

Abstract
Background
The genome of multiple myeloma (MM) cells is extremely unstable, characterized by a complex combination of structure and numerical abnormalities. It seems that there are several “myeloma subgroups” which differ in expression profile, clinical manifestations, prognoses and treatment response. In our previous work, the list of 35 candidate genes with a known role in carcinogenesis and associated with centrosome structure/function was used as a display of molecular heterogeneity with an impact in myeloma pathogenesis. The current study was devoted to establish a risk stratification model based on the aforementioned candidate genes.
Methods
A total of 151 patients were included in this study. CD138+ cells were separated by magnetic-activated cell sorting (MACS). Gene expression profiling (GEP) and Interphase FISH with cytoplasmic immunoglobulin light chain staining (cIg FISH) were performed on plasma cells (PCs). All statistical analyses were performed using freeware R and its additional packages. Training and validation cohort includes 73 and 78 patients, respectively.
Results
We have finally established a model that includes 12 selected genes (centrosome associated gene pattern, CAGP) which appears to be an independent prognostic factor for MM stratification. We have shown that the new CAGP model can sub-stratify prognosis in patients without TP53 loss as well as in IMWG high risk patients’ group.
Conclusions
We assume that newly established risk stratification model complements the current prognostic panel used in multiple myeloma and refines the classification of patients in relation to the disease risks. This approach can be used independently as well as in combination with other factors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-016-0906-9) contains supplementary material, which is available to authorized users.
Keywords: Multiple myeloma, Gene expression profiling, Risk stratification
Background
Multiple myeloma (MM) is a lymphoproliferative disease characterized by the clonal expansion of neoplastic plasma cells within the bone marrow. The genome of the malignant plasma cells is extremely unstable, characterized by a complex combination of structure and numerical abnormalities. These abnormalities serve as background for large variability in clinical course and outcome of MM patients [1]. A multistep process of malignant transformation can explain the presence of these genetic and clinical heterogeneities, which is admittedly associated with cell cycle deregulation.
Although several staging systems based on clinical and laboratory tests have been developed for MM [2, 3], standard prognostic factors, such as β2-microglobulin, albumin and C-reactive protein, account for only 15–20 per cent of outcome heterogeneity [4]. Previously, conventional cytogenetics was recognized as a relevant prognostic tool in multiple myeloma. Nevertheless, in spite of advances in molecular cytogenetics, many others undefined abnormalities forming genetic complexity in MM may still exist [5].
In our previous studies, we have used gene expression profiling to analyze a set of genes involved in formation of centrosome abnormalities in MM. Taking into consideration that centrosome amplification is common in all stages of plasma cell neoplasia [6] and is therefore an early event in MM and can serve as a source of genomic instability [7, 8]. Centrosome associated molecular signature is related to overall survival as well as to clinical parameters and ISS staging in MM [9]. We have identified a gene pattern, which was used as a display of molecular heterogeneity with an impact on myeloma pathogenesis [10].
We believe that the analysis of molecular signature will supplement existing prognostic models based on screening of chromosomal aberrations in plasma cells. Thus, the objective of our study was to create and validate risk stratification model based on previously described centrosome associated genes pattern in MM.
Design and methods
Patients and sample preparation
A total of 151 patients with MM enrolled in University Hospital Brno, Czech Republic, University Hospital Olomouc, Czech Republic and University Hospital Bratislava, Slovakia, were included in this study. The study was approved by the Ethical Committee of the Faculty of Medicine, Masaryk University (chairman: Josef Kure, PhD; ref number: 14/2009 and 29/2011), and the study was conducted according to the Helsinki declaration. All patients provided written informed consent. Training and validation cohort includes 73 and 78 patients, respectively. Patients’ baseline characteristics are summarized in Table 1.
Table 1.
Patients’ baseline characteristic
| Training cohorta | Validation cohorta | |
|---|---|---|
| No. of patients | 73 | 78 |
| Follow-up median (min–max) [month] | 23.6 (0.3–97.0) | 18.6 (0.1–250.0) |
| Gender: males–females | 49.3–50.7 % | 55.1–44.9 % |
| Age median (range) [years] | 69 (38–84) | 66 (40–90) |
| ISS stage: I–II–III | 28.8 %–27.3 %–43.9 % | 24.7 %–35.1 %– 40.3 % |
| Durie-Salmon stage: I–II–III | 4.3 %–14.3 %–81.4 % | 3.8 %–29.5 %–66.7 % |
| Durie-Salmon substage: A–B | 81.4 %–18.6 % | 75.6 %–24.4 % |
| Ig isotype: IgG–IgA–FLC-Non-secr. | 60.3 %–23.5 %–16.2 % | 57.7 %–28.2 %–1.3 %–12.8 % |
| Light chains: kapp–lambda | 58.0 %–42.0 % | 53.8 %–46.2 % |
| Plasma cell infiltration in bone marrow | 34.4 % (0.8 %–93.6 %) | 36.0 % (2.2 %–81.2 %) |
| No. of previous treatment lines | ||
| None (first line treatment) | 57.7 % (41/71) | 64.1 % (50/78) |
| One | 19.7 % (14/71) | 15.4 % (12/78) |
| Two | 8.5 % (6/71) | 10.3 % (8/78) |
| More (>2) | 14.1 % (10/71) | 10.3 % (8/78) |
| Treatment regimen | ||
| Bortezomib-based | 47.8 % (32/67) | 64.9 % (50/77) |
| Thalidomide-based | 14.9 % (10/67) | 10.4 % (8/77) |
| Lenalidomide-based | 25.4 % (17/67) | 18.2 % (14/77) |
| Others | 11.9 % (8/67) | 6.5 % (5/77) |
| Treatment response | ||
| CR-VGPR-PR-MR-SD-PG | 12.3 %–29.8 %–22.8 %–5.3 %–5.3 %–26.3 % | 10.8 %–20.0 %–27.7 %–7.7 %–4.6 %–29.2 % |
| Biochemical parameters | ||
| Hemoglobin (g/l) | 105.5 (67.0–151.0) | 95.5 (65.9–146.0) |
| Thrombocytes (×109) | 192.0 (33.0–416.0) | 188.5 (55.0–485.0) |
| Calcium (mmol/l) | 2.29 (1.74–23.37) | 2.32 (1.75–2.78) |
| Albumin (g/l) | 38.2 (21.1–54.1) | 35.7 (17.4–52.2) |
| Creatinine (umol/l) | 98.5 (53.0–783.0) | 94.5 (30.0–849.0) |
| β2-microglobulin (mg/l) | 4.70 (1.79–42.60) | 4.61 (1.62–50.0) |
| Lactate dehydrogenase (ukat/l) | 3.72 (1.53–22.92) | 3.36 (1.14–7.77) |
| C-reactive protein (mg/l) | 3.6 (0.0–174.3) | 4.0 (0.0–149.3) |
| Monoclonal Ig (g/l) | 29.8 (0.0–92.6) | 30.2 (0.0–85.6) |
| Chromosomal abnormality | ||
| Deletion 13q14 | 49.2 % (30/61) | 60.0 % (39/65) |
| Deletion 17p13 | 8.3 % (5/60) | 13.8 % (9/65) |
| Translocation t(4;14) | 46.9 % (15/32) | 44.4 % (16/36) |
| Amplification 1q21 | 56.9 % (37/65) | 54.5 % (36/66) |
| Hyperdiploidy (H-MM) | 45.5 % (30/66) | 47.0 (17/46) |
CR complete response; VGPR very good partial response; PR partial response; MR minimal response; SD stable disease; PG progression
aBoth cohorts have no significant difference in basic clinical parameters
The bone marrow of patients was obtained during routine diagnostic procedure. Plasma cells in mononuclear cell fraction were enriched by anti-CD138+ immunomagnetic beads and sorted using AutoMACS (Miltenyi Biotec). Purity of CD138 + fraction was measured by flowcytometry and/or cytospin and samples with >80 % plasma cells were provided for total RNA isolation.
Gene expression profiling (GEP)
Total RNA was isolated using QIAGEN RNeasy Mini Kit. RNA isolation. purification, and microarray hybridization has been reported previously [11]. Total RNA with purity ratio 260/280 >1.7 and integrity (RIN) >7.5 (as measured by Agilent 2100 Bioanalyzer) was transcribed into cDNA (Ambion WT Expression Kit), labeled and hybridized to the Affymetrix GeneChip® Human Gene ST 1.0 array and further processed through R/Bioconductor framework by oligo package. RMA normalized data at gene level were statistically analyzed. Generated CEL files of patients included in this study have been deposited in the ArrayExpress Archive database under accession number E-MTAB-1038 for training set and E-MTAB-4032 for validation set. Both are available online (http://www.ebi.ac.uk/arrayexpress/).
Fluorescence in situ hybridization (FISH)
FISH was performed as a part of routine diagnostic procedure as previously described [12]. The following aberrations were studied: 1q21 gain, 13q14 deletion, 17p13 deletion and translocation t(4;14). Hyperdiploidy status was determined with commercial probes mapping to chromosome 5 (LSI D5S23/D5S721), 9 (CEP9) and 15 (CEP15) (Abbott Molecular, Des Plaines, IL, USA).
Statistical analysis
All statistical analyses were performed using freeware R and its additional packages: oligo, affy, survival, nnet and pROC [13–15]. Training and validation cohort includes 73 and 78 patients, respectively. Both cohorts have no significant differences in basic clinical parameters and all continuous variables were tested by nonparametric Mann–Whitney test. For categorical variables, the Fisher exact test was used. Overall survival (OS) was calculated from the date of diagnosis to death; progression free survival (PFS) was defined by the date of diagnosis and the date of disease progression or any death; time to progression (TTP) was defined by the date of diagnosis and the date of disease progression or disease-related death. Survival rates were estimated using the Kaplan–Meier method. Differences in survival of patients’ subgroups were compared using the log-rank test. p values below 0.05 were considered statistically significant.
Results
In our previous study [9], a set of 111 genes with a known role in oncogenesis associated with centrosomal structure/function abnormalities corresponding to their proteins were selected for hierarchical clustering of gene expression (RMA-normalized, log 2 transformed expression level) profiles on CD138+ plasma cells from 73 patients with multiple myeloma. Furthermore, clustering analysis revealed a pattern of 35 genes. These genes and patients were later re-clustered to reveal three subgroups of patients according to different expression patterns of the chosen genes: “high expressed,” “medium expressed” and “low expressed,” respectively.
The list of 35 initially identified candidate genes were utilized for the generation of 3-subgroup predictor. To discover the gene signature that is able to predict subgroup membership of each sample, the bidirectional stepwise selection procedure with multinomial logistic regression model was performed. The best model was selected due to the Akaike information criteria. To apply such procedure, 12 candidate genes (BUB1, BUB1B|PAK6, RAD51, PLK1, BRCA1, CENPA, BARD1, AURKA, MAD2L1, CENPH, XRCC2 and CDC25C|FAM53C) were identified and coefficients of multinomial regression model were estimated. The suggested regression model allows to predict the grouping of samples from both training and validation cohort into one of the subgroups (“high expressed,” “medium expressed” or “low expressed”). The pseudocode of predictive analyses and expression level of candidate genes are available in Additional files 1 and 2.
Based on this centrosome associated genes pattern (CAGP) model, patients were stratified into three groups—“High expressed”, “Medium expressed” and “Low expressed”. The overall survival of patients in “High expressed” and “Medium expressed” subgroups was significantly worse than in patients in “Low expressed” subgroup for both training and validation cohorts (Fig. 1) and was in concordance with previously published results [9]. Analysis of progression free survival (PFS) and time to progression (TTP) in the three CAGP expression groups did not reveal any significant differences (data not shown).
Fig. 1.
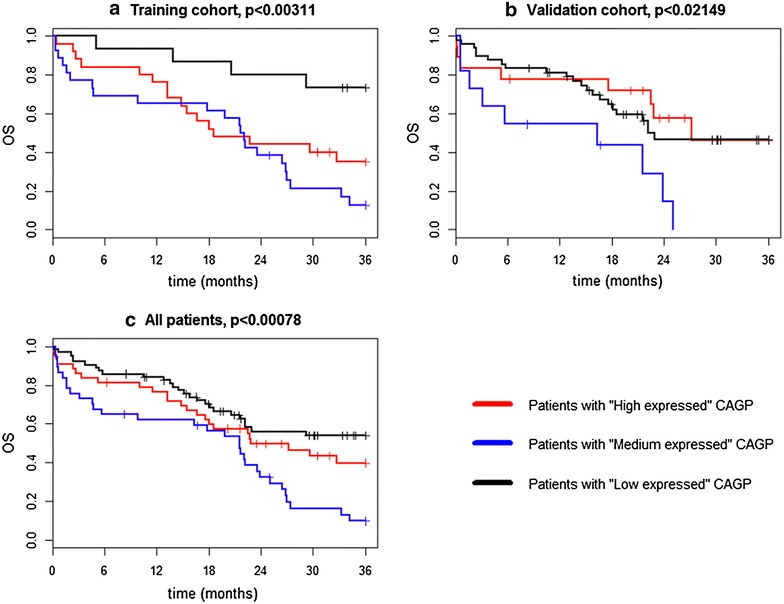
Overall survival of three CAGP expression groups of MM patients. a Kaplan–Meier curves for OS of training patients’ cohort (n = 73) stratified by centrosome associated gene pattern (CAGP). b Kaplan–Meier curves for OS of validation patients’ cohort (n = 78) stratified by centrosome associated gene pattern (CAGP). c Kaplan–Meier curves for OS of jointed training and validation patients’ cohort (n = 151) stratified by centrosome associated gene pattern (CAGP)
Additionally, we combined patients with worse overall survival and clinical characteristics in High- and Medium- expressed subgroups as “HR CAGP” (high risk centrosome associated genes pattern). Patients with Low expressed were defined as “LR CAGP” (low risk centrosome associated genes pattern), respectively. Significantly worse prognosis was found for HR CAGP group (HR = 1.8; 3-year OS = 25.1 %) compared to LR CAGP group (3-year OS = 53.8 %, p < 0.01). Analysis of PFS and TTP in two CAGR risk groups did not reveal any significant differences (Fig. 2).
Fig. 2.
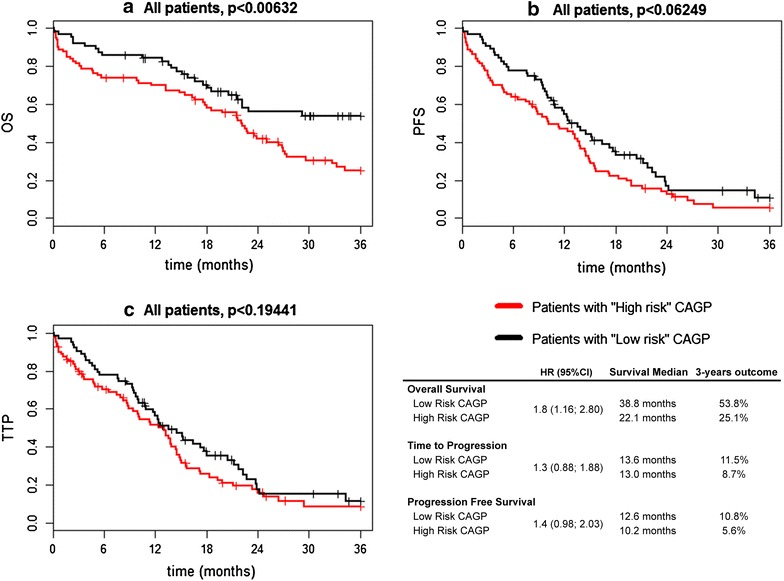
Overall survival, progression free survival and time to progression in CAGP risk groups of MM patients. a Kaplan–Meier curves for OS of combined training and validation patients’ cohort (n = 151) stratified by centrosome associated gene pattern (CAGP). b Kaplan–Meier curves for PFS of combined training and validation patients’ cohort (n = 151) stratified by centrosome associated gene pattern (CAGP). c Kaplan–Meier curves for TTP of combined training and validation patients’ cohort (n = 151) stratified by centrosome associated gene pattern (CAGP)
To characterize the prognostic significance of CAGP model, multivariate Cox proportional hazards survival model was used. Besides CAGP, the following parameters were used for multivariate Cox proportional hazards survival model: ISS stage, β2-microglobulin, del 17p13, t(4;14), amp 1q21. The variables in the multivariate model were the only variables, which remained statistically significant when potential predictors were combined together including CAGP, which was forced into the model. Among all subsequently tested combinations of predictors, the best results in risk of death assessment were obtained for CAGP combined with del 17p13 (p < 0.001). It is worth to mention that both prognostic factors were independent. “HR” CAGP subgroup as well as TP53 deletion had significantly higher risk of death assessment (HR = 1.8 and HR = 2.5, respectively; p < 0.005). Survival characteristics for different risk sub-groups are presented in Table 2. Apparent controversies in sub-stratified TP53 deletion groups probably associated with small cohort size (7 patients in each TP53+/low risk CAGP and TP53+/high risk CAGP sub-groups).
Table 2.
Survival characteristics for different TP53 risk groups sub-stratified with CAGP model
| Stratification group | N | HR (95 % CI) | Survival median (months) | 3-years outcome (%) | p value (log-rank) |
|---|---|---|---|---|---|
| Overall survival | |||||
| Low risk CAGP | 66 | 38.8 | 53.8 | <0.00799 | |
| High risk CAGP | 84 | 1.8 (1.16; 2.80) | 22.1 | 25.1 | |
| TP53− | 110 | 25.1 | 35.6 | <0.00281 | |
| TP53+ | 14 | 2.5 (1.35; 4.73) | 12.8 | 12.2 | |
| TP53−/low risk CAGP | 47 | 38.8 | 61.1 | <0.00099 | |
| TP53−/high Risk CAGP | 63 | 2.5 (1.41; 4.30) | 22.2 | 20.1 | |
| TP53+/low Risk CAGP | 7 | 12.8 | 0 | <0.3621 | |
| TP53+/high risk CAGP | 7 | 0.5 (0.14; 2.05) | 16.7 | 21.4 | |
| Time to progression | |||||
| Low risk CAGP | 66 | 13.6 | 11.5 | <0.1939 | |
| High risk CAGP | 84 | 1.3 (0.88; 1.88) | 13.0 | 8.7 | |
| TP53− | 110 | 13.9 | 11.6 | <0.01378 | |
| TP53+ | 14 | 2.1 (1.15; 3.79) | 8.7 | 0 | |
| TP53−/low risk CAGP | 47 | 16.4 | 15.3 | <0.08914 | |
| TP53−/high risk CAGP | 63 | 1.5 (0.94; 2.34) | 13.2 | 8.0 | |
| TP53+/low risk CAGP | 7 | 7.6 | 0 | <0.676 | |
| TP53+/high risk CAGP | 7 | 0.8 (0.23; 2.61) | 9.9 | 0 | |
| Progression free survival | |||||
| Low risk CAGP | 66 | 12.6 | 10.8 | <0.06232 | |
| High risk CAGP | 84 | 1.4 (0.98; 2.03) | 10.2 | 5.6 | |
| TP53− | 110 | 13.2 | 8.8 | <0.04351 | |
| TP53+ | 14 | 1.8 (1.01; 3.29) | 8.7 | 0 | |
| TP53−/low risk CAGP | 47 | 15.2 | 14.1 | <0.02102 | |
| TP53−/high risk CAGP | 63 | 1.7 (1.07; 2.55) | 11.3 | 4.6 | |
| TP53+/low risk CAGP | 7 | 7.6 | 0 | <0.676 | |
| TP53+/high risk CAGP | 7 | 0.8 (0.23; 2.61) | 9.9 | 0 | |
Low risk CAGP” group includes patients with “Low expressed” centrosome associated gene pattern. “High risk CAGP” group includes patients with united “High and medium expressed” centrosome associated gene pattern. “TP53+” group includes patients with deletion 17p13; “TP53−” group includes patients without deletion 17p13 (positivity cut-off >20 %)
Further analysis includes comprising of CAGP model and International Myeloma Working Group (IMWG) risk stratification model [16, 17]. Briefly, IMWG consensus recommendations includes the following makers: serum β2-microglobulin, serum albumin, t(4;14), 17p13 and 1q21 by FISH. Using this combination, high-risk patients will survive less than 2 years despite novel agents, and low-risk patients can survive for more than 10 years [16]. In total, 70 patients had sufficient clinical data to be included to the analysis. To the IMWG High and Standard Risk groups belong 28 and 41 patients respectively. The only one patient belongs to IMWG Low Risk group was excluded from the further analysis.
Fisher exact test did not show significance of the association (contingency) between the two kinds of risk classification (p = 0.55). Thus, CAGP model can be used to sub-stratify IMWG risk groups. Survival characteristics for Standard and High Risk IMWG groups sub-stratified with CAGP model are presented in Table 3.
Table 3.
Survival characteristics for standard and high risk IMWG groups sub-stratified with CAGP model
| Stratification group | N | HR (95 % CI) | Survival median (months) | 3-years outcome (%) | p value (log-rank) |
|---|---|---|---|---|---|
| Overall survival | |||||
| Low risk CAGP | 66 | 38.8 | 53.8 | <0.00799 | |
| High risk CAGP | 84 | 1.8 (1.16; 2.80) | 22.1 | 25.1 | |
| IMWG standard risk | 41 | 22.8 | 32.1 | <0.1371 | |
| IMWG high risk | 28 | 1.6 (0.86; 2.91) | 15.8 | 23.5 | |
| IMWG standard risk/low risk CAGP | 18 | 29.2 | 42.9 | <0.4532 | |
| IMWG standard risk/high risk CAGP | 23 | 1.4 (0.59; 3.25) | 22.8 | 27.4 | |
| IMWG high risk/low risk CAGP | 14 | 16.6 | 49.9 | <0.02836 | |
| IMWG high risk/high risk CAGP | 14 | 2.8 (1.07; 7.45) | 11.5 | 8.2 | |
| Time to progression | |||||
| Low risk CAGP | 66 | 13.6 | 11.5 | <0.1939 | |
| High risk CAGP | 84 | 1.3 (0.88; 1.88) | 13.0 | 8.7 | |
| IMWG standard risk | 41 | 13.2 | 6.9 | <0.7824 | |
| IMWG high risk | 28 | 1.1 (0.62; 1.89) | 12.0 | 0 | |
| IMWG Standard risk/low risk CAGP | 18 | 12.3 | 0 | <0.9978 | |
| IMWG standard risk/high risk CAGP | 23 | 1 (0.48; 2.07) | 14.5 | 9.6 | |
| IMWG high risk/low risk CAGP | 14 | 17.7 | 0 | <0.05469 | |
| IMWG high risk/high risk CAGP | 14 | 2.3 (0.96; 5.55) | 8.0 | 0 | |
| Progression free survival | |||||
| Low risk CAGP | 66 | 12.6 | 10.8 | <0.06232 | |
| High risk CAGP | 84 | 1.4 (0.98; 2.03) | 10.2 | 5.6 | |
| IMWG standard risk | 41 | 13.2 | 6.3 | <0.8121 | |
| IMWG high risk | 28 | 1.1 (0.62; 1.84) | 11.4 | 0 | |
| IMWG standard risk/low risk CAGP | 18 | 12.3 | 0 | <0.9395 | |
| IMWG standard Risk/high risk CAGP | 23 | 1 (0.48; 1.97) | 13.9 | 9.1 | |
| IMWG high risk/low risk CAGP | 14 | 17.7 | 0 | <0.0325 | |
| IMWG high risk/high risk CAGP | 14 | 2.5 (1.05; 5.89) | 7.7 | 0 | |
“Low risk CAGP” group includes patients with “Low expressed” centrosome associated gene pattern. “High risk CAGP” group includes patients with united “High and medium expressed” centrosome associated gene pattern. “IMWG standard risk” group includes patients with ISS III and no adverse FISH or ISS I and t(4;14)/17p13 del; “IMWG high risk” group includes patients with ISS II/III and t(4;14)/17p13 del
Data shows that proposed CAGP model can be used to sub-stratify IMWG High risk group. This statement is relevant for overall survival, progression-free survival and time to progression.
Discussion
The role of genes is a potential for the determination of molecular signature as a genetic based prognostic or predictive marker, but it is still currently unclear. High- and low-risk groups defined with cytogenetic prognostic models that is based on the most important chromosomal abnormalities such as deletion 17p13 (TP53 gene), translocation t (4;14) and gain 1q21, are still heterogeneous [18, 19]. Heterogeneity determined by molecular variability also determines the diversity of disease at clinical level. It seems that there are several “myeloma subgroups” which differ in expression profile and in clinical manifestations, i.e., prognosis and response for treatment [20]. Until now, approximately 10 % of low-risk patients relapse in 2 years whereas a reverse tendency can be observed in high-risk group as 5–10 % do not reach early relapse (our unpublished data). In spite of substantial progress in therapeutics, the outcome for patients requiring therapy is still highly variable.
In our previous work, we identified the list of 35 candidate genes that play a known role in carcinogenesis and associated with centrosome structure/function, which was used to display molecular heterogeneity with an impact on myeloma pathogenesis [9]. The current study was devoted to create and validate risk stratification model based on these centrosome associated candidate genes. Finally, the created model including 12 selected genes (centrosome associated gene pattern) appears to be an independent prognostic factor for MM stratification.
Nowadays, however, it is needed to use an integrated genomics approach to develop a comprehensive model for risk stratification [21], as GEP-based signature alone appears to have limited power for prognosis in MM [22]. We believe that stratification models reflecting RNA level (gene expression profiling) can supply stratification models reflecting DNA level (interphase fluorescence in situ hybridization).
We have shown that the combination of two independent risk factors such as expression of centrosome associated related genes pattern (CAGP) with TP53 loss depicts the best results in death assessment risk stratification. We suppose that the stated combination of risk factors has become pathogenetically relevant. It may be logically explained by affecting the centrosome associated mitotic damage, which appears catastrophic in the absence of p53-dependent checkpoint response [23, 24].
Summarizing our previously published studies [25], we suggest that centrosome dysfunction accomplished with safe apoptotic system will reset cell cycle and make such clone more sensitive to pro-apoptotic signals. In contrast, in case of an affected apoptotic response, centrosome dysfunction will cause severe genomic instability, which evade apoptosis despite being induced, and may eventually develop a clone with even more aggressive phenotype. Probably, systemic study of apoptotic response in concordances with integrative “omics” study of centrosome machinery will elucidate biological background of revealed controversies in sub-stratified TP53 loss groups.
In conclusion, we have created a new GEP-based model for classifying every patient into one of two prognostic subgroups (high- and low risk CAGP). It seems that CAGP model is able to sub-stratify TP53 negative subgroup: patients with high expressed CAGP attribute to the group with higher risk of death assessment, while patients with low expressed CAGP attribute to the group with lower risk of death assessment within aforementioned subgroup.
Basic laboratory tests including serum albumin and β2-microglobulin for ISS staging, and FISH for t(4;14), del 17p13 and 1q21 gain are markers risk stratification themselves [17, 26]. International Myeloma Working Group consensus on risk stratification in multiple myeloma recommends combination of ISS and FISH for risk stratification. This risk model includes serum β2-microglobulin, serum albumin, t (4;14), 17p13 and 1q21 by FISH [16]. IMWG High Risk group has surviving less than 2 years despite novel agents and includes patients with ISS II/III and t (4;14)/17p13 del. IMWG Standard Risk group includes patients with ISS III and no adverse FISH or ISS I and t (4;14)/17p13 del. IMWG Low Risk group can survive for more than 10 years and includes ISS I/II without adverse FISH [16, 17]. In our study, cohort of patients from IMWG high risk group has overall survival median 15.8 month with 3-year survival 23.5 % and corresponding with previously published data [16]. We found no significant association between the IMWG and CAGP risk classifications. Moreover, CAGP model can be used to sub-stratify IMWG High Risk group: patients with high expressed CAGP attribute to the group with higher risk of death assessment, shorter overall survival, progression-free survival and time to progression.
Conclusions
We assume that the newly established prognostic stratification model complements the current prognostic panel used in multiple myeloma and refines the classification of patients in relation to the risk of disease. This approach can be used independently as well as in combination with other factors. Thus, the new model can sub-stratify prognosis in patients without TP53 loss as well as in IMWG high risk patients’ group. These findings need to be confirmed on a larger cohort with longer follow-up.
Authors’ contributions
FK designed the research, analyzed data, wrote the paper; PN, EK analyzed data, wrote the paper; LR, performed statistical analyses; JM, ZS, LP contributed to sample collection; SO contributed to text overview, RH was responsible for paper proof-reading and overall research coordination. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
Supported by the Ministry of Health (NT13190 and NT14310); the Ministry of Education, Youth and Sports (project no. IRP201550, SGS01/LF/2014-2015, SGS02/LF/2014-2015, SGS03/LF/2015-2016); the Moravian-Silesian Region (MSK 02680/2014/RRC and MSK 02692/2014/RRC) and by MH CZ-DRO-FNOs/2014.
Abbreviations
- MM
multiple myeloma
- GEP
gene expression profiling
- FISH
fluorescent in situ hybridization
- CAGP
centrosome associated gene pattern
- IMWG
International Myeloma Working Group
- OS
overall survival
- PFS
progression free survival
- TTP
time to progression
- ISS
international staging system
Additional files
10.1186/s12967-016-0906-9 Centrosome associated gene pattern (CAGP) expression subgroups.
10.1186/s12967-016-0906-9 Pseudocode of predictive analyses.
Contributor Information
Fedor Kryukov, Email: f.kryukov@gmail.com.
Pavel Nemec, Email: geniusmaximusoptimus@gmail.com.
Lenka Radova, Email: avodar@gmail.com.
Elena Kryukova, Email: eldemenntyeva@gmail.com.
Samuel Okubote, Email: sam3_bote@hotmail.com.
Jiri Minarik, Email: jiri.minarik@fnol.cz.
Zdena Stefanikova, Email: stefanikova@pe.unb.sk.
Ludek Pour, Email: l.pour@seznam.cz.
Roman Hajek, Email: rom.hajek@seznam.cz.
References
- 1.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi M, Minvielle S, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210–2221. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greipp PR, San Miguel J, Durie BG, Crowley JJ, Barlogie B, Blade J, Boccadoro M, Child JA, AvetLoiseau H, Kyle RA, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 3.Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842–854. doi: 10.1002/1097-0142(197509)36:3<842::AID-CNCR2820360303>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.Harousseau JL, Shaughnessy J, Richardson P. Multiple myeloma. Hematology Am Soc Hematol Educ Program. 2004;25:237–256. doi: 10.1182/asheducation-2004.1.237. [DOI] [PubMed] [Google Scholar]
- 5.Matsui S, Yamanaka T, Barlogie B, Shaughnessy JD, Jr, Crowley J. Clustering of significant genes in prognostic studies with microarrays: application to a clinical study for multiple myeloma. Stat Med. 2008;27:1106–1120. doi: 10.1002/sim.2997. [DOI] [PubMed] [Google Scholar]
- 6.Chng WJ, Ahmann GJ, Henderson K, Santana-Davila R, Greipp PR, Gertz MA, Lacy MQ, Dispenzieri A, Kumar S, Rajkumar SV, et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood. 2006;107:3669–3675. doi: 10.1182/blood-2005-09-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dementyeva E, Nemec P, Kryukov F, Muthu Raja KR, Smetana J, Zaoralova R, Greslikova H, Kupska R, Kuglik P, Hajek R. Centrosome amplification as a possible marker of mitotic disruptions and cellular carcinogenesis in multiple myeloma. Leuk Res. 2010;34:1007–1011. doi: 10.1016/j.leukres.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Kryukov F, Dementyeva E, Smetana J, Sevcikova S, Kuglik P, Hajek R. Association of aneuploidy category with centrosome amplification in multiple myeloma. Leuk Lymphoma. 2011;52:2020–2022. doi: 10.3109/10428194.2011.587561. [DOI] [PubMed] [Google Scholar]
- 9.Kryukov F, Nemec P, Dementyeva E, Kubiczkova L, Ihnatova I, Budinska E, Jarkovsky J, Sevcikova S, Kuglik P, Hajek R. Molecular heterogeneity and centrosome-associated genes in multiple myeloma. Leuk Lymphoma. 2013;54:1982–1988. doi: 10.3109/10428194.2013.764416. [DOI] [PubMed] [Google Scholar]
- 10.Dementyeva E, Kryukov F, Kubiczkova L, Nemec P, Sevcikova S, Ihnatova I, Jarkovsky J, Minarik J, Stefanikova Z, Kuglik P, Hajek R. Clinical implication of centrosome amplification and expression of centrosomal functional genes in multiple myeloma. J Transl Med. 2013;11:77. doi: 10.1186/1479-5876-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sevcikova S, Nemec P, Pour L, Hajek R. Genomics in multiple myeloma research. Klin Onkol. 2011;24(Suppl):S34–S38. [PubMed] [Google Scholar]
- 12.Nemec P, Zemanova Z, Kuglik P, Michalova K, Tajtlova J, Kaisarova P, Oltova A, Filkova H, Holzerova M, Balcarkova J, et al. Complex karyotype and translocation t(4;14) define patients with high-risk newly diagnosed multiple myeloma: results of CMG2002 trial. Leuk Lymphoma. 2011;53:920. doi: 10.3109/10428194.2011.634042. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 15.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S, Munshi N, Palumbo A, Miguel JS, Sonneveld P, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28:269–277. doi: 10.1038/leu.2013.247. [DOI] [PubMed] [Google Scholar]
- 17.Avet-Loiseau H, Durie BG, Cavo M, Attal M, Gutierrez N, Haessler J, Goldschmidt H, Hajek R, Lee JH, Sezer O, et al. Combining fluorescent in situ hybridization data with ISS staging improves risk assessment in myeloma: an International Myeloma Working Group collaborative project. Leukemia. 2013;27:711–717. doi: 10.1038/leu.2012.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, Hollmig K, Zangarri M, Pineda-Roman M, van Rhee F, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–1732. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, Leyvraz S, Michallet M, Yakoub-Agha I, Garderet L, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–3495. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 20.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23:6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 21.Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, Palumbo A, Durie B, Fonseca R, Stewart AK, Harousseau JL, Dimopoulos M, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood. 2011;117:4696–4700. doi: 10.1182/blood-2010-10-300970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amin SB, Yip WK, Minvielle S, Broyl A, Li Y, Hanlon B, Swanson D, Shah PK, Moreau P, van der Holt B, et al. Gene expression profile alone is inadequate in predicting complete response in multiple myeloma. Leukemia. 2014;28:2229–2234. doi: 10.1038/leu.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uetake Y, Sluder G. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a “tetraploidy checkpoint”. J Cell Biol. 2004;165:609–615. doi: 10.1083/jcb.200403014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 25.Kryukova E, Kryukov F, Hajek R. Centrosome amplification and clonal evolution in multiple myeloma: Short review. Crit Rev Oncol Hematol. 2016;98:116–121. doi: 10.1016/j.critrevonc.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Avet-Loiseau H, Attal M, Campion L, Caillot D, Hulin C, Marit G, Stoppa AM, Voillat L, Wetterwald M, Pegourie B, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30:1949–1952. doi: 10.1200/JCO.2011.36.5726. [DOI] [PubMed] [Google Scholar]