et al. GABAergic but not anti‐cholinergic agents re‐induce clinical deficits after stroke. J Neurol Sci 2010;292:72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jin K, Wang X, Xie L, et al. Transgenic ablation of doublecortin‐expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc Natl Acad Sci U S A 2010;107:7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jin K, Wang X, Xie L, et al. Evidence for stroke‐induced neurogenesis in the human brain. Proc Natl Acad Sci U S A 2006;103:13198–13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Macas J, Nern C, Plate KH, Momma S. Increased generation of neuronal progenitors after ischemic injury in the aged adult human forebrain. J Neurosci 2006;26:13114–13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ekonomou A, Johnson M, Perry RH, et al. Increased neural progenitors in individuals with cerebral small vessel disease. Neuropathol Appl Neurobiol 2012;38:344–353. [DOI] [PubMed] [Google Scholar]
- 75. Huttner HB, Bergmann O, Salehpour M, et al. The age and genomic integrity of neurons after cortical stroke in humans. Nat Neurosci 2014;17:801–803. [DOI] [PubMed] [Google Scholar]
- 76. Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular‐associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 2006;24:1–14. [DOI] [PubMed] [Google Scholar]
- 77. Macklis JD. Human adult olfactory bulb neurogenesis? Novelty is the best policy. Neuron 2012;74:595–596. [DOI] [PubMed] [Google Scholar]
- 78. Sanin V, Heeß C, Kretzschmar HA, Schüller U. Recruitment of neural precursor cells from circumventricular organs of patients with cerebral ischaemia. Neuropathol Appl Neurobiol 2013;39:510–518. [DOI] [PubMed] [Google Scholar]
- 79. Zheng X, Schlaug G. Structural white matter changes in descending motor tracts correlate with improvements in motor impairment after undergoing a treatment course of tDCS and physical therapy. Front Hum Neurosci 2015;9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wan CY, Zheng X, Marchina S, et al. Intensive therapy induces contralateral white matter changes in chronic stroke patients with Broca's aphasia. Brain Lang 2014;136:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rosenzweig S, Carmichael ST. Age‐dependent exacerbation of white matter stroke outcomes: a role for oxidative damage and inflammatory mediators. Stroke 2013;44:2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Miyamoto N, Pham LD, Hayakawa K, et al. Age‐related decline in oligodendrogenesis retards white matter repair in mice. Stroke 2013;44:2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barreto GE, Sun X, Xu L, Giffard RG. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One 2011;6:e27881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by Notch‐1 in the peri‐infarct area after stroke. Stroke 2011;42:3231–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Benner EJ, Luciano D, Jo R, et al. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 2013;497:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Faiz M, Sachewsky N, Gascón S, et al. Adult neural stem cells from the subventricular zone give rise to reactive astrocytes in the cortex after stroke. Cell Stem Cell 2015;17:624–634. [DOI] [PubMed] [Google Scholar]
- 87. Dziewulska D. Age‐dependent changes in astroglial reactivity in human ischemic stroke. Immunohistochemical study. Folia Neuropathol 1997;35:99–106. [PubMed] [Google Scholar]
- 88. Hinman JD, Lee MD, Tung S, et al. Molecular disorganization of axons adjacent to human lacunar infarcts. Brain 2015;138:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schofield R. The relationship between the spleen colony‐forming cell and the haemopoietic stem cell. Blood Cells 1978;4:7–25. [PubMed] [Google Scholar]
- 90. Scadden DT. The stem‐cell niche as an entity of action. Nature 2006;441:1075–1079. [DOI] [PubMed] [Google Scholar]
- 91. Belin S, Nawabi H, Wang C, et al. Injury‐induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 2015;86:1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Makwana M, Serchov T, Hristova M, et al. Regulation and function of neuronal GTP‐Ras in facial motor nerve regeneration. J Neurochem 2009;108:1453–1463. [DOI] [PubMed] [Google Scholar]
- 93. Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008;322:963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sun F, Park KK, Belin S, et al. Sustained axon regeneration induced by co‐deletion of PTEN and SOCS3. Nature 2011;480:372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature 2005;436:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lino MM, Merlo A. PI3Kinase signaling in glioblastoma. J Neurooncol 2011;103:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 2013;15:807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Culig Z. Suppressors of cytokine signalling‐3 and −1 in human carcinogenesis. Front Biosci (Schol Ed) 2013;5:277–283. [DOI] [PubMed] [Google Scholar]
- 99. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med 2013;19:1450–1464. [DOI] [PubMed] [Google Scholar]
- 100. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dajas‐Bailador F, Bonev B, Garcez P, et al. microRNA‐9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat Neurosci 2012;15:697–699. [DOI] [PubMed] [Google Scholar]
- 102. Xin H, Li Y, Chopp M. Exosomes/miRNAs as mediating cell‐based therapy of stroke. Front Cell Neurosci 2014;8:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Drakaki A, Hatziapostolou M, Polytarchou C, et al. Functional microRNA high throughput screening reveals miR‐9 as a central regulator of liver oncogenesis by affecting the PPARA‐CDH1 pathway. BMC Cancer 2015;15:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu Y, Zhang X, Zhang Y, et al. Identification of miRNomes in human stomach and gastric carcinoma reveals miR‐133b/a‐3p as therapeutic target for gastric cancer. Cancer Lett 2015;369:58–66. [DOI] [PubMed] [Google Scholar]
- 105. Wang H, Zhang W, Zuo Y, et al. miR‐9 promotes cell proliferation and inhibits apoptosis by targeting LASS2 in bladder cancer. Tumour Biol 2015;36:9631–9640. [DOI] [PubMed] [Google Scholar]
- 106. Lee SR, Kim HY, Rogowska J, et al. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci 2006;26:3491–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. L Wan, K Pantel, Y Kang. Tumor metastasis: moving new biological insights into the clinic. Nat Med 2013;19:1450–1464. [DOI] [PubMed] [Google Scholar]
- 108. Ehrenreich H, Weissenborn K, Prange H, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 2009;40:e647–e656. [DOI] [PubMed] [Google Scholar]
- 109. Ringelstein EB1, Thijs V, Norrving B, et al. Granulocyte colony‐stimulating factor in patients with acute ischemic stroke: results of the AX200 for Ischemic Stroke trial. Stroke 2013;44:2681–2687. [DOI] [PubMed] [Google Scholar]
- 110. Bogousslavsky J, Victor SJ, Salinas EO, et al. Fiblast (trafermin) in acute stroke: results of the European‐Australian phase II/III safety and efficacy trial. Cerebrovasc Dis 2002;14:239–251. [DOI] [PubMed] [Google Scholar]
- 111. Naguib A, Trotman LC. PTEN plasticity: how the taming of a lethal gene can go too far. Trends Cell Biol 2013;23:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ma J, Tian WM, Hou SP, et al. An experimental test of stroke recovery by implanting a hyaluronic acid hydrogel carrying a Nogo receptor antibody in a rat model. Biomed Mater 2007;2:233–240. [DOI] [PubMed] [Google Scholar]
- 113. Carmichael ST, Archibeque I, Luke L, et al. Growth‐associated gene expression after stroke: evidence for a growth‐promoting region in peri‐infarct cortex. Exp Neurol 2005;193:291–311. [DOI] [PubMed] [Google Scholar]
- 114. Hobohm C, Günther A, Grosche J, et al. Decomposition and long‐lasting downregulation of extracellular matrix in perineuronal nets induced by focal cerebral ischemia in rats. J Neurosci Res 2005;80:539–548. [DOI] [PubMed] [Google Scholar]
- 115. Levelt CN, Hübener M. Critical‐period plasticity in the visual cortex. Annu Rev Neurosci 2012;35:309–330. [DOI] [PubMed] [Google Scholar]
- 116. Southwell DG, Nicholas CR, Basbaum AI, et al. Interneurons from embryonic development to cell‐based therapy. Science 2014;344:1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cramer SC, Chopp M. Recovery recapitulates ontogeny. Trends Neurosci 2000;23:265–271. [DOI] [PubMed] [Google Scholar]
- 118. Kelley‐Loughnane N, Sabla GE, Ley‐Ebert C, et al. Independent and overlapping transcriptional activation during liver development and regeneration in mice. Hepatology 200;35:525–534. [DOI] [PubMed] [Google Scholar]
- 119. Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development 2004;131:3021–3034. [DOI] [PubMed] [Google Scholar]
- 120. Kawakami T, Ren S, Duffield JS. Wnt signalling in kidney diseases: dual roles in renal injury and repair. J Pathol 2013;229:221–231. [DOI] [PubMed] [Google Scholar]
- 121. Hadjiargyrou M, O'Keefe RJ. The convergence of fracture repair and stem cells: interplay of genes, aging, environmental factors and disease. J Bone Miner Res 2014;29:2307–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bosse F, Hasenpusch‐Theil K, Küry P, Müller HW. Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury‐dependent and reactivated developmental processes. J Neurochem 2006;96:1441–1457. [DOI] [PubMed] [Google Scholar]
- 123. Wang J, Conboy I. Embryonic vs. adult myogenesis: challenging the 'regeneration recapitulates development' paradigm. J Mol Cell Biol 2010;2:1–4. [DOI] [PubMed] [Google Scholar]
- 124. Nacu E, Tanaka EM. Limb regeneration: a new development? Annu Rev Cell Dev Biol 2011;27:409–440. [DOI] [PubMed] [Google Scholar]