

Summary
The hepatic iron-regulatory hormone hepcidin and its receptor, the cellular iron exporter ferroportin, constitute a feedback-regulated mechanism that maintains adequate plasma concentrations of iron-transferrin for erythropoiesis and other functions, ensures sufficient iron stores, and avoids iron toxicity and iron-dependent microbial pathogenesis. In chronic kidney disease, inflammation and impaired renal clearance increase plasma hepcidin, inhibiting duodenal iron absorption and sequestering iron in macrophages. These effects of hepcidin can cause systemic iron deficiency, decreased availability of iron for erythropoiesis, and resistance to endogenous and exogenous erythropoietin. Together with impaired renal production of erythropoietin, hepcidin-mediated iron restriction contributes to anemia of chronic kidney disease.
Keywords: Anemia, inflammation, iron deficiency, renal failure
This article reviews the basics of pathophysiology of iron, focusing on features that contribute to clinical problems in chronic kidney disease. For complementary perspectives the reader is referred to other comprehensive reviews.1–3
IRON HOMEOSTASIS
Biological Role of Iron
Iron is an essential trace element that is highly abundant in nature, predominantly in its poorly soluble ferric form. Because iron readily participates in oxidation and reduction chemistry, it has evolved to have an important role in oxygen transport (in hemoglobin), oxygen storage (in myoglobin), energy metabolism (cytochromes), and intermediary metabolism (as a component of many enzymes). Either iron deficiency or iron excess (overload) can have adverse consequences on organ function and tissue integrity. Iron deficiency is the most common cause of anemia worldwide, adversely impacting population health and economic productivity predominantly in developing countries. Severe iron excess can lead to deposition of toxic forms of iron in the liver, heart, and other vital organs, with resulting organ damage, and, in the case of the liver, carcinogenesis.
Iron Distribution and Flux
In the average adult male there is approximately 4 g of iron, of which 2.5 g is in the hemoglobin of erythrocytes and approximately 1 g is in storage in hepatocytes. The remainder is distributed among other cell types. Iron stores in women are much smaller, even as a proportion of body size. Faced with poor water solubility of iron and its low bioavailability, animals including human beings conserve and recycle iron. Thus, in human beings, iron that is absorbed in the intestine and distributed among tissues is retained and reused, and cannot be excreted actively. Relatively small iron losses (1–2 mg/d) are caused almost entirely by desquamation of the skin and intestinal epithelia, representing a loss of less than 0.1% of total body iron content daily. Under normal circumstances, the iron content of the human body thus is regulated entirely at the point of iron absorption, in enterocytes in the proximal duodenum. In steady state, iron balance is maintained by absorption of 1 to 2 mg of iron per day. However, the daily dietary iron requirement is approximately 10-fold higher because only a fraction of dietary iron is absorbed in the intestine. Clinically significant pathologic iron loss is nearly always a consequence of blood loss because each milliliter of packed erythrocytes contains 1 mg of iron, a vastly higher iron concentration than in other cell types or body fluids. In comparison, blood plasma contains a thousand times lower concentration of iron.
Iron Delivery to Cells
Once absorbed, iron is bound in plasma to the iron carrier protein transferrin, which distributes iron to most tissues and cells in the body. Each cell meets its individual iron needs by selective uptake of iron transferrin via the transferrin receptor, extracting the iron in acidified vacuoles and recycling the receptor and iron-free transferrin back to the extracellular fluid where the iron-free transferrin is released to extracellular fluid for another cycle of iron binding and delivery. At any time, less than 0.1% of the total body iron (or approximately 2–3 mg) is associated with transferrin. The daily demand for iron is approximately 20 to 25 mg, mainly for erythropoiesis, causing the transferrin-associated iron to turn over every 2 to 3 hours. Within cells, iron is stored in ferritin. The number of transferrin receptors and the amount of iron-storing ferritin in each cell are reciprocally controlled by iron-regulated posttranscriptional mechanisms, thus meeting the iron needs of each individual cell.
Iron Deficiency
Iron deficiency4,5 develops when the absorption of iron from the diet is insufficient to compensate for iron losses. In adult men in the developed world, with diets that usually contain sufficient iron to balance normal losses, iron deficiency is almost always a pathologic consequence of excessive blood loss. In women, iron deficiency also can develop as a result of menstrual blood losses and iron deficits incurred during pregnancy and childbirth. Iron requirements are greatly increased in children during periods of rapid growth and this can make their iron balance tenuous. The best known and most common manifestation of iron deficiency is microcytic hypochromic anemia, but iron deficiency also can cause neurobehavioral changes, impair muscle function, and may cause epithelial pathologies, including skin, nail, hair, tongue, and esophageal abnormalities. Erythroid heme synthesis, globin chain synthesis, as well as erythroid maturation appear to be inhibited by even moderate iron deficiency through feedback regulatory mechanisms.6 Slowing down erythropoiesis in iron deficiency may spare enough iron to protect other tissues from dysfunction, at the cost of causing anemia.
Iron Toxicity
Transferrin-bound iron appears to be chemically nonreactive and its uptake and utilization is normally under tight control of each cell. In disorders of iron homeostasis in which iron absorption is excessive, or large amounts of iron are delivered parenterally as drugs or in the form of erythrocyte transfusions, the capacity of transferrin to bind iron may be exceeded (100% transferrin saturation), causing iron to bind to metabolic intermediates that function as iron chelators, mainly citrate. Depending on how these species are measured, they are referred to collectively as nontransferrin bound iron or labile plasma iron.7 These iron forms are taken up extremely rapidly by hepatocytes, cardiac myocytes, and endocrine cells via specific iron transporters,8 overwhelming normal cellular homeostatic mechanisms, and causing cellular toxicity through oxidative stress.
Extracellular Iron Homeostasis
The concentration of iron in extracellular fluid and plasma is controlled systemically, so that the concentration of iron in plasma is normally approximately 10 to 30 μmol/L, leading to 20% to 45% transferrin saturation. The amount of iron in storage, predominantly in macrophages and hepatocytes, also is subject to systemic control. The hormone responsible for regulating extracellular iron concentration and the amount of iron in storage is hepcidin,3 a small peptide produced by hepatocytes. In turn, the production of hepcidin is regulated transcriptionally by plasma iron concentration, iron stores in the liver, erythropoietic activity, and inflammation. The feedback regulation of iron by hepcidin is a classic endocrine mechanism, analogous to the regulation of glucose by insulin.
The Mechanism of Hepcidin’s Effect on Iron
Hepcidin acts by binding to the cellular iron exporter ferroportin,9 which mediates all the major flows of iron into plasma and extracellular fluid: the transfer of dietary iron from duodenal enterocytes to plasma, the release of recycled iron from splenic and hepatic macrophages, and the release of stored iron from hepatocytes. After hepcidin binds to ferroportin, ferroportin undergoes endocytosis and intracellular proteolysis. The loss of ferroportin from cell surfaces then proportionally decreases the delivery of cellular iron to blood plasma. The ongoing consumption of iron by erythroid precursors and other cells rapidly depletes the relatively small extracellular iron compartment, resulting in hepcidin-induced lowering of plasma iron concentration (hypoferremia). Hepcidin is a hypoferremia-inducing hormone, acting analogously to the hypoglycemic effect of insulin.
Regulation of Hepcidin Secretion by Iron
Systemic hepcidin production appears to be solely transcriptionally regulated. Hepcidin gene transcription is stimulated by the dual effect of liver iron stores and the concentration of plasma holotransferrin (iron-saturated transferrin), conveyed through iron-regulated production of bone morphogenetic proteins (BMP) acting on BMP receptors and the associated Smad pathways.10 The hepcidin gene promoter contains BMP-responsive elements that bind nuclear Smad complexes to potently increase transcription.
The concentration of the BMP ligand (in mice mainly BMP-6) appears to be regulated by hepatic iron stores. Increased holotransferrin concentrations also potentiate the BMP receptor signaling via a partly defined mechanism involving transferrin receptors 1 and 2 and HFE (a membrane protein that interacts with transferrin receptor 1). The important role of HFE was highlighted by the finding that it is mutated in the most common form of hereditary hemochromatosis, a disease in which iron homeostasis is dysregulated. BMP signaling also is dependent on hemojuvelin, a glycophosphatidylinositol-linked, iron-related co-receptor for BMPs. Hemojuvelin and transferrin receptor 2 genes are mutated in rare forms of hereditary hemochromatosis, again showing the importance of these molecules for human iron homeostasis. A membrane protease matriptase 2 (also called transmembrane serine protease 6) negatively regulates BMP signaling by cleaving hemojuvelin,11,12 with this inhibitory influence mainly manifested during iron deficiency.
Regulation of Hepcidin Secretion by Inflammation
During infections or other inflammatory conditions hepcidin production is stimulated intensely, causing the characteristic hypoferremia of inflammation. Accumulating evidence confirms that this response has a host defense function, essential for resistance to certain microbes, particularly “siderophilic” bacteria whose pathogenicity is strongly enhanced by iron13 (eg, Vibrio species and Yersinia enterocolitica). Suppressive effects of inflammatory hypoferremia on other pathogenic microbes also are likely but remain to be documented. Inflammation induces hepcidin transcriptionally, mainly through interleukin-6,14 its receptor, and the Jak2-Stat3 pathway, but a second inflammatory pathway recently was identified, stimulating hepcidin through activin B, the BMP receptor, and its Smad signaling mechanism.15
Regulation of Hepcidin Secretion by Erythroid Activity
In human beings who hemorrhage or breathe air with low oxygen content, hypoxic renal interstitial fibroblast-like cells secrete erythropoietin, which stimulates compensatory erythropoiesis. To replace a 10% loss of erythrocytes or to increase oxygen-carrying capacity by 10% in the average adult requires approximately 250 mg of iron, representing a major burden for the iron-homeostatic system. Within hours after a hemorrhagic or hypoxic event, hepcidin is profoundly suppressed, allowing increased absorption of iron from the diet and, even more importantly, releasing iron from stores in macrophages and hepatocytes into blood plasma. The rapid suppression of hepcidin and the resulting increased iron availability for erythropoiesis now are known to be mediated by erythroferrone,16 a protein secreted by erythroblasts in response to stimulation by erythropoietin. Pharmacologic stimulation of erythropoiesis by erythropoietin also causes erythroferrone secretion and suppression of hepcidin.16,17 Hepcidin suppression is mediated by a direct effect of erythroferrone on hepatocytes but the specific pathways have not yet been identified.
Hypoxia and Hepcidin Regulation
Hypoxia may affect the whole organism, as in exposure to air with low oxygen content, or may affect specific tissues in which oxygen tension may decrease because of low regional blood flow or decreased oxygen carrying capacity of blood. At the cellular level, oxygen is sensed by prolyl hydroxylases 1 to 3, oxygen- and iron-dependent enzymes that hydroxylate the hypoxia-inducible factors (HIFs)1α or 2α, and thereby target them for degradation.18,19 During hypoxia, decreased degradation of HIFs causes them to accumulate and increase the transcription of many hypoxia-inducible genes, prominently including erythropoietin and vascular endothelial factor. The hydroxylating activity of prolyl hydroxylases on HIFs is dependent not only on local oxygen tension but also on iron concentration and the concentration of a metabolic cofactor, the citrate cycle intermediate α-ketoglutarate (2-oxoglutarate). Local hypoxia promotes iron absorption in the duodenum, mainly by the HIF-dependent transcriptional activation of the apical iron transporter divalent metal transporter 1 and the basolateral transporter ferroportin.20 This effect of hypoxia is hepcidin-independent. Systemic hypoxia potently but indirectly modulates hepcidin and iron homeostasis by inducing the transcription of erythropoietin,21 thereby stimulating erythroferrone production and hepcidin suppression.
Hepcidin Clearance
Hepcidin is a 2.7-kD peptide that is not strongly bound to plasma proteins and is cleared rapidly by the normal kidneys where it is largely reabsorbed and degraded by the proximal tubular mechanism that metabolizes other peptides. A small proportion of hepcidin escapes this mechanism and can be detected in urine,22 where daily excretion is approximately proportional to serum hepcidin concentrations.23 Hepcidin also undergoes endocytosis with its receptor/iron transporter ferroportin and both molecules then are proteolysed.24 In mice, injected radiolabeled hepcidin is cleared within minutes, with most of the radioactivity appearing rapidly in the kidneys and the bladder, and smaller amounts found at sites where ferroportin is highly expressed (the duodenum, spleen, and the liver).25 Another degradation mechanism is dependent on as yet uncharacterized N-terminal peptidases that convert the 25 amino acid mature hepcidin-25 into mainly 22 amino acid and 20 amino acid N-terminally truncated forms, hepcidin-22 and hepcidin-20. These have severely impaired ability to induce ferroportin internalization, and are thought to be inactive degradation products.26 They are much more prominent in urine than in serum, and hepcidin-20 appears to increase out of proportion to total hepcidin in chronic kidney disease.27,28 Neither the mechanisms by which the smaller hepcidin forms are generated nor their modulation by renal impairment are understood.
IRON DYSREGULATION IN CHRONIC KIDNEY DISEASE
Iron Disorders in Chronic Kidney Disease
In advanced kidney diseases iron metabolism becomes severely disrupted through multiple mechanisms.29 Although a detailed study of the time course in individual patients has not been reported, iron deficiency is already found in a majority of non–dialysis-dependent patients with chronic kidney disease.30 Iron deficiency is probably a consequence of decreased iron absorption31 caused by high hepcidin concentrations,32 as well as increased iron losses, mostly from gastrointestinal bleeding.33 High hepcidin concentrations are caused in part by inflammation involved in the pathogenesis of many kidney diseases, as indicated by the correlation of serum hepcidin concentrations with C-reactive protein, and in part by decreased clearance of hepcidin by the diseased kidneys, as reflected by the anticorrelation of hepcidin with estimated glomerular filtrate rate.34–36 Once patients are treated with hemodialysis or peritoneal dialysis, additional inflammatory stimuli come into play, including intermittent infections and exposure of blood to foreign materials, such as catheters and dialysis membranes, further increasing hepcidin levels.32 During hemodialysis, plasma hepcidin concentration decreases, suggesting that the peptide is cleared through the membrane,37 but the high rate of hepcidin synthesis causes a rebound of hepcidin concentrations within an hour after the completion of dialysis.38 Hemodialysis also causes increased blood loss from discarded residual blood in the dialysis equipment, from the hemorrhagic effects of anticoagulation, and from the blood requirements of more frequent laboratory examinations,29 greatly increasing iron requirements.
Erythropoiesis-Stimulating Agents
As chronic kidney disease progresses, the kidneys of most patients eventually fail to produce sufficient erythropoietin to maintain adequate erythropoiesis. When patients develop symptomatic anemia, they may be treated by intermittent administration of erythropoiesis-stimulating agents, which further challenges iron homeostasis. Surges of erythropoiesis after each dose of erythropoietin require an increased flow of iron to the marrow. In patients with systemic inflammation and high hepcidin concentrations, even when iron stores are adequate and sufficient for baseline erythropoiesis, iron cannot be released from stores rapidly enough to meet the needs of pharmacologically stimulated erythropoiesis. This condition is referred to as “functional iron deficiency.”39 Functional iron deficiency limits the effectiveness of erythropoiesis-stimulating agents, contributing to “erythropoietin resistance.”40 Increased doses of erythropoietin, likely acting by inducing the production of erythroferrone, may suppress hepcidin, release iron from stores, and overcome resistance to erythropoietin. However, high doses of erythropoietin have been associated with adverse effects and are no longer recommended.41 In addition, economic incentives have favored decreased use of erythropoietin and related drugs. It is now well established that the administration of intravenous iron potentiates the effectiveness of lower doses of erythropoiesis-stimulating agents,42 most likely by greatly increasing the iron-loading of macrophages and hence the rate at which iron is released from these cells to meet the requirements of stimulated erythropoiesis.
BIOMARKERS OF IRON STATUS IN CHRONIC KIDNEY DISEASE
Iron and Transferrin Saturation
Although the use of biomarkers for patient management is addressed elsewhere in this issue of Seminars in Nephrology, it is useful to review how the various biomarkers are affected by disturbances in iron homeostasis. Erythroid precursors use almost exclusively transferrin-bound iron as a source of iron for heme and hemoglobin synthesis. Once the concentration of transferrin-bound iron decreases to less than approximately 10 μmol/L, erythroid heme, hemoglobin synthesis, and erythropoiesis are progressively inhibited.43,44 Depending on the concentration of transferrin, this threshold typically corresponds to 15% to 20% transferrin saturation. Low serum iron concentration and low transferrin saturation therefore are useful markers of an inadequate iron supply to erythropoiesis. Because transferrin-bound iron turns over every few hours and its concentration is rapidly affected by iron ingestion, parenteral iron treatment, and diurnal variation, it is considered too labile to be used as the sole parameter for diagnosis.
Serum Transferrin Receptor
Transferrin receptor 1 (TfR1) mediates the uptake of iron transferrin by erythrocyte precursors, and these cells collectively account for most of the iron flow and most of the TfR1 molecules in the body. When iron supply becomes inadequate for erythropoiesis, erythrocyte precursors compensate by increased production of TfR1, achieved by iron-regulated stabilization of TfR1 messenger RNA. Because the receptors shed into plasma, plasma TfR1 concentrations reflect the total number of TfR1 molecules in the body.45 Accordingly, increased serum transferrin-receptor concentrations are seen when erythrocyte precursors become iron-deficient and/or the total number of erythrocyte precursors is increased. Because inflammation suppresses TfR1 synthesis, iron limitation from a systemic iron deficiency causes a greater increase in serum transferrin–receptor concentrations than iron limitation caused by an inflammatory iron sequestration, allowing some discrimination between the two conditions.
Serum Ferritin
Intracellular iron is stored in the cavities of ferritin nanoparticles, which serve as an iron depot after cellular iron uptake, and a source of iron for the synthesis of iron-containing proteins, including hemoglobin. A poorly understood byproduct of this storage form is plasma ferritin, which is secreted from iron-storing cells, predominantly macrophages and hepatocytes, in proportion to the amount of stored iron. In the absence of inflammation, serum ferritin is a reliable indicator of iron stores. During inflammation, ferritin production is stimulated and, under the influence of hepcidin, iron is sequestered in macrophages, which are prolific secretors of serum ferritin.46 Accordingly, the highest serum ferritin concentrations are seen in patients with macrophage activation syndromes47 and genetic defects in ferroportin that interfere with macrophage iron export.48 Serum ferritin concentrations therefore reflect at least three parameters of iron metabolism: iron stores, their distribution between macrophages and hepatocytes, and the intensity of inflammation. Nevertheless, low serum ferritin levels are specific indicators of iron deficiency even in chronic kidney disease but are not sensitive because inflammation can drive up ferritin levels and mask iron deficiency.
Red Cell Indices
Under normal circumstances, human erythrocytes live for approximately 120 days, thus blood hemoglobin levels and mean corpuscular volume reflect the average conditions during this entire timespan. During periods of inadequate iron supply for erythropoiesis, the newly produced erythrocytes are hypochromic and microcytic, but when the iron availability improves, more normal erythrocytes are released from the marrow. Therefore, the percentage of hypochromic erythrocytes represents a record of episodes of iron deficiency over the erythrocyte lifespan. In contrast, measurement of hemoglobin content in reticulocytes, transitional cells released from the marrow in the preceding few days, reflects a snapshot of recent iron availability for hemoglobin synthesis.49
Erythrocyte Zinc Protoporphyrin
The terminal step in heme synthesis is the incorporation of iron into the protoporphyrin molecule by the mitochondrial enzyme ferrochelatase. If iron supply to the mitochondria is inadequate, zinc may be substituted for iron, yielding zinc protoporphyrin, an intensely fluorescent product that can be quantified in red cells. Increased erythrocyte zinc protoporphyrin levels are seen both in iron deficiency and in anemia of inflammation.50
Therapeutic implications
Advances in understanding of iron homeostasis and of its disturbances in chronic kidney disease have suggested potential interventions to correct iron physiology and facilitate a more effective treatment of anemia in this condition. On the iron supply side, the current strategy of administering relatively large amounts of intravenous iron could be moderated by minimizing treatment-related inflammation or counteracting it. Less inflammatory stimulation will decrease hepcidin concentrations and increase the availability of iron for erythropoiesis. Alternatively, experimental therapies to decrease hepcidin are under development.51 On the erythroid demand side, new anemia therapies that exert prolonged erythroid-stimulatory and hepcidin-suppressive effect may require less iron supplementation.52,53
Figure 1.
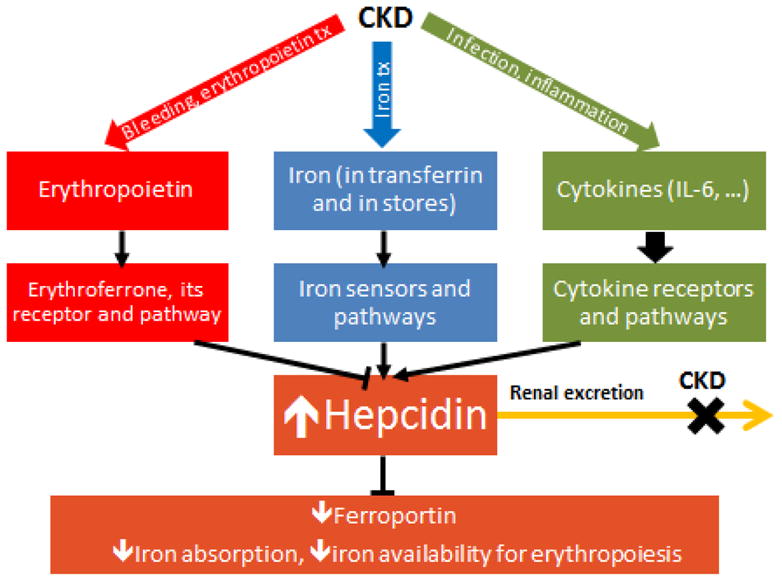
The pathogenesis of iron dysregulation in chronic kidney disease (CKD). The diagram shows the hepcidin-stimulatory effects of inflammation and iron treatment, the hepcidin-increasing effect of decreased glomerular filtration rate, and the opposing suppressive effects of erythropoietin and erythroferrone on hepcidin production. Plasma hepcidin concentrations are usually high, decreasing ferroportin on cell membranes, and thus inhibiting duodenal iron absorption and diminishing iron availability for erythropoiesis. IL, interleukin.
Acknowledgments
Financial support: Supported in part by the Will Rogers Fund and National Institutes of Health grants.
Footnotes
Conflict of interest statement: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112:219–30. doi: 10.1182/blood-2007-12-077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hentze MW, Muckenthaler MU, Galy B, et al. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93:1721–41. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 4.Cook JD. Diagnosis and management of iron-deficiency anaemia. Best Pract Res Clin Haematol. 2005;18:319–32. doi: 10.1016/j.beha.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 5.Cogswell ME, Looker AC, Pfeiffer CM, et al. Assessment of iron deficiency in US preschool children and nonpregnant females of childbearing age: National Health and Nutrition Examination Survey 2003–2006. Am J Clin Nutr. 2009;89:1334–42. doi: 10.3945/ajcn.2008.27151. [DOI] [PubMed] [Google Scholar]
- 6.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breuer W, Ronson A, Slotki IN, et al. The assessment of serum nontransferrin-bound iron in chelation therapy and iron supplementation. Blood. 2000;95:2975–82. [PubMed] [Google Scholar]
- 8.Jenkitkasemwong S, Wang CY, Coffey R, et al. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 2015;22:138–50. doi: 10.1016/j.cmet.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 10.Core AB, Canali S, Babitt JL. Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front Pharmacol. 2014;5:104. doi: 10.3389/fphar.2014.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–11. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang AS, Anderson SA, Wang J, et al. Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein. Blood. 2011;117:1687–99. doi: 10.1182/blood-2010-06-287292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arezes J, Jung G, Gabayan V, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe. 2015;17:47–57. doi: 10.1016/j.chom.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin B by inflammatory stimuli upregulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120:431–9. doi: 10.1182/blood-2012-02-411470. [DOI] [PubMed] [Google Scholar]
- 16.Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–84. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95:505–8. doi: 10.3324/haematol.2009.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salahudeen AA, Bruick RK. Maintaining mammalian iron and oxygen homeostasis: sensors, regulation, and cross-talk. Ann N Y Acad Sci. 2009;1177:30–8. doi: 10.1111/j.1749-6632.2009.05038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–26. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 20.Shah YM, Xie L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology. 2014;146:630–42. doi: 10.1053/j.gastro.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Davidoff O, Niss K, et al. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122:4635–44. doi: 10.1172/JCI63924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–10. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 23.Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood. 2008;112:4292–7. doi: 10.1182/blood-2008-02-139915. [DOI] [PubMed] [Google Scholar]
- 24.Preza GC, Pinon R, Ganz T, et al. Cellular catabolism of the iron-regulatory peptide hormone hepcidin. PLoS One. 2013;8:e58934. doi: 10.1371/journal.pone.0058934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera S, Nemeth E, Gabayan V, et al. Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood. 2005;106:2196–9. doi: 10.1182/blood-2005-04-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemeth E, Preza GC, Jung CL, et al. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood. 2006;107:328–33. doi: 10.1182/blood-2005-05-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroot JJ, Laarakkers CM, Geurts-Moespot AJ, et al. Immunochemical and mass-spectrometry-based serum hepcidin assays for iron metabolism disorders. Clin Chem. 2010;56:1570–9. doi: 10.1373/clinchem.2010.149187. [DOI] [PubMed] [Google Scholar]
- 28.Addo L, Ikuta K, Tanaka H, et al. The three isoforms of hepcidin in human serum and their processing determined by liquid chromatography-tandem mass spectrometry (LC-tandem MS) Int J Hematol. 2015 doi: 10.1007/s12185-015-1885-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 29.Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23:1631–4. doi: 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minutolo R, Locatelli F, Gallieni M, et al. Anaemia management in non-dialysis chronic kidney disease (CKD) patients: a multicentre prospective study in renal clinics. Nephrol Dial Transplant. 2013;28:3035–45. doi: 10.1093/ndt/gft338. [DOI] [PubMed] [Google Scholar]
- 31.Goch J, Birgegard G, Danielson BG, et al. Iron absorption in patients with chronic uremia on maintenance hemodialysis and in healthy volunteers measured with a simple oral iron load test. Nephron. 1996;73:403–6. doi: 10.1159/000189100. [DOI] [PubMed] [Google Scholar]
- 32.Zaritsky J, Young B, Wang HJ, et al. Hepcidin--a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4:1051–6. doi: 10.2215/CJN.05931108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sohal AS, Gangji AS, Crowther MA, et al. Uremic bleeding: pathophysiology and clinical risk factors. Thromb Res. 2006;118:417–22. doi: 10.1016/j.thromres.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Atkinson M, Kim J, Roy C, et al. Hepcidin and risk of anemia in CKD: a cross-sectional and longitudinal analysis in the CKiD cohort. Pediatr Nephrol. 2015;30:635–43. doi: 10.1007/s00467-014-2991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mercadel L, Metzger M, Haymann JP, et al. The relation of hepcidin to iron disorders, inflammation and hemoglobin in chronic kidney disease. PLoS One. 2014;9:e99781. doi: 10.1371/journal.pone.0099781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Troutt JS, Butterfield AM, Konrad RJ. Hepcidin-25 concentrations are markedly increased in patients with chronic kidney disease and are inversely correlated with estimated glomerular filtration rates. J Clin Lab Anal. 2013;27:504–10. doi: 10.1002/jcla.21634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaritsky J, Young B, Gales B, et al. Reduction of serum hepcidin by hemodialysis in pediatric and adult patients. Clin J Am Soc Nephrol. 2010;5:1010–4. doi: 10.2215/CJN.08161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuragano T, Shimonaka Y, Kida A, et al. Determinants of hepcidin in patients on maintenance hemodialysis: role of inflammation. Am J Nephrol. 2010;31:534–40. doi: 10.1159/000312381. [DOI] [PubMed] [Google Scholar]
- 39.Cavill I, Macdougall IC. Functional iron deficiency. Blood. 1993;82:1377. [PubMed] [Google Scholar]
- 40.Johnson DW, Pollock CA, Macdougall IC. Erythropoiesis-stimulating agent hyporesponsiveness. Nephrology (Carlton) 2007;12:321–30. doi: 10.1111/j.1440-1797.2007.00810.x. [DOI] [PubMed] [Google Scholar]
- 41.Fishbane S, Besarab A. Mechanism of increased mortality risk with erythropoietin treatment to higher hemoglobin targets. Clin J Am Soc Nephrol. 2007;2:1274–82. doi: 10.2215/CJN.02380607. [DOI] [PubMed] [Google Scholar]
- 42.Fishbane S, Frei GL, Maesaka J. Reduction in recombinant human erythropoietin doses by the use of chronic intravenous iron supplementation. Am J Kidney Dis. 1995;26:41–6. doi: 10.1016/0272-6386(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 43.Bullock GC, Delehanty LL, Talbot AL, et al. Iron control of erythroid development by a novel aconitase-associated regulatory pathway. Blood. 2010;116:97–108. doi: 10.1182/blood-2009-10-251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han AP, Yu C, Lu L, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–18. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skikne BS. Serum transferrin receptor. Am J Hematol. 2008;83:872–5. doi: 10.1002/ajh.21279. [DOI] [PubMed] [Google Scholar]
- 46.Cohen LA, Gutierrez L, Weiss A, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116:1574–84. doi: 10.1182/blood-2009-11-253815. [DOI] [PubMed] [Google Scholar]
- 47.Moore C, Jr, Ormseth M, Fuchs H. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. J Clin Rheumatol. 2013;19:324–8. doi: 10.1097/RHU.0b013e31829ce01f. [DOI] [PubMed] [Google Scholar]
- 48.Pietrangelo A. The ferroportin disease. Blood Cell Mol Dis. 2004;32:131–8. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol. 2013;20:222–30. doi: 10.1097/MOH.0b013e32835f5933. [DOI] [PubMed] [Google Scholar]
- 50.Braun J. Erythrocyte zinc protoporphyrin. Kidney Int Suppl. 1999;69:S57–S60. doi: 10.1046/j.1523-1755.1999.055suppl.69057.x. [DOI] [PubMed] [Google Scholar]
- 51.Nemeth E. Anti-hepcidin therapy for iron-restricted anemias. Blood. 2013;122:2929–31. doi: 10.1182/blood-2013-08-522466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonomini M, Del Vecchio L, Sirolli V, et al. New treatment approaches for the anemia of CKD. Am J Kidney Dis. 2016;67:133–42. doi: 10.1053/j.ajkd.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 53.Besarab A, Chernyavskaya E, Motylev I, et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015030241. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]