

Abstract
Multisite phosphorylation is required for the biological function of serine-arginine (SR) proteins, a family of essential regulators of mRNA splicing. These modifications are catalyzed by serine-arginine protein kinases (SRPKs) that phosphorylate numerous serines in arginine-serine-rich (RS) domains of SR proteins using a directional, C-to-N-terminal mechanism. The present studies explore how SRPKs govern this highly biased phosphorylation reaction and investigate biological roles of the observed directional phosphorylation mechanism. Using nuclear magnetic resonance (NMR) spectroscopy with two separately expressed domains of SRSF1 we showed that several residues in the RNA-binding motif 2 (RRM2) interact with the N-terminal region of the RS domain (RS1). These contacts provide a structural framework that balances the activities of SRPK1 and the protein phosphatase PP1, thereby regulating the phosphoryl content of the RS domain. Disruption of the implicated intramolecular RRM2–RS1 interaction impairs both the directional phosphorylation mechanism and the nuclear translocation of SRSF1 demonstrating that the intrinsic phosphorylation bias is obligatory for SR protein biological function.
Keywords: kinetics, NMR, phosphorylation, RS Domain, SR Protein
Graphical Abstract
Introduction
The splicing of precursor mRNA is dependent on an essential group of splicing factors known as SR proteins¶ that guide the selection of exon-intron boundaries, thereby defining appropriate splice sites in genes. SR proteins are composed of two fundamental domain types: N-terminal RNA recognition motifs (RRMs) that bind exonic splicing enhancer sequences (ESEs) and orchestrate the assembly of the spliceosome and a C-terminal arginine-serine-rich (RS) domain that regulates the former domains. The SRPK family of protein kinases efficiently phosphorylates consecutive Arg-Ser dipeptide repeats in the RS domains of SR proteins 1. This modification by cytoplasmic SRPKs facilitates interaction of SR proteins with an SR-specific transportin (TRN-SR) that transfers the splicing factor to the nucleus where it engages in splicing 2, 3. SR proteins largely reside in dynamic, membrane-free domains known as speckles. Additional phosphorylation mobilizes SR proteins from speckles to pre-mRNA and components of the spliceosome 4–6. Although these phosphorylation-dependent interactions are important for establishing the 5′-3′ splice sites, SR protein dephosphorylation is necessary for maturation of the spliceosome and splicing catalysis 7. SR protein dephosphorylation has also been implicated in the export of SR proteins and processed mRNA to the cytoplasm for protein translation 8. Thus, RS domain phosphorylation-dephosphorylation is vital for numerous steps in SR protein subcellular localization and splicing control.
Although multisite phosphorylation of SR proteins was first acknowledged as an essential step for splicing more than twenty years ago 9, the mechanism underlying this modification has only been revealed in recent years 1. Using protease footprinting experiments we showed that SRPK1 efficiently phosphorylates numerous serines (8–10) in the N-terminal region of the SRSF1 RS domain using a directional, C-to-N-terminal mechanism 10. In this process, the active site of SRPK1 initially binds near the C-terminal end of the Arg-Ser stretch in RS1 and then proceeds to add phosphates sequentially in an N-terminal direction until the entire dipeptide stretch is modified (Fig. 1A). Insights into what drives this directional mechanism later came from the X-ray structure of the kinase domain of SRPK1 with a portion of SRSF1 bound. The N-terminal Arg-Ser dipeptides from RS1 were found in a docking groove in the large lobe of the kinase domain away from the active site 11. Cross-linking studies then showed that these dipeptides leave the docking groove as a function of phosphorylation suggesting that the directional mechanism involves movement of the Arg-Ser stretch from this groove to the active-site pocket 11. Confirmation that the docking groove is an essential structural element came from the observation that mutations in this groove disconnect the directional bias resulting in a random phosphorylation mechanism 12. These findings led to a simple “yardstick model” in which the close apposition of the docking groove and active site measures out a specific number of Arg-Ser repeats in a mandatory orientation for initiation and subsequent directional phosphorylation (Fig. 1A).
Figure 1. RRM2 Induces Directional Phosphorylation of RS1 in SRSF1.

A) Model of the SRPK1-mediated directional phosphorylation of SRSF1. SRPK1 contains N- and C-terminal lobes that recognize Arg-Ser repeats. SRSF1 is composed of two RRMs and a C-terminal RS domain divided into RS1 and RS2 segments. For clarity, only the Arg-Ser repeats from RS1 of the RS domain are shown. B–D) LysC cleavage of SRPK1-phosphorylated cl-SRSF1 (B), cl-SR(ΔRRM1) (C), and cl-nRS (D) at low and high ATP concentrations monitored by autoradiography. *RRM2 represents an RRM2 variant in which five lysines are mutated to arginines (K138R, K165R, K174R, K179R & K193R). A cartoon of each construct is shown with indication of the sizes of the two fragments resulting from LysC treatment. E) Bar graph showing N/C ratios for the cleavage substrates at low and high ATP concentrations. Ratios for cl-nRS at high ATP are not shown due to the lack of reactivity with LysC (see text).
All presently available data cannot readily be rationalized with this simple “yardstick model” and there are new experiments to check on predictions from this hypothesis. For example, if the docking groove and active site in SRPK1 measures out Arg-Ser repeats of a particular length then directionality should be encoded solely in the RS domain and not dependent on the neighboring RRMs in the SR protein. In prior footprinting of a cleavage form of SRSF1 that contained the RRM2 at the C-terminal end of the RS domain, we found that swapping the positions of these domains led to a random phosphorylation mechanism, suggesting that RRM2 might play a role in phosphate addition 12. However, placing a bulky domain on the C-terminal end of the RS domain could disrupt enzyme-substrate docking owing to potential steric conflicts, so that additional studies are needed. In a different approach, an X-ray structure of substrate-bound SRPK1 suggested that contacts between RRM2 and the kinase domain might orient RS1 for C-terminal phosphorylation initiation 11, but mutation of the presumed binding contacts in RRM2 had no effect on the directional mechanism or the affinity of the substrate for the kinase, suggesting that RRM2-kinase interactions are not important for the preferred phosphorylation direction 12. Furthermore, previous mass spectrometric studies showed that the RRMs play no role in controlling the total phosphoryl content of the RS domain 13. In rapid quench experiments, SRPK1 rapidly phosphorylates 8–10 serines in RS1 whether or not the RRMs are present 14. Thus, while the docking groove is an essential element for directional phosphorylation, the role of the RRMs in this process remained unclear.
In the present study we addressed whether the RRMs serve a role in guiding the unique phosphorylation mechanism of SRPK1 and whether such a mechanism plays a role in SR protein biological function. Based on footprinting experiments, we showed that the isolated RS domain gets randomly phosphorylated. This result, along with other deletion studies, suggests that RRM2 controls the directional phosphorylation of the RS domain in SRSF1. To obtain structural information that would bear on intramolecular long-range contacts in the SR protein, we used NMR spectroscopy to show that an Arg-Ser dipeptide stretch in the RS domain interacts with three negatively charged residues in RRM2. These distal contacts present the C-terminal dipeptides to the active site for initiation of directional phosphorylation. Although wild-type SRSF1 is localized exclusively in the nucleus of cells, breaking the RRM2-RS1 interactions results in increased cytoplasmic SR protein. The improper localization of SRSF1 upon mutation is correlated with a misbalance between kinase and phosphatase activities affecting the RS domain. The available data indicate that the directional phosphorylation mechanism of the RS domain is tied in obligatory ways to the biological function of SR proteins.
Results
RRM2 Regulates Directional Phosphorylation of the SRSF1 RS Domain
In prior studies we showed using a protease footprinting technique that SRPK1 phosphorylates the RS1 segment of the RS domain in the SR protein SRSF1 (residues 204–221) at 8–10 serines using a C-to-N directional mechanism 10, 13 (Fig. 1A). In this method, a single Arg-to-Lys mutation in the center of RS1 (R214K) along with several Lys-to-Arg mutations in RRM2 (*RRM) are inserted into SRSF1. This cleavage substrate, cl-SRSF1, is then phosphorylated using SRPK1 under single turnover conditions (SRPK1>cl-SRSF1) and then cleaved using the lysine-specific protease LysC to obtain two phosphorylated fragments easily identified by SDS-PAGE autoradiography (Fig. 1B). These fragments, containing the N- and C-terminal halves of RS1, can be used to assess the preferred order of phosphate addition by SRPK1. Using this technology we wished to define potential roles for the neighboring RRMs. We first confirmed previous results that whereas SRPK1 phosphorylates both the N- and C-terminal halves of cl-SRSF1 to levels representing the natural serine distribution in RS1 at high ATP (N/C = 0.7), SRPK1 strictly phosphorylates only the C-terminal fragment at 0.2 μM ATP when no more than one phosphate is present on the RS domain (Fig. 1B and E). Based on the N/C ratio at low ATP, SRPK1 prefers to phosphorylate the C- versus the N-terminus by about 30-fold, a value in line with previous reports 10, 13. We then showed, using a cleavage construct lacking the N-terminal domain, that RRM1 does not significantly influence this directional mechanism (Fig. 1 C and E).
To address the role of RRM2, we designed and expressed a new cleavage substrate that lacks both RRMs but contains the single Arg-to-Lys mutation in the center of RS1 and a His tag at the N-terminus (cl-nRS) (Fig. 1D). In kinetic progress curves, we showed that this substrate is phosphorylated at many sites (Fig. S1), confirming previous findings that the RRMs do not influence total phosphoryl content of the RS domain 13. At 0.2 μM ATP, two fragments of cl-nRS were obtained upon LysC cleavage that correspond to the N- and C-terminal halves of RS1 (Fig. 1D). Since both fragments are phosphorylated under conditions where a total of only one serine is modified, the N/C ratio of 0.62 indicates that SRPK1 does not show a strong preference for phosphorylating the C-terminus (Fig. 1D). LysC did not cleave cl-nRS at high ATP concentrations even at longer incubation times (4 hrs) suggesting that the RS domain adopts a protease-resistant conformation upon full phosphorylation in the absence of the RRMs. In this context, the increased conformational rigidity observed by Xiang et. al. for the phosphorylated RS domain 15 may also play a role in reducing LysC activity. In a prior study we found that an SRSF1 cleavage construct containing the RS domain with RRM2 attached to the C-terminal end (and lacking RRM1) was also phosphorylated with no N- or C-directional preference 13. Taken together, these findings indicate that RRM2 is necessary for directional phosphorylation of RS1 in SRSF1.
Monitoring Interactions Between RS1 and RRM2 of SRSF1 Using NMR
Given the implicated role of RRM2 in guiding the phosphorylation of RS1, we wished to attain structural information on this interaction. Structural studies including RS domains, either in isolated form or in intact SR proteins, have until now been scarce, due to the limited stability of Arg-Ser dipeptide repeats at pH above 3.0 and sufficiently high concentrations for structure determination. To overcome these limitations, we fused RS1 with Glutathione S-transferase (GST-RS1), which yielded a fusion protein that is stable in the 5.5–7.5 pH range, can be phosphorylated by SRPK1 at all Arg-Ser dipeptides (Fig. S2) and is amenable to NMR studies. In view of the difficulties encountered in attempts to structurally characterize RS domains in intact SR proteins, the present approach with studies of separately expressed RRM2 and RS1 provides key information that can be extrapolated to intramolecular interactions in intact SRSF1.
Tintaru et. al. had previously determined the NMR structure of RRM2 in the presence of arginine/glutamic acid buffer (50 mM) 16. However, to avoid competition between free arginine molecules in the buffer and arginines from the RS domain, we determined the NMR structure of RRM2 in phosphate buffer at 25 °C following the J-UNIO protocol 17. The standard set of three APSY-NMR experiments 18 in combination with the automated algorithm UNIO-MATCH 19 yielded 86% of the backbone resonance assignments, and complete polypeptide backbone chemical shift assignments were obtained after interactive validation and extension of these assignments with the aid of 3D 15N- and 13C-resolved [1H,1H]-NOESY experiments. The same NOESY experiments were used as input for UNIO-ATNOS-ASCAN 20, which automatically yielded 72% of the proton side chain assignments. Interactive validation then resulted in chemical shift assignments for 90% of the expected side chain signals. The input for the structure calculation with CYANA3.0 obtained on the basis of these chemical shift assignments with UNIO-ATNOS-CANDID 21 is listed in Table 1, which also presents the statistics of the structure calculation. The structure of the SRSF1 RRM2 in phosphate buffer superimposes with the NMR structure determined in arginine/glutamic acid buffer (PDB id 2O3D) 16 with a backbone RMSD value of 1.95 Å and exhibits a canonical RRM fold with two α-helices and five β-strands arranged in the sequence order β1-α1-β2-β3-α2-β4-β5. The β-strands form an antiparallel sheet and are flanked on one side by the two α-helices (Fig. 3A).
Table 1.
Input for the structure calculation and characterization of the bundle of 20 energy-minimized CYANA conformers representing the NMR structure of SRSF1-RRM2.
| Quantitya | SRSF1-RRM2 |
|---|---|
| NOE upper distance limits | 1414 |
| Intraresidual | 317 |
| Short range | 394 |
| Medium range | 204 |
| Long range | 499 |
| Dihedral angle constraints | 479 |
| Residual target function value (Å2) | 0.89 ± 0.13 |
| Residual NOE violations | |
| Number ≥ 0.1 A | 2 ± 2 |
| Maximum (Å) | 0.15 |
| Residual dihedral angle violations | |
| Number ≥ 2.5° | 1 ± 1 |
| Maximum (°) | 0.98 |
| Amber energies (kcal/mol) | |
| Total | −2880 ± 132 |
| Van der Waals | −251 ± 12 |
| Electrostatic | −4075 ± 178 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.0078 ± 0.0001 |
| Bond angles (°) | 1.66 ± 0.007 |
| RMSD to the mean co-ordinates (Å)b | |
| bb (120 195) | 0.68 ± 0.13 |
| ha (120 195) | 1.51 ± 0.14 |
| Ramachandron plot statistics (%) c | |
| Most favoured regions | 82.1 |
| Additional allowed regions | 11.3 |
| Generously allowed regions | 5.1 |
| Disallowed regions | 1.5 |
Except for the top six entries, which describe the input generated in the final cycle of the ATNOS/CANDID/CYANA calculation, the last entries refer to the 20 best CYANA conformers after energy minimization with OPALp (see text). Where applicable, the average value for the bundle of 20 conformers and the standard deviation are given.
bb indicates the backbone atoms N, Cα, and C′; ha stands for all heavy atoms. Numbers in parentheses indicate the residues for which the RMSD was calculated
As determined by PROCHECK 46
Figure 3. Mapping Electrostatic Contacts Between RRM2 and RS1.

A) RRM2 structure with several solvent–accessible, negatively charged residues shown as sticks. B) Pull-down assays for wild-type and several mutant forms of RRM2 using GST-RS1. I=input; PD=pull down. C) Alignment of affected sequences in RRM2’s of several SR proteins. Red boxes highlight residues Glu143, Asp146, and Asp151 in SRSF1 and the positions of conserved residues in four other SR proteins. D) GST-tagged RS1 deletion constructs. E) Pull-down assays for deletion constructs using wild-type RRM2. I=input; PD=pull down. F) Pull-down assays using GST-RS1(N) and RRM2 mutants defective in binding GST-RS1. I=input; PD=pull down. G) Time-dependent incubation of cSR(R210K) and cSR(R218K) with LysC. Rate constants for cSR(R210K) are 0.26 and 0.0062 min−1 and those for cSR(R218K) are 0.91 and 0.018 min−1. H) Model of RRM2 with the RSRSRSRS octapeptide bound. The surface of RRM2 in the same orientation as in Fig. 2C, using the same color code. Residues Glu143, Asp146 and Asp151 found to influence binding affinity are colored red and the non-responsive acidic residues Glu120, Glu166 and Asp167 are grey. The RSRSRSRS octapeptide is presented as a space-filling model of the backbone with carbon, nitrogen and oxygen atoms shown in brown, red and blue, respectively. For clarity the side chain atoms used in the docking model are not shown. The structure was generated with AUTODOCK 44 and the drawing was prepared with pyMOL 45.
In addition to the NMR structure determination of RRM2, we prepared phosphorylated GST-RS1 (GST-RS1pho). NMR chemical shift mapping was then used to identify RRM2 residues involved in the interaction with GST-RS1 and GST-RS1pho. 1.2 equivalents of either GST-RS1 or GST-RS1pho were added into 400 μM solutions of uniformly 15N-labeled RRM2, and changes in the signals from the amide groups were monitored using [15N,1H]-HSQC experiments 22. GST-RS1 induced significant changes in the spectra, including chemical shift perturbations and line broadening, while no differences were observed with its phosphorylated derivative (Fig. 2A and B). Based on sequence-specific polypeptide backbone resonance assignments, we mapped the residues experiencing perturbations on the structure of RRM2. These clustered near the N-terminal of helix α1, the loops connecting α1 with β2 and β3 with α2, and residues in the strands β1, β2 and β3 in close proximity to α2, forming a well-defined patch in one side of the protein (Fig. 2C and D). Binding to RS1 is thus localized to a discrete surface region of RRM2.
Figure 2. NMR observation of intermolecular complex formation between SRSF1-RRM2 and the solubilized SRSF1-RS domain.

A) Superposition of the [15N,1H]-HSQC spectra of 15N-labeled SRSF1-RRM2 with (red) and without (black) addition of 1.2 equivalents of GST-RS1. Residues of SRSF1-RRM2 experiencing chemical shifts and/or line broadening upon binding of GST-RS1 are indicated. B) Superposition of the [15N,1H]-HSQC spectra of 15N-labeled SRSF1-RRM2 with (red) and without (black) addition of 1.2 equivalents of phosphorylated GST-RS1. There are no detectable chemical shift perturbations; This spectrum also documents the high purity/homogeneity of the protein preparation. C,D) Front and back surface views of SRSF1-RRM2, with residues affected by the presence of GST-RS1 (see panel A) colored cyan. Some amino acid positions are indicated to guide the eye.
Probing Residues In RRM2 That Bind RS1
In a search for RRM2 residues involved in binding to RS1, we selected six negatively charged solvent-accessible amino acids in the cluster identified by the NMR experiments (Figs. 2C and 3A). These residues were replaced with alanine either in single or double mutants. To evaluate whether these amino acid replacements affect the binding of RRM2 to RS1, we performed pull-down assays using GST-RS1. GST-RS1 pulled down wild-type RRM2 (Fig. 3B), which is in keeping with the NMR data. The RRM2-RS1 interaction appears to be very specific since RRM1 was not pulled down by GST-RS1 (Fig. S3A) and addition of GST-RS1 did not induce changes in the [15N,1H]-HSQC spectrum of 15N-labeled SRSF1-RRM1 (Fig. S4). Whereas RRM2E166,D167A and RRM2E120A were readily pulled down by GST-RS1, RRM2E143,D146A and RRM2D151A were not (Fig. 3B). To evaluate whether both residues in RRM2E143,D146A are important for binding, we performed pull-down assays using the single mutants RRM2E143A and RRM2D146A and found that neither were pulled down by GST-RS1 (Fig. S3B). These findings suggest that Glu143, Asp146 and Asp151 are important for RS1 binding. In control experiments we showed that RRM2 does not interact with the g-agarose resin in the absence of GST-RS1 (data not shown). We overlaid the sequences of SR proteins that have an RRM2 and found that the three binding-related residues are in a conserved region, supporting that they may be part of an epitope governing function-related RS domain interactions (Fig. 3C). Furthermore, Glu-143 and Asp-146 are replaced by lysine and alanine in RRM1, which likely explains the poor interactions observed between GST-RS1 and RRM1 (Fig. S3A).
RRM2 Binds to Arg-Ser Repeats in RS1
Having identified potential binding contacts for RS1 on RRM2, we next wished to detect which region of RS1 interacts with RRM2. The RS1 segment of the SRSF1 RS domain contains two structural elements: an Arg-Ser dipeptide repeat and a more diverse linker segment, PRSPSYG, which connects it to RRM2. To identify which residues are most important for binding, we made two deletions in GST-RS1, removing respectively, the C-terminal part of the Arg-Ser stretch (GST-RS1N) and the entire Arg-Ser stretch (GST-RS1L) (Fig. 3D). In pull-down assays, we found that whereas GST-RS1 and GST-RS1N interact robustly with wild-type RRM2, GST alone and GST-RS1L do not (Fig. 3E). The lack of affinity for the diverse linker segment was also confirmed by NMR, where addition of two equivalents of the peptide GPRSTSYG did not induce significant changes in the [15N,1H]-HSQC spectrum of RRM2 (Fig. S4). Although we observed similar results for GST-RS1 and GST-RS1N, it is possible that additional Arg-Ser repeats could participate in stabilizing RS1 on RRM2. To address this possibility, we performed concentration-dependent pull-down experiments and from plots of fraction bound versus RRM2 we obtained similar apparent Kd values for GST-RS1 and GST-RS1N (Fig. S5). Taken together, these findings indicate that the N-terminal portion of the Arg-Ser stretch (residues 203–211) is sufficient for RRM2 binding whereas the linker segment is not. These results indicate that only 4 Arg-Ser repeats are needed to reproduce the affinity of the full-length RS1. We also found that GST-RS1N did not interact with either RRM2E143,D146A or RRM2D151A, supporting the idea that these residues in RRM2 interact with a limited Arg-Ser repeat (Fig. 3F). To assess how RS1 interacts with RRM2, we constructed two forms of SRSF1 in which LysC cleavage sites are placed either in (cSR(R210K)) or outside (cSR(R218K)) the predicted docking region in RS1 (Fig. 3G). We found that cSR(R218K) is cleaved at a faster rate than cSR(R210K), suggesting that the C-terminal portion is more solvent exposed than the N-terminal portion of the Arg-Ser stretch. This is consistent with the idea that the C-terminal repeats in RS1 are more accessible to SRPK1 (Fig. 3G). Taken together, these results indicate that RRM2 binds to the Arg-Ser repeats in RS1, with three negatively charged residues in RRM2 having a key role in binding.
Model of the RRM2-RS1 interaction in SRSF1
We combined the binding information from NMR and biochemistry studies with a new docking protocol in AutoDock to generate a model of the complex of RRM2 with an RSRSRSRS octapeptide. The sequence of RS repeats and the large range of rotational angles that are energetically accessible for linear peptides required the following strategy for the computation. A coarse-grain model of the essential chemical moieties present in the amino acids was used, which also account for flexibility. It was then possible to use the simplified peptide structure to sample its conformational flexibility over the protein surface. In addition, the ligand was docked using different energy rewards to favor interactions with key residues identified by NMR and by mutagenesis (2-fold and 4-fold energy rewards, respectively), generating 200K docked poses. In the best binding mode found, the peptide is in close proximity to Asp151 and Asp146, extends along the strand β2, and contacts residues which experienced large chemical shift changes in the presence of GST-RS1 (Fig. 3H). The RS peptide is also near the C-terminus of RRM2 where it is expected to reside in the full-length SR protein. Comparison of the modeled RRM2-RS1 complex with the NMR structure of SRSF1-RRM2 in complex with RNA UGAAGAC 23 revealed a partial overlap. This observation suggests that RS-RRM interactions may regulate RNA binding and is in line with reports showing that the RS domain in SRSF1 requires phosphorylation for efficient RNA interactions 24, 25.
Docking Residues Facilitate the Association and Turnover Rates of SRSF1 With SRPK1
We next wished to ask whether docking of the Arg-Ser stretch in RS1 onto RRM2 has an impact on SRSF1 phosphorylation efficiency. To answer this question, we made an SRSF1 mutant that replaces Glu143, Asp146 and Asp151 in RRM2 with alanine (SRSF13M) and used NMR spectroscopy to show that the structural integrity of the isolated RRM was not compromised by these amino acid replacements (Fig. S6). We found that the steady-state kinetic parameters for SRSF13M were altered relative to wild-type SRSF1 (Fig. 4A). We found that the kcat was reduced 2.5-fold from 1 to 0.4 sec−1 and the Km was increased 4-fold from 180 to 720 nM for SRSF13M compared to wild-type SRSF1. This results in a 10-fold decrease in kcat/Km for SRSF13M when compared to wild-type SRSF1 (Fig. 4A). To evaluate the cause of these changes, we performed steady-state viscosometric experiments and found that the addition of 30% sucrose decreased both kcat and kcat/Km by about 2.2-fold for SRSF13M and 2.6-fold for wild-type SRSF1 (Fig. 4B,C). If an enzymatic kinetic parameter is limited by a diffusion-controlled step(s), then the parameter will decline with increasing buffer viscosity according to the Stokes-Einstein equation 26. We observed that the kinetic parameters for SRSF1 and SRSF13M decreased in direct proportion to changes in relative buffer viscosity (2.5-fold) suggesting that diffusion-controlled steps limit these parameters. In prior studies we showed that these decreases for SRSF1 indicate that kcat is limited by product release (i.e.-ADP release) and kcat/Km is limited by the association rate constant for the protein substrate 14. Since we observe similar viscosometric effects for SRSF13M and SRSF1, the effects of mutation are the result of a 10-fold reduction in the association constant for substrate and a 2.5-fold reduction in the release of the reaction product (ADP) (Fig. 4C). These findings indicate that internal contacts between the RS1 segment and RRM2 within SRSF1 increases both the encounter probability and turnover efficiency of the SR protein to SRPK1.
Figure 4. Residues in RRM2 modulate SRPK1 association with SRSF1.

A) Steady-state kinetic profiles for SRSF1 and SRSF13M. Bar graphs represent kinetic parameters from fitting plots of enzyme-normalized velocity versus substrate. B,C) Effects of 30% sucrose on the steady-state kinetic parameters for SRSF1 (B) and SRSF13M (C).
RRM2-RS1 Interactions Regulate Nuclear Import & Phosphatase Sensitivity of SRSF1
Owing to the role of SRPK1 for the nuclear import of SR proteins, we wished to determine whether effects on the phosphorylation mechanism could impact the phosphoryl content of SRSF1 and its cytoplasmic-nuclear distribution. We first monitored the phosphorylation of SRSF1 by SRPK1 (75 nM) in the absence and presence of PP1 (300 nM) using excess ATP (50 μM). We found that PP1 lowered the observed phosphoryl content of SRSF1 relative to the control reaction lacking PP1 by about 25% after total phosphorylation levels approached an equilibrium (Fig. 5A and D). In this experiment, only 3 μM ATP (6% of total) was used to generate phospho-SRSF1 in the control, so that the apparent reaction endpoints in the kinetic traces after 30 minutes are not limited by ATP depletion and reflect the equilibrium phosphoryl contents of the SR protein in the presence of both kinase and phosphatase. By comparison, PP1 lowered the observed phosphoryl content of SRSF13M by about 85%, suggesting that RRM2-RS1 contacts impact the lability of the phosphates on RS1 (Fig. 5B and D). To ensure that other residues in RRM2 or RRM1 do not play a role in this phenomenon, we measured the phosphorylation of a construct lacking both RRMs and found that the observed phosphoryl content of the RS domain was lowered by about 85% in the presence of PP1 (Fig. 5C and D). The similarity of the effects for the isolated RS domain and SRSF13M suggest that replacement of the three contact residues identified by NMR has the same consequence as removal of the RRM2 domain.
Figure 5. Interactions of RRM2 With RS1 Affect Cytoplasmic-Nuclear Distribution and Dephosphorylation of SRSF1.

A–C) Phosphorylation of 1 μM SRSF1 (A), SRSF13M (B) and RS (C) by SRPK1 (75 nM) in the absence (●) and presence (▲) of PP1 (300 nM). D) Bar graph showing relative phosphorylation levels of SR proteins in the presence of both SRPK1 and PP1. E) Confocal imaging of GFP-SRSF1 and GFP-SRSF13M in HeLa cells. F) Subcellular fractionation of HeLa cells expressing GFP-SRSF1 and GFP-SRSF13M. WT=GFP-SRSF1; Mut=GFP-SRSF13M.
To evaluate whether these differences in phosphoryl content impact the subcellular location of SRSF1, we next expressed a GFP-tagged form of SRSF1 in HeLa cells and found that it localized exclusively to the nucleus, residing mostly in speckles (Fig. 5E). In contrast, a GFP-tagged form of SRSF13M (GFP-SRSF13M) was visible in both the cytoplasm and the nucleus (Fig. 5E). Also, the nuclear fraction of GFP-SRSF13M appeared more homogeneous, with fewer speckled patterns. We next performed cell fractionation studies and found that GFP-SRSF1 was present exclusively in the nucleus as expected (Fig. 5F). In contrast, the full-length GFP-SRSF13M was present in both the cytoplasm and nucleus (Fig. 5F). Subcellular fractions were confirmed by immunoblotting for GAPDH (cytoplasm) and Histone (nucleus). Unlike GFP-SRSF1, some GFP-SRSF13M was degraded, suggesting that the three mutations in RRM2 increased the susceptibility of the SR protein to proteolysis. This finding is consistent with the observation that mutations in RRM2 lead to dissociation of RS1, rendering the SR protein more susceptible to proteolysis. Furthermore, some of the GFP-SRSF13M fragments are unique in both cellular compartments, possibly suggesting varying conformations or interacting proteins in the nucleus versus the cytoplasm. Overall, the data indicate that localization of SRSF1 is dependent on RRM2-RS1 contacts that regulate phosphate balance through alterations in kinase-phosphatase activities.
Regulating Directional Phosphorylation Through a Negatively Charged Cluster in RRM2
Deletion analyses had indicated that RRM2 regulates the directional (C-to-N) phosphorylation of SRSF1 (Fig. 1). Having identified residues in RRM2 that interact with RS1 (Fig. 3), we wished to determine whether these residues alone regulate the directional mechanism. We inserted the three mutations from SRSF13M into the cleavage substrate cl-SRSF1 to assess changes in the phosphorylation mechanism but found that the new mutant substrate, cl-SRSF13M, was extensively degraded in E. coli relative to the control substrate cl-SRSF1, generating a phosphorylated 15 kDa fragment consistent with cleavage near the RRM1-RRM2 linker (Fig. 6A). These results are in line with the observed proteolysis of GFP-SRSF13M in HeLa cells (Fig. 5F). Since we were able to express and purify SRSF13M as an N-terminally His-tagged protein in a pure, non-degraded form, we explored the possibility that an N-terminal His-tagged cleavage substrate might be more amenable to LysC footprinting. Also, moving the His-tag to the N-terminal side of the substrate allowed us to investigate whether it influences the directional mechanism. cl-nSRSF1 contained all the mutations in RRM2 and the RS domain of cl-SRSF1, with the His tag moved to the N-terminus (Fig. 6B). The parent substrate, phosphorylated to varying extents using limiting ATP, was cleaved with LysC to obtain the N- and C-terminal fragments (Fig. 6C). At minimal phosphorylation (0.2 μM ATP, <1 site), SRPK1 favored phosphorylation of the C-terminal compared to the N-terminal portion of RS1 by 25-fold (N/C=0.04) (Fig. 6C). This value is similar to that for the C-terminally-tagged cleavage substrate (Fig. 1B). At 100 μM ATP, when all the sites are phosphorylated, we obtained an N/C ratio consistent with the natural distribution of serines in RS1 (N/C 0.8). These findings indicate that the new cleavage substrate is phosphorylated in a C-to-N manner and the position of the His tag does not influence this phosphorylation pattern.
Figure 6. Residues in RRM2 Regulate Directional Phosphorylation of RS1 in SRSF1.

A) Phosphorylation of cl-SRSF1 & cl-SRSF13M monitored by autoradiography. A proteolytic fragment of cl-SRSF13M migrates at 15 kDa. B) N-terminal His-tagged cleavage substrate, cl-nSRSF1. LysC cleavage generates two fragments splitting RS1 in half. C) SRPK1 phosphorylates cl-nSRSF1 in a C-to-N manner. cl-nSRSF1 is phosphorylated by excess SRPK1 using varying 32P-ATP and then treated with LysC to generate the N- & C-terminal fragments. The ratios of the N- & C-terminal fragments (N/C) are plotted as a function of the total number of phosphorylation sites in cl-nSRSF1. D) N/Cs ratio of cl-SRSF1D151A and cl-SRSF1D143A,E146A phosphorylated with SRPK1 at low ATP (0.2 μM) and treated with LysC. E) Bar graph showing SRPK1 specificity (C/N ratio) for C-terminus of RS1 for cl-SRSF1, cl-SRSF1D151A, cl-SRSF1D143A,E146A & cl-nRS.
To determine whether residues in RRM2 identified by our binding studies affect the directional phosphorylation mechanism, we inserted the three mutations from SRSF13M into cl-nSRSF1. This new substrate was not well expressed in E.coli. However, we were able to express two forms that contain either a single alanine mutation at Asp151 (cl-nSRSF1D151A) or a double alanine mutant at Glu143 and Asp146 (cl-nSRSF1E143A,D146A) with no evidence of proteolytic fragmentation. To determine whether these mutants impact the directional mechanism, we minimally phosphorylated cl-nSRSF1D151A and cl-nSRSF1E143A,D146A (< 1 site) and then obtained the N- and C-terminal fragments using LysC cleavage (Fig. 6D). In three separate experiments, we found that the N/C ratios for cl-nSRSF1D151A and cl-nSRSF1E143A,D146 were consistently 5- to 9-fold higher than for cl-nSRSF1 at a similar phosphoryl content (Fig. 6D). To address the role of these residues for C-terminal specificity control, we measured the C/N ratios of cl-nSRSF1, cl-nSRSF1D151A, cl-nSRSF1E143A,D146, and cl-nRS (Fig. 6E). This analysis showed that SRPK1 prefers to phosphorylate at the C- rather than the N-terminus of RS1 by 25-fold, a value more than 10-fold higher than that for the isolated RS domain (cl-nRS). Although we could not measure the role of all three residues together, we found that either Asp151 or the Glu143/Asp146 pair is responsible for providing approximately 50% of the C-terminal specificity preference observed in cl-nSRSF1. Since the observed decreases in C/N ratios for the mutants are multiplicatively related to the total specificity of the wild-type substrate cl-nSRSF1, the aforementioned three charged residues appear to function in an additive manner to control C-to-N directionality. Thus, this cluster of negatively charged residues in RRM2 appears to be largely responsible for regulating the directional phosphorylation mechanism.
Discussion
Enzymes have evolved highly efficient reaction mechanisms to convert substrates to products. The principle strategy is to lower the energy barriers for the transition states of individual steps so that the net flux through a given biochemical pathway is optimized 27. The specific tactics used to manipulate substrates vary significantly and are most likely defined by the nature of the reaction chemistry, the stabilities of internal intermediates and available positioning of amino acids in the active site. In the case of the SR kinase family, we found that SRPK1 has evolved a directional (C-to-N) mechanism to phosphorylate a series of eight Arg-Ser dipeptides in the RS domain of its physiological substrate SRSF1 (Fig. 1). This transformation occurs in the cytoplasm and is important for physical coupling to the transport machinery and localization of SRSF1 in nuclear speckles 3, 28. Although the transporter, TRN-SR, recognizes the product of SRPK1-mediated phosphorylation, it is unclear whether the nature of the phosphorylation mechanism plays any role in the subcellular localization of SR proteins. Using structural, kinetic and cell imaging methods, we now show how the directional mechanism catalyzed by SRPK1 is regulated by RRM2 and how a charge cluster in this RRM interacts with a portion of the RS domain controlling the phosphoryl content and subcellular localization of an SR protein.
A New Strategy for Defining SR Protein Conformations
While some steps have been made in identifying contacts between RS domains and interacting proteins 11, 29, there is still little information on the structure of an SR protein. Poor solubility of the SR protein due to its disordered nature and physicochemical properties still presents a significant problem for structural characterization of RS domain complexes at the atomic level. In contrast, the RRMs from SR proteins are very soluble and many X-ray and NMR structures are now available 30. To circumvent the problem of poor solubility of intact RS domains and full-length SR proteins, we took a different strategy by attaching a solubility tag (GST) to different SRSF1 variants. Using this approach, we produced protein samples with soluble RS domains at nearly physiological conditions. These were used to perform NMR binding experiments revealing that GST-RS1 interacts with a well-defined epitope on RRM2 (Fig. 2). We then identified Glu143, Asp146, and Asp151 in RRM2, and multiple Arg-Ser repeats in RS1 as interacting elements (Fig. 3). Phosphorylation of RS1 breaks these contacts further suggesting that charge-charge contacts drive RRM2-RS1 binding. With these core interactions identified, we are now able to connect aspects of SR protein conformation with the phosphorylation mechanism and biological function in an unprecedented manner.
Long-Range Contacts In An SR Protein Guide the Phosphorylation Mechanism
In the present study we show that the phosphorylation mechanism catalyzed by SRPK1 relies on the conformation of the SR protein. We propose that discrete contacts between the N-terminal portion of RS1 and RRM2 present the C-terminal Arg-Ser dipeptides for initiation of phosphorylation (Fig. 7). We base this conclusion on two findings. First, only four Arg-Ser repeats are necessary for binding RRM2 leaving the remainder of the RS1 segment solvent exposed. Second, the C-terminus of RS1 shows increased sensitivity to LysC cleavage suggesting that this region is more accessible to SRPK1. Once SRPK1 binds at the C-terminus of RS1, we predict that the N-terminal dipeptides begin to dissociate from RRM2 and the docking groove then stabilizes the remaining RS1 segment for directional phosphorylation. In this manner, both the conformation of the SR protein and the docking groove work to support a directional mechanism. We suspect that these structural elements function synergistically since either breaking the internal contacts within the apo-SR protein or mutation of the docking groove in SRPK1 disrupts the C-to-N phosphorylation mechanism 12. Our results imply that contacts between two domains in SRSF1 offer a biased presentation of RS1 for SRPK1 that sets up a cascade of ensuing interactions leading to directional phosphorylation. The present study with separately expressed RRM2 and RS1 cannot discriminate between intramolecular (monomer) or intermolecular (homodimers or higher order aggregates) interactions. However, the proposed mechanism in context of an SRSF1 monomer (Fig. 7) is also conceivable for multiple self-associated SRSF1 molecules, since both forms could display the RS1 segment in a manner promoting directional phosphorylation. Finally, we observed that the cluster of negatively charged residues identified by NMR is conserved in all RRM2’s in the SR protein family suggesting that RS-RRM contacts not only may play a role in regulating phosphorylation and cellular localization, but also may be involved in the recruitment of other splicing factors during spliceosome complex assembly.
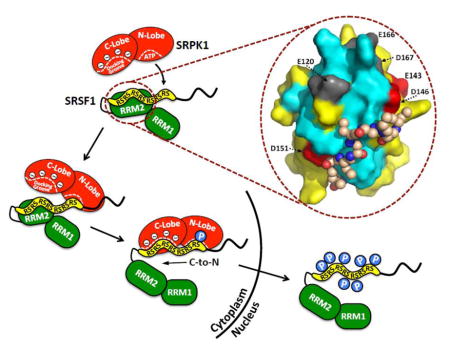
Figure 7. Model for SRPK1-Dependent Directional Phosphorylation and SRSF1 Nuclear Import.

RRM2-RS1 interactions direct SRPK1 to C-terminal end of RS1 for efficient, directional phosphorylation driven by the docking groove. Severing RRM2-RS1 interactions by mutation leads to non-directional phosphorylation, increased accessibility of RS domain to protein phosphatases (PPases) and enhanced protease sensitivity (box).
Phosphorylation Mechanism & Subcellular Localization Are Coupled
Upon translation, all SR proteins enter the nucleus where they reside in speckles or, upon further phosphorylation, engage in splicing activity. Several SR proteins including SRSF1 shuttle continuously between the cytoplasm and nucleus 31. Whereas SRPK-dependent phosphorylation promotes transport into the nucleus, dephosphorylation by PPases supports SR protein export to the cytoplasm 32. This shuttling mechanism is vital for transferring processed mRNA from the nucleus to the cytoplasm for translation. Thus, it is expected that the SRPK1 phosphorylation mechanism is optimized for efficient subcellular transport of SR proteins. Indeed, although SRSF1 is a large protein substrate with an intrinsically disordered domain, kcat/Km is still diffusion controlled falling within a medium range for protein-protein interactions (107 M−1s−1) 33. Disruption of the RRM2-RS1 contacts through mutation not only interrupts the directional phosphorylation mechanism but also lowers the encounter and turnover rates for the substrate lacking the interacting residues. We suspect that RRM2 interactions arrange the RS1 segment for efficient, directional phosphorylation by SRPK1, avoiding unproductive conformers expected from a disordered domain. The reduction in association kinetics is likely to participate in the elevated PP1 activity toward the RS domain and subsequent decrease in SRSF1 nuclear transport (Fig 7). These findings indicate that the directional mechanism for SRPK1 is likely to have evolved as a means to efficiently transport SR proteins into the nucleus and, once present, stabilize its conformation for function in the spliceosome
Materials & Methods
Materials
Adenosine triphosphate (ATP), 3-(N-morpholino)propanesulphonic acid (MOPS), Tris (hydroxymethyl) aminomethane (Tris), MgCl2, NaCl, EDTA, acetic acid, Lysozyme, DNAse, RNAse, Phenix imaging film, BSA, and liquid scintillant were obtained from Fisher Scientific. γ32P-ATP was obtained from NEN Products, a division of Perkin-Elmer Life Sciences. Protease inhibitor cocktail and LysC were obtained from Roche. 10x MnCl2 and 10x PMP buffer (500 mM HEPES, 100 mM NaCl, 20 mM DTT, 0.1% Brij 35, pH 7.5) was obtained from NEB. GFP and Histone antibodies were obtained from Cell Signaling. GAPDH antibody was obtained from Genescript.
Expression and Purification of Recombinant Proteins
Human SRPK1 and all forms of His-tagged SRSF1 were expressed and purified from pET19b vectors containing an N-terminal His tag as previously described 34. All forms of GST-SRSF1 and GST-RS1 were expressed and purified from a pGEX vector as previously described 34. Uniformly 15N- and 13C,15N-labeled RRM2 was expressed in the E. coli strain BL21(DE3) (Novagen), using M9 minimal growth medium containing 15NH4Cl (1 g/L) and either unlabeled or [13C6]-D-glucose (4 g/L) as the sole sources of nitrogen and carbon, respectively. Cell cultures were grown at 37 °C and induced with 1 mM IPTG after reaching an optical density OD600 0.6–0.8. Cells were allowed to grow for 16 h at 18 °C and harvested by centrifugation. Cells were then resuspended in extraction buffer (0.2 M NaCl, 10 mM imidazole, 20 mM Na2HPO4/NaH2PO4 buffer, pH 7.5) containing Complete EDTA-free protease inhibitor cocktail tablets (Roche) and lysed by sonication. Following centrifugation at 30,000g for 30 min, the lysate was loaded onto a HisTrap HP Ni-affinity column (GE Healthcare) pre-equilibrated with purification buffer (0.2 M NaCl, 10 mM imidazole, 20 mM Na2HPO4/NaH2PO4, pH 7.5). The imidazole concentration was increased, first to 30 mM to remove non-specifically bound proteins and then to 500 mM to elute the target protein. TEV protease cleavage was performed overnight at room temperature, and the resulting protein solution was loaded onto a HiPrep 26/10 desalting column (GE Healthcare) and eluted with purification buffer. The protein fractions were then passed through a HisTrap HP column (GE Healthcare) equilibrated with buffer A to remove the His-tagged TEV protease and the cleaved His-tag. Fractions containing RRM2 were pooled and loaded onto a size exclusion column HiLoad 26/60 Superdex75 (GE Healthcare) equilibrated with NMR buffer (50 mM NaCl, 20 mM Na2HPO4/NaH2PO4 buffer, pH 6.0). The fractions containing the target protein were concentrated to 550 μL, with a final protein concentration of 1.1 mM, using 3 kDa-cut-off centrifugal filter devices (Millipore). For the NMR measurements, these concentrated protein solutions were supplemented with 5 % 2H2O (v/v) and 4.5 mM NaN3
NMR Data Acquisition
All NMR experiments were recorded at 298 K. A Bruker AVANCE 600 MHz spectrometer equipped with a 5 mm z-gradient cryoprobe was used to record the 4D APSY-HACANH, 5D APSY-CBCACONH and 5D APSY-HACACONH NMR experiments 35, 36 and a Bruker AVANCE 800 MHz spectrometer equipped with a room temperature TXI-HCN probehead was used to record the 3D 15N-resolved, 3D 13C(aliphatic)-resolved and 3D 13C(aromatic)-resolved [1H,1H]-NOESY experiments with a mixing time of 65 ms. Proton chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonic acid sodium salt (DSS). The 13C and 15N chemical shifts were referenced indirectly to DSS, using the absolute frequency ratios 37. Acquisition of 2D [15N,1H]-HSQC spectra for the study of interactions between SRSF1-RRM2 and all RS domain containing constructs was carried out on a Bruker AVANCE 700 MHz spectrometer equipped with a 1.7 mm z-gradient room temperature microcoil probehead.
NMR Structure Determination
The NMR structure determination followed the J-UNIO protocol 17. Automated routines 19, 20 yielded 95% of the backbone assignments, and 82% of the side-chain assignments. These assignments were validated and interactively extended to 96%, and then used as input for UNIO-ATNOS/CANDID 22, 38 in combination with the torsion angle dynamics algorithm CYANA-3.0 39. The 40 conformers with the lowest residual target function values were energy–minimized in a water shell with the program OPALp40, 41, using the AMBER force field 42. The 20 best conformers, as identified during structure validation 17, were selected to represent the NMR structure of SRSF1-RRM2, and the program MOLMOL 41 was used for structure analysis. The atomic coordinates of the bundle of 20 conformers have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb) with accession code 2M7S.
LysC Proteolysis Experiments
Substrate phosphorylation by SRPK1 was carried out in the presence of 50 mM Mops (pH 7.4), 10 mM free Mg2+, and [γ-32P]ATP (600–1000 cpm/pmol) at 25 °C. Reactions were initiated with the addition of varying amounts of [32P]-ATP (0.2–100 μM) into a total reaction volume of 10 μL. After 20 min at 25 °C, proteolysis with 20 ng of LysC was carried out in a digestion buffer of 25 mM Tris–HCl (pH 8.5) and 1 mM EDTA for 0, 1, or 4 hours at 37 °C. Reactions were quenched with 10 μL of SDS-PAGE loading buffer and loaded onto a 10% SDS-PAGE gel. Dried gels were then exposed with Kodak imaging film (Biomax MR), and protein bands corresponding to phosphorylated SR protein or digested fragments were excised and quantitated on the 32P channel in liquid scintillant.
Viscosometric Experiments
Viscosity experiments were carried out in the presence of 100 mM MOPS (pH 7.4), 10 mM Mg2+ and 5 mg/ml BSA at 25 °C for 1.5 minutes, using 15 nM enzyme, 50 μM 32P-ATP (4000–8000 cpm/pmol) and varying SRSF1 [30–1000 nM] in either the absence or presence of 30% Sucrose. All reactions were carried out in a total volume of 10 μl and quenched with 10 μl of SDS/PAGE loading buffer. Phosphorylated SR proteins were separated from unreacted 32P-ATP by SDS-PAGE (18% gel), cut from the dried gel and quantified on the 32P channel in liquid scintillant.
SRPK-PP1 Kinetic Experiments
Time-course experiments were carried out in the presence of 100 mM MOPS (pH 7.4), 50 mM HEPES, 100 mM NaCl, 2 mM DTT, 0.01% Brij 35, 1 mM Mn2+, 10 mM Mg2+ and 5 mg/ml BSA at 37°C, using 75 nM SRPK1, 50 μM 32P-ATP (4000–8000 cpm/pmol), and 1 μM SRSF1 with or without 300 nM PP1γ. All reactions were carried out in a total volume of 10 μl and quenched with 10 μl of SDS/PAGE loading buffer. Phosphorylated SR proteins were separated from unreacted 32P-ATP by SDS-PAGE (18% gel), cut from the dried gel and quantified on the 32P channel in liquid scintillant.
Pull-Down Assays
GST-tagged proteins (10 μM) were incubated with His-tagged proteins (10 μM) in binding buffer (0.1% NP40 (Nonidet P40), 20 mM Tris/HCl (pH 7.5) and 75 mM NaCl) in a total volume of 40 μL for 30 min before incubating with 25 μl of glutathione–agarose resin for 30 min at room temperature. In all cases, the resin was washed 4X with 200 μl of binding buffer, and the bound proteins were eluted with SDS quench buffer and boiled for 5 min. Retained protein was resolved by SDS-PAGE (18% gel) and visualized by Instant Blue Coomassie stain.
Confocal Imaging & Cell Fractionation
HeLa cells were transfected with 1μg of GFP-SRSF1 and GFP-SRSF13M using Fugene for 24 h. Cells were then imaged using an Olympus FV1000 with a 488 laser line according to a previous described procedure 43. For cell fractionation studies, HeLa cells were plated in 60mm plates and transfected in duplicates with 2μg of GFP-SRSF1 and GFP-SRSF13M using Fugene for 24 h. Transfected cells were harvested and the cell lysates were fractionated into cytoplasmic and nuclear fractions using the Lamond Lab protocol (http://www.lamondlab.com/pdf/CellFractionation.pdf). The sub-cellular fractions were probed by western analysis using GFP rabbit polyclonal antibody for over-expressed GFP-SRSF1 and GFP-SRSF13M in both cytoplasm and nucleus. GAPDH was used as marker for cytoplasm and histone was used as control for nuclear fractions.
Peptide Docking
A coarse grain model was developed to simplify the description of the octapeptide and dock it on the entire protein surface. In particular, the peptide backbone was described by three beads representing amino, C-alpha and carbonyl moieties, respectively. For side chains, one bead was used for serine (C-beta and O-gamma), and three for arginine (C-beta, C-gamma/C-delta, and guanidinium). The beads have been implemented by modifying the standard AutoDock force field parameter file and the conversion from the full atomic model to the coarse grain model was performed using a Python script. Comparing to the full atomic model, the coarse grain model reduced the number of atoms from 78 to 40 and the number of torsions from 51 to 32, speeding up the calculation and simplifying the conformational space to be searched. The first model of the PDB entry 2m8d has been used as target structure and prepared using the standard protocol 44. Interactions with key NMR and mutagenesis residues have been favored by multiplying their atomic contributions to the score (2-fold and 4-fold respectively). A grid box of 101x82x116 points was defined and centered approximately on the center of mass of the protein. The standard GA parameters have been used for the dockings, and 200K poses have been generated.
Supplementary Material
Highlights.
SRPK1 catalyzes multisite phosphorylation of SRSF1 using a directional mechanism.
Several charged residues in RRM2 interact with RS domain in SRSF1.
RRM2-RS domain interactions control directional phosphorylation.
Directional phosphorylation regulates subcellular localization of SRSF1.
Phosphorylation mechanism is tied to biological function of an SR protein.
Footnotes
This work was supported by NIH grants GM67969 and GM67969-S1.
The abbreviations used are: NMR, nuclear magnetic resonance; RRM, RNA recognition motif; RS domain, domain rich in arginine-serine dipeptide repeats; SR protein, splicing factor containing arginine-serine dipeptide repeats; SRPK1, serine-arginine-specific protein kinase 1; SRSF1, SR protein splicing factor 1 (aka ASF/SF2).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2011;278:587–97. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai MC, Lin RI, Huang SY, Tsai CW, Tarn WY. A human importin-beta family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J Biol Chem. 2000;275:7950–7. doi: 10.1074/jbc.275.11.7950. [DOI] [PubMed] [Google Scholar]
- 3.Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci U S A. 2001;98:10154–9. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohtz JD, Jamison SF, Will CL, Zuo P, Luhrmann R, Garcia-Blanco MA, Manley JL. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–24. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 5.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–70. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 6.Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, Ghosh G. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1–70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mermoud JE, Cohen PT, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO J. 1994;13:5679–88. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai MC, Tarn WY. Hypophosphorylated ASF/SF2 binds TAP and is present in messenger ribonucleoproteins. J Biol Chem. 2004;279:31745–9. doi: 10.1074/jbc.C400173200. [DOI] [PubMed] [Google Scholar]
- 9.Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–82. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 10.Ma CT, Velazquez-Dones A, Hagopian JC, Ghosh G, Fu XD, Adams JA. Ordered multi-site phosphorylation of the splicing factor ASF/SF2 by SRPK1. J Mol Biol. 2008;376:55–68. doi: 10.1016/j.jmb.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 11.Ngo JC, Giang K, Chakrabarti S, Ma CT, Huynh N, Hagopian JC, Dorrestein PC, Fu XD, Adams JA, Ghosh G. A sliding docking interaction is essential for sequential and processive phosphorylation of an SR protein by SRPK1. Mol Cell. 2008;29:563–76. doi: 10.1016/j.molcel.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagopian JC, Ma CT, Meade BR, Albuquerque CP, Ngo JC, Ghosh G, Jennings PA, Fu XD, Adams JA. Adaptable molecular interactions guide phosphorylation of the SR protein ASF/SF2 by SRPK1. J Mol Biol. 2008;382:894–909. doi: 10.1016/j.jmb.2008.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma CT, Hagopian JC, Ghosh G, Fu XD, Adams JA. Regiospecific phosphorylation control of the SR protein ASF/SF2 by SRPK1. J Mol Biol. 2009;390:618–34. doi: 10.1016/j.jmb.2009.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aubol BE, Adams JA. Applying the brakes to multisite SR protein phosphorylation: substrate-induced effects on the splicing kinase SRPK1. Biochemistry. 2011;50:6888–900. doi: 10.1021/bi2007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang S, Gapsys V, Kim HY, Bessonov S, Hsiao HH, Mohlmann S, Klaukien V, Ficner R, Becker S, Urlaub H, Luhrmann R, de Groot B, Zweckstetter M. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure. 2013;21:2162–74. doi: 10.1016/j.str.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Tintaru AM, Hautbergue GM, Hounslow AM, Hung ML, Lian LY, Craven CJ, Wilson SA. Structural and functional analysis of RNA and TAP binding to SF2/ASF. EMBO Rep. 2007;8:756–62. doi: 10.1038/sj.embor.7401031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano P, Pedrini B, Mohanty B, Geralt M, Herrmann T, Wuthrich K. The J-UNIO protocol for automated protein structure determination by NMR in solution. J Biomol NMR. 2012;53:341–54. doi: 10.1007/s10858-012-9645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dutta SK, Serrano P, Proudfoot A, Geralt M, Pedrini B, Herrmann T, Wuthrich K. APSY-NMR for protein backbone assignment in high-throughput structural biology. J Biomol NMR. 2015;61:47–53. doi: 10.1007/s10858-014-9881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volk J, Herrmann T, Wuthrich K. Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR. 2008;41:127–38. doi: 10.1007/s10858-008-9243-5. [DOI] [PubMed] [Google Scholar]
- 20.Fiorito F, Herrmann T, Damberger FF, Wuthrich K. Automated amino acid side-chain NMR assignment of proteins using (13)C- and (15)N-resolved 3D [ (1)H, (1)H]-NOESY. J Biomol NMR. 2008;42:23–33. doi: 10.1007/s10858-008-9259-x. [DOI] [PubMed] [Google Scholar]
- 21.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. Journal of molecular biology. 2002;319:209–27. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002;24:171–89. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 23.Clery A, Sinha R, Anczukow O, Corrionero A, Moursy A, Daubner GM, Valcarcel J, Krainer AR, Allain FH. Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc Natl Acad Sci U S A. 2013;110:E2802–11. doi: 10.1073/pnas.1303445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tacke R, Chen Y, Manley JL. Sequence-specific RNA binding by an SR protein requires RS domain phosphorylation: creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci U S A. 1997;94:1148–53. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–67. [PMC free article] [PubMed] [Google Scholar]
- 26.Blacklow SC, Raines RT, Lim WA, Zamore PD, Knowles JR. Triosephosphate isomerase catalysis is diffusion controlled. Appendix: Analysis of triose phosphate equilibria in aqueous solution by 31P NMR. Biochemistry. 1988;27:1158–67. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 27.Albery WJ, Knowles JR. Efficiency and evolution of enzyme catalysis. Angew Chem Int Ed Engl. 1977;16:285–93. doi: 10.1002/anie.197702851. [DOI] [PubMed] [Google Scholar]
- 28.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 29.Maertens GN, Cook NJ, Wang W, Hare S, Gupta SS, Oztop I, Lee K, Pye VE, Cosnefroy O, Snijders AP, KewalRamani VN, Fassati A, Engelman A, Cherepanov P. Structural basis for nuclear import of splicing factors by human Transportin 3. Proc Natl Acad Sci U S A. 2014;111:2728–33. doi: 10.1073/pnas.1320755111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daubner G, Cléry A, Allain F. RRM–RNA recognition: NMR or crystallography…and new findings. Curr Opin Struct Biol. 2013;23:100–8. doi: 10.1016/j.sbi.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Caceres JF, Screaton GR, Krainer AR. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998;12:55–66. doi: 10.1101/gad.12.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci U S A. 2004;101:9666–70. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlosshauer M, Baker D. Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci. 2004;13:1660–9. doi: 10.1110/ps.03517304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aubol BE, Plocinik RM, Keshwani MM, McGlone ML, Hagopian JC, Ghosh G, Fu XD, Adams JA. N-terminus of the protein kinase CLK1 induces SR protein hyperphosphorylation. Biochem J. 2014;462:143–52. doi: 10.1042/BJ20140494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiller S, Fiorito F, Wuthrich K, Wider G. Automated projection spectroscopy (APSY) Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10876–81. doi: 10.1073/pnas.0504818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiller S, Wider G, Wuthrich K. APSY-NMR with proteins: practical aspects and backbone assignment. J Biomol NMR. 2008;42:179–95. doi: 10.1007/s10858-008-9266-y. [DOI] [PubMed] [Google Scholar]
- 37.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–40. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 38.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–27. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 39.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. Journal of molecular biology. 1997;273:283–98. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 40.Luginbuhl P, Guntert P, Billeter M, Wuthrich K. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J Biomol NMR. 1996;8:136–46. doi: 10.1007/BF00211160. [DOI] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 42.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. Journal of the American Chemical Society. 1995;117:5179–97. [Google Scholar]
- 43.Keshwani MM, Aubol BE, Fattet L, Ma CT, Qiu J, Jennings PA, Fu XD, Adams JA. Conserved proline-directed phosphorylation regulates SR protein conformation and splicing function. Biochem J. 2015;466:311–22. doi: 10.1042/BJ20141373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.