

Abstract
Atypical teratoid rhabdoid tumors (AT/RT) are characterized by mutations and subsequent inactivation of SMARCB1 (INI1, hSNF5), a predilection for very young children and an unfavorable outcome. The European Registry for rhabdoid tumors (EU‐RHAB) was established to generate a common European database and to establish a standardized treatment regimen as the basis for phase I/II trials. Thus, genetic analyses, neuropathologic and radiologic diagnoses, and a consensus treatment regimen were prospectively evaluated. From 2005 to 2009, 31 patients with AT/RT from four countries were recruited into the registry study Rhabdoid 2007 and treated with systemic and intraventricular chemotherapy. Eight patients received high‐dose chemotherapy, 23 radiotherapy, and 17 maintenance therapy. Reference evaluations were performed in 64% (genetic analyses, FISH, MLPA, sequencing) up to 97% (neuropathology, INI1 stain). Germ‐line mutations (GLM) were detected in 6/21 patients. Prolonged overall survival was associated with age above 3 years, radiotherapy and achievement of a complete remission. 6‐year overall and event‐free survival rates were 46% (±0.10) and 45% (±0.09), respectively. Serious adverse events and one treatment‐related death due to insufficiency of a ventriculo peritoneal shunt (VP‐shunt) and consecutive herniation were noted. Acquisition of standardized data including reference diagnosis and a standard treatment schedule improved data quality along with a survival benefit. Treatment was feasible with significant but manageable toxicity. Although our analysis is biased due to heterogeneous adherence to therapy, EU‐RHAB provides the best available basis for phase I/II clinical trials.
Keywords: AT/RT, EU‐RHAB Registry, pediatric brain tumor, Rhabdoid 2007
Introduction
The development of specific diagnostic measures including immunohistochemistry and molecular genetics has led to a precise definition of the entity rhabdoid tumors 1. The majority of rhabdoid tumors demonstrate mutations in SMARCB1 (INI1, hSNF5) 2, 3 and rarely SMARCA4 (BRG1) 4. Survival remains unsatisfactory due to the aggressive nature of the disease and ranges from 15% to 40% even with intensive multimodality therapy 5, 6. Dufour et al. report 58 nonuniformly treated patients with atypical teratoid rhabdoid tumor (AT/RT, 1998–2008) with a median overall survival (OS) of 9 months 7. Chi et al. demonstrated a promising 2‐year OS of 70% treated in a prospective protocol 8.
The implementation of the European registry for rhabdoid tumors (EU‐RHAB) – was intended to prospectively collect comprehensive data on consistently treated children with AT/RT across several European countries. Data collection was initiated in 2005 with centers mainly from Germany and Austria but also Scandinavia and Spain. The registry provides basic clinical data and a system of high‐quality reference diagnostics and expert counseling for diagnostic and therapeutic measures. Thus, EU‐RHAB was an innovation in AT/RT research at the time of initiation despite the fact that the quality of resulting data may not entirely be comparable to a controlled clinical trial. Children with rare diseases and a risk profile, which makes a controlled clinical trial difficult, have the same rights for a standardized treatment with the best available scientific evidence of activity. The treatment schedule Rhabdoid 2007, specifically designed for rhabdoid tumors, comprised elements including anthracyclines, intraventricular methotrexate and early start of radiotherapy (RT), all of which have previously been described as promising tools in this entity 6, 9.
Materials and Methods
From 06/2005 to 03/2009 a total of 35 patients with a centrally confirmed diagnosis of AT/RT were registered to EU‐RHAB. Thereof 31 patients were treated according to the Rhabdoid 2007 schedule. Eight patients received high‐dose chemotherapy (HDCT) with autologous stem cell transplantation following induction chemotherapy at the treating physicians discretion. Results for these eight patients have partly been reported 10. Inclusion criteria were diagnosis of an AT/RT as confirmed by negative INI1 staining and typical neuropathological features as confirmed by a reference neuropathologist, age below 18 years of age and the presence of informed consent by the legal guardians to contribute patient‐associated data.
EU‐RHAB including Rhabdoid 2007 has received continuous approval by the Ethics Committee of the University of Münster (ID 2009‐532‐f‐S).
Diagnostic measures
Reference neuropathology including immunohistochemistry of SMARCB1 was performed according to WHO criteria on formalin‐fixed paraffin‐embedded (FFPE) tumor tissue 1. Staging procedures such as evaluation of lumbar cerebrospinal fluid (CSF) was in accordance with the Chang classification 11. Neuroradiologic response was evaluated according to criteria of the German National Reference Center for Neuroradiology in Würzburg, Germany 12. DNA was derived from FFPE tumor material and peripheral blood cells, and analyzed for somatic and germ‐line genetic changes in SMARCB1 (SMARCA4) to identify tumor associated and constitutional aberrations. Fluorescence‐in situ hybridization (FISH), multiplex ligation‐dependent probe amplification (MLPA), and sequencing were performed at reference centers in Hamburg and Kiel as described 13, 14. FISH was performed on fixed unstained slides of peripheral blood smears and tumor sections of FFPE material.
Treatment
Rhabdoid 2007 comprised postoperative chemotherapy with a total of nine courses including five alternating courses of vincristine, cyclophosphamide, doxorubicin (VCD) and four of ifosfamide, carboplatinum, etoposide (ICE) every 21 days (Fig. 1). Age‐adjusted intraventricular methotrexate (MTX) or triple chemotherapy (MTX/cytarabine/hydro‐cortisone) applied via Ommaya or Rickham reservoir was recommended with systemic chemotherapy until the start of radiotherapy. The suggested maintenance therapy (MT) consisted of eight cycles of trofosfamide/idarubicin (TI) and trofosfamide/etoposide (TE) every 3 weeks.
Figure 1.
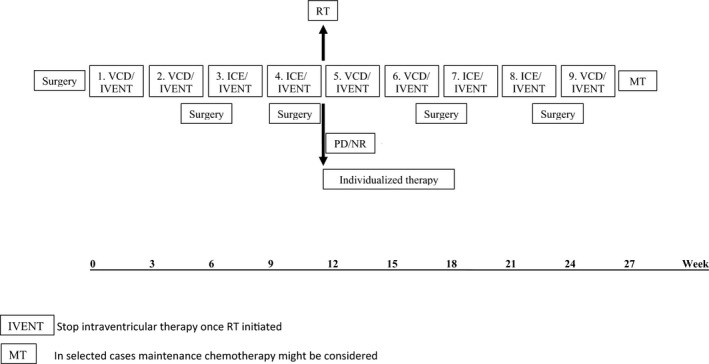
Rhabdoid 2007 treatment recommendation for patients with AT/RT.
Conformal radiotherapy of the primary tumor region (encompassing the postsurgical tumor bed including any residual mass plus a 10–15 mm margin) was recommended at 54 Gy for patients over 18 months of age preferably after the 4th course of chemotherapy. In patients with CSF dissemination at diagnosis (M1 through M4 according to Chang) craniospinal radiotherapy with 24 Gy plus an additional boost to the primary tumor region and any central nervous system (CNS) deposits was recommended for ages 3 years and older. Circumscribed spinal deposits could be boosted up to 49.2 years. Innovative technologies such as IMRT or proton beam therapy were encouraged.
Toxicity
Toxicity was assessed according to version 3.0. of the Common Terminology Criteria for Adverse Events. Reporting of serious adverse events (SAEs) to the registry office was requested but not monitored.
Statistics
Overall (OS) and event‐free survival (EFS) were determined according to Kaplan–Meier estimates. OS was defined as the time from diagnosis until death of any cause or the last visit. EFS was defined as the time from diagnosis until first progression, relapse, death of any cause, or last contact.
The potential impact of prognostic factors on OS and EFS was analyzed as follows: congenital.
Kaplan–Meier analyses were performed for basic elements such as age and metastases, tumor location, extent of resection, germ‐line mutation (GLM), etc. Time‐dependent factors, for example, radiotherapy (RT), complete remission (CR), and MT were evaluated using Cox regression for time‐dependent covariates. P‐values were significant for P ≤ 0.05. Results were considered exploratory, not confirmatory. Adjustment for multiple testing was not performed. An overall significance level was not determined and could not be calculated due to small numbers.
Results
Clinical characteristics of patients with AT/RT
Thirty‐one patients were registered and treated according to Rhabdoid 2007 (Table 1). This accounts for approximately 51% of all eligible cases from Germany during the corresponding time (2005–2009 total incidence n = 61). The median age at diagnosis of 12 females and 19 males was 20 months (range 0–120). Seventy‐four percent of patients were below 3 years at diagnosis (12 < 1 years, 11 = 1 – 3 years, 8 > 3 years). Three congenital cases were registered. One was symptomatic with visible multiple synchronous extracerebral tumors (thorax) in addition to an AT/RT at (no. 15). A second presented with a visible periorbital tumor plus AT/RT at birth (no. 19), the third demonstrated frequent vomiting and hydrocephalus leading to imaging (no. 28). Tumor localization was infratentorial (n = 13), supratentorial (n = 15), or encompassed both compartments (s.t. and i.t. n = 3). Imaging and CSF cytology revealed six patients with metastases at diagnosis. Four patients presented with CSF dissemination (M1), one patient with additional intracerebral metastases (M2), and one patient with soft tissue metastases (M4). Analysis of CSF was completed in 28 patients (no analysis in no. 3, 6, 15). Patients no. 3 and no. 6 were not stable enough for a lumbar puncture at diagnosis, no. 15 had multiple congenital synchronous tumors accompanied by soft tissue metastases (M4).
Table 1.
Patient characteristics, therapeutic measures, and outcome.
| No. | Agea | INI1 | Germline mutation | Genetic analysisb | Diagnosis | Location | M‐stagec | Surgery | Chemotherapyd | Intraventricular therapy | HDCT | Radiotherapy | RT dose (GY) | Age at RT (months) | Maintenance therapy | Complete remission during therapy | Dead of disease | Survival (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | neg | No | WT | AT/RT | Cerebellum | M0 | Total | 10 | Yes | No | Local | 54 | 15 | No | Yes | Yes | – |
| 2 | 56 | neg | No | WT | AT/RT | Cerebral hemisphere | M0 | Subtotal | 8 | Yes | No | Local | 54 | 57 | Yes | Yes | No | 85 |
| 3 | 32 | neg | n.a. | n.a. | AT/RT | Supratentorial n.s. | n.a. | Partial | 6 | No | Yes | No | – | No | No | Yes | – | |
| 4 | 5 | neg | Yes | het del, dupl 838insT | AT/RT | Cerebellar hemisphere, IV. ventricle | M2 | Total | 5 | Yes | No | No | – | No | No | Yes | – | |
| 5 | 7 | neg | No | n.a. | AT/RT | Fossa cranii posterior | M0 | Subtotal | 4 | Yes | No | No | – | No | No | Yes | – | |
| 6 | 29 | neg | n.a. | WT | AT/RT | Tempo‐parietal | n.a. | Partial | 3 | No | No | No | – | No | No | Yes | – | |
| 7 | 14 | neg | No | n.a. | AT/RT | Cerebellum | M0 | Total | 7 | Yes | No | Local | 54 | 21 | Yes | Yes | No | 75 |
| 8 | 23 | neg | No | het del | AT/RT | Cerebral hemisphere frontal | M1 | Subtotal | 6 | Yes | Yes | No | – | No | Yes | Yes | – | |
| 9 | 67 | neg | No | 1148delC ex9 | AT/RT | Pineal gland, mesencephalon, thalamus | M0 | Total | 7 | Yes | No | Local | 54 | 70 | Yes | Yes | No | 77 |
| 10 | 11 | neg | No | del chromosome 22 | AT/RT | Cerebellum | M0 | Subtotal | 9 | Yes | No | Local | 54 | 19 | Yes | Yes | No | 95 |
| 11 | 19 | neg | No | het del | AT/RT | Intracerebral | M0 | Subtotal | 9 | Yes | No | Local | 54 | 26 | Yes | Yes | Yes | – |
| 12 | 120 | neg | No | het del, 1148delC ex9 | AT/RT | Pineal gland, mesencephalon, III. ventricle | M0 | Total | 7 | Yes | No | Local | 54 | 122 | Yes | Yes | No | 79 |
| 13 | 38 | neg | n.a. | n.a. | AT/RT | Cerebellar hemisphere, IV. ventricle | M0 | Total | 9 | Yes | No | Local | 54 | 44 | No | Yes | No | 66 |
| 14 | 5 | neg | Yes | 170delTG ex2 | AT/RT | Cerebellum | M1 | Subtotal | 8 | Yes | No | Local | 54 | 12 | Yes | Yes | Yes | – |
| 15 | 0 | neg | Yes | homo del | AT/RT+MRT | Cerebellopontine angle, thorax | M4 | Biopsy | 5 | No | No | No | – | No | No | Yes | – | |
| 16 | 26 | neg | n.a. | n.a. | AT/RT | Cerebral hemisphere | M0 | Subtotal | 9 | Yes | No | Local | 54 | 30 | Yes | Yes | Yes | – |
| 17 | 22 | neg | n.a. | n.a. | AT/RT | Fossa cranii posterior | M0 | Total | 7 | Yes | No | Local | 54 | 29 | Yes | Yes | No | 79 |
| 18 | 10 | neg | No | WT | AT/RT | Basal ganglia, lateral ventricle | M0 | Partial | 4 | Yes | Yes | Local | 54 | 15 | No | Yes | No | 96 |
| 19 | 0 | neg | Yes | het und homo del | AT/RT+MRT | Orbi and periorbital, temporal head/neck | M0 | Total | 8 | Yes | Yes | No | – | Yes | Yes | No | 70 | |
| 20 | 11 | neg | n.a. | n.a. | AT/RT | Infratentorial n.s. | M0 | Partial | 8 | Yes | Yes | Local | 54 | 20 | Yes | Yes | Yes | – |
| 21 | 18 | neg | No | n.a. | AT/RT | Cerebral hemisphere, mesencephalon, pons, medulla | M0 | Partial | 2 | Yes | No | Local | 54 | 25 | Yes | No | Yes | – |
| 22 | 6 | neg | No | n.a. | AT/RT | Basal ganglia, pons | M0 | Partial | 6 | Yes | No | Local | 54 | 13 | Yes | No | Yes | – |
| 23 | 18 | neg | No | Not evaluable | AT/RT | Mesencephalon, pons | M0 | Total | 9 | Yes | No | Local | 54 | 21 | Yes | Yes | No | 89 |
| 24 | 11 | neg | No | 511insC ex5 | AT/RT | Fossa cranii posterior | M0 | Subtotal | 9 | Yes | Yes | Local | 54 | 29 | Yes | Yes | No | 66 |
| 25 | 88 | neg | Yes | del, het dupl ex6 | AT/RT | Fossa cranii posterior, cerebellum, medulla IV. ventricle | M0 | Subtotal | 11 | Yes | No | Local | 54 | 92 | No | Yes | No | 87 |
| 26 | 13 | neg | No | WT | AT/RT | Cerebellum | M0 | Partial | 4 | Yes | Yes | Local | 54 | 21 | No | Yes | No | 60 |
| 27 | 49 | neg | No | 1148delC ex9 | AT/RT | Cerebral hemisphere | M1 | Partial | 9 | Yes | No | Craniospinal | 54 | 52 | Yes | Yes | No | 60 |
| 28 | 0 | pos | Yes | del ex17 SMARCA4 | AT/RT | Cerebral hemisphere | M0 | Subtotal | 6 | Yes | No | No | – | No | No | Yes | – | |
| 29 | 105 | neg | No | homo del | AT/RT | Cerebral hemisphere | M0 | Total | 9 | Yes | No | Local | 54 | 110 | No | Yes | No | 64 |
| 30 | 60 | neg | No | WT | AT/RT | Cerebral hemisphere | M1 | Partial | 8 | Yes | Yes | Local + CSI | 54 + 30 | 63 | No | Yes | Yes | – |
| 31 | 22 | neg | No | del ex5 | AT/RT | Cerebral hemisphere, basal ganglia | M0 | Partial | 9 | Yes | No | Local | 54 | 27 | Yes | Yes | Yes | – |
n.a., not analyzed; WT, wild‐type sequence; het, heterozygous; del, deletion; dupl, duplication; ins, insertion; ex, exon; homo, homozygous; n.s., not specified; neg, negative; pos, positive.
Age at diagnosis in months.
FISH, MLPA, sequencing in tumor.
Metastases according to Chang classification.
Courses.
The diagnosis was centrally confirmed on tumor tissue in all cases except one (no. 28) with an underlying SMARCA4 GLM 4.
GLM were detected in six out of 21 evaluated patients. Five mutations affected the SMARCB1 and one the SMARCA4 locus (Table 2). All three patients with congenital tumors displayed a GLM (no. 15, 19, and 28).
Table 2.
Safety – serious adverse events recorded while treatment according to the “Rhabdoid 2007” protocol.
| Serious adverse events (13 SAE in 12/31 patients) | n |
|---|---|
| Necrotizing leukoencephalopathy (causing therapy discontinuation) | 1 |
| Radiogenic gliosis | 1 |
| Sepsis (Ommaya infection n=2, post‐op n=1) | 7 |
| Recurrent infections (causing therapy discontinuation) | 1 |
| Coxitis with candida | 1 |
| Pneumonia (causing discontinuation of maintenance) | 1 |
| Shunt insufficiency causing death (not due to disease) | 1 |
Response to chemotherapy
All patients received chemotherapy according to Rhabdoid 2007 (Fig. 1). Nine of them completed the recommended schedule of nine courses. Two patients received additional courses (no. 1, no. 25). The reasons were (no. 1) bridging until initiation of radiotherapy (RT) and (no. 25) combination with RT for expected synergistic effects. Seven patients received HDCT following induction (after a different number of courses [range 4–8]), and one patient (no. 24) after completed chemotherapy (9 courses).
Thirteen patients received 2–8 courses of conventional chemotherapy courses. Seven patients discontinued chemotherapy due to disease progression, two due to toxicities (no. 7, 9), and one due to shunt insufficiency (no. 22). Three patients had started therapy according to protocols for different entities until definitive reference diagnosis was available (no. 2, 12, 17).
Twenty‐eight patients received strict intraventricular chemotherapy. Twenty‐three of these had no sign of M+ disease in the CSF, five patients were positive (no. 4, 8, 14, 27, 30). Reasons for not applying intraventricular therapy were initiation of concomitant radiotherapy, insufficient circulation of CSF and/or CNS infections.
MT was applied to 17 patients, 14 patients did not receive it due to early disease progression or decision of the treating physician.
Radiotherapeutic approach
Radiotherapy was applied locally to the primary tumor region in 23 of 31 patients. Six patients did not receive RT due to early disease progression, one patient due to age <18 months (no. 19), and one patient (no. 28) due to necrotizing leukoencephalopathy. The age of patients at the time of radiotherapy ranged from 1 to 10 years (Table 1). Fifteen patients were younger than 36 months old, and four (no. 1, 14, 18, 22) younger than 18 months at RT (15, 12, 15, 13 months). Two of the four patients died from progressive disease and one following VP‐shunt insufficiency.
Of 23 patients who had received radiotherapy, 20 were diagnosed with M0 and 3 with M1 disease (no. 14, 27, 30). One (no. 14) patient with M1 disease received only local radiotherapy, another patient (no. 27) craniospinal, and the third patient (no. 30) craniospinal and boost radiotherapy. One had a relapse while still on therapy, the second is alive 5 years from diagnosis and the third died 6 years following diagnosis. Three of six patients with M+ (no. 4, 8, 15) did not receive radiotherapy due to progression.
Proton beam therapy to the tumor region was applied to 4 patients (no. 7, 11, 14, 30) at 21, 26, 12, 63 months of age.
Response to therapy and outcome
A CR was achieved in 23/31 patients. Four patients presented with M1 at diagnosis (no. 8, 14, 27, 30). In 10 CR was achieved by neurosurgical resection (in no. 26 after second look surgery) and in 13 by additional chemo‐and radiotherapy. Five patients achieved CR after two courses of induction, two after three courses, one after four courses, one after five courses, four patients thereafter.
Follow‐up of survivors comprises 61–96 months FUP (follow‐up period) with a median of 77 months. Fifteen patients survived, another 15 died due to disease progression/relapse and one (no. 22) following shunt insufficiency without signs of relapse on imaging. Only one patient with metastatic disease survived (no. 27), but also one with synchronous tumor and GLM (no. 19). Ten patients received a total resection and were in CR; another one (no. 26) after second look surgery. One of these relapsed (no. 1) and one (no. 4) progressed despite total resection and died. Among 20 patients with residual disease following surgery one (no. 22) died following shunt insufficiency without relapse, six eventually demonstrated progressive disease. Of 13 patients who achieved CR on chemotherapy seven died due to relapse 1–23 months after the end of therapy. Whenever feasible RT was performed as a rescue measure.
Six‐year overall and event‐free survival rates are 46% (±0.10) and 45% (±0.09) resp. (Fig. 2). The latest relapse was 37 months following diagnosis. This patient (no. 11) succumbed 5 years after initial diagnosis from relapse despite having achieved a CR to initial therapy. The latest death thus far occurred 72 months from diagnosis (no. 31).
Figure 2.
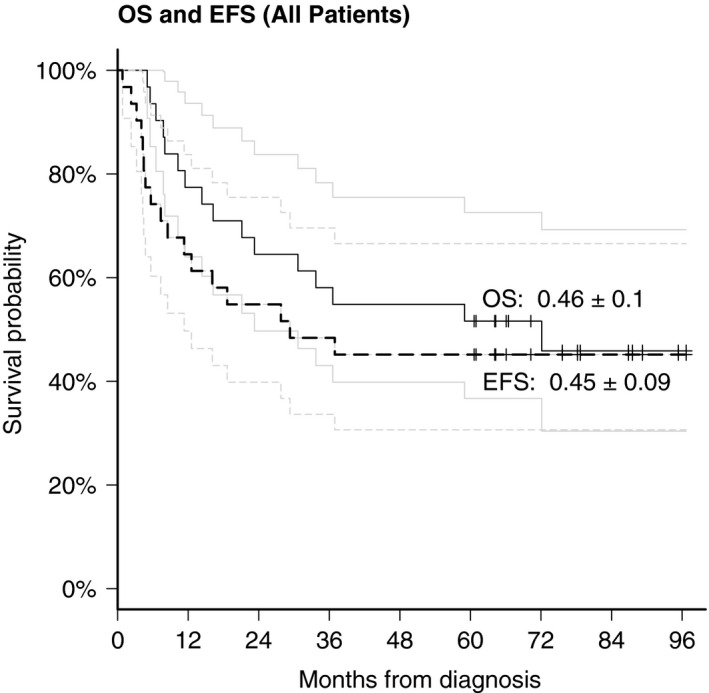
Overall survival and event‐free survival of the Rhabdoid 2007 cohort n = 23.
Prognostic factors in the cohort Rhabdoid 2007
The Kaplan–Meier estimates for factors influencing OS demonstrated a significant prognostic benefit for age > 3y at diagnosis (P < 0.015) (Fig. 3). Of the eight patients who were older than 3 years at diagnosis only one relapsed and died (no. 30), seven are alive 61–85 months following diagnosis. Cox regression analysis for time‐dependent covariates demonstrated a significant impact of radiotherapy (P < 0.005) and achievement of a CR (P < 0.002) on OS (Fig. 4).
Figure 3.
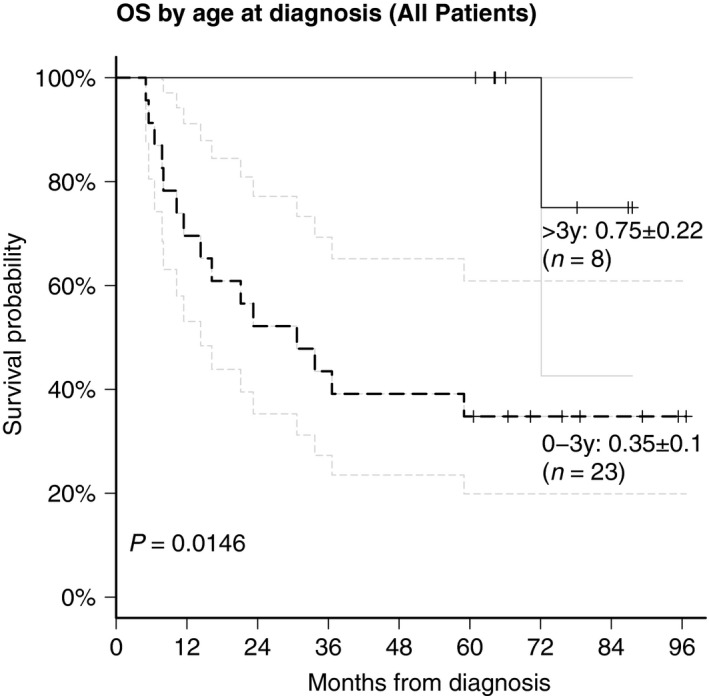
Statistical analysis of prognostic factors: influence of age on survival.
Figure 4.
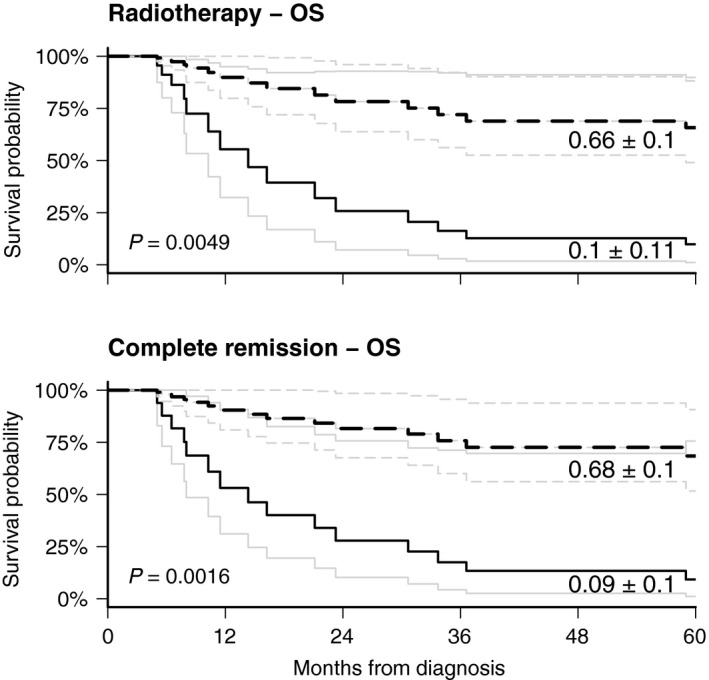
Influence of radiation therapy and achievement of complete remission on survival, cox model with time‐dependent variates.
Presence of metastases at diagnosis, complete resection, and GLM did not influence outcome in this series. Intraventricular MTX or MT did not show any beneficial effect on OS.
Patterns of failure in AT/RT
Sixteen of 31 patients experienced treatment failure and all but one (death due to ventriculo‐peritoneal shunt insufficiency) died due to disease. The median time to progression or relapse was 6.5 months (range 1–37 months). In nine patients early progression or relapse occurred while on therapy. The latest relapse with intracranial metastases outside the proton radiotherapy field was recorded 37 months after diagnosis (no. 11). Pattern of progression/relapse included seven patients with progress disease, six local, and two combined (local + distant) failures.
Toxicities of therapy according to Rhabdoid 2007
A total of 13 SAEs were reported in 12/31 patients (Table 2). One treatment‐related death due to insufficiency of a VP‐shunt and subsequent herniation of the brain was noted (no. 22). Three SAE led to therapy discontinuation due to pneumonia, recurrent infections, or necrotizing leukoencephalopathy associated with intraventricular MTX (no. 9, 23, 28).
Discussion
Challenges in the care of patients with AT/RT
The prognosis of patients with AT/RT remains dismal despite intensive multimodal therapy. Reinhard et al. reported a 5‐year OS of 29% for 25 patients with AT/RT 5. Dufour et al. presented data of 58 AT/RT patients treated from 1998 to 2008 with a median OS of 9 months 7. Comparable unfavorable results of a median OS of 13.5 months were reported by the Canadian Pediatric Brain Consortium 16. Recently, data of a first clinical trial in 20 patients with AT/RT indicated a promising 2‐year OS of 70%, but no follow‐up data are available yet 8. It appears that 5‐year OS rates around 40–50% are achievable (S. N. Chi, pers. comm.). Rather provocative data have been published by Slavc et al. with OS rates of 100% in nine patients. Independent confirmation is currently missing 15.
Reasons for the rather limited data are the rare incidence, high vulnerability of very young infants and children, localization in critical anatomical structures, potential for synchronous tumors and likelihood for chemoresistance. Thus, rhabdoid tumors are far from being ideal subjects for controlled clinical trials. Interest by pharmaceutical companies in performing drug trials with novel compounds has been sparse due to the inherent risk associated with mentioned factors.
The highest level of evidence will certainly be obtained by controlled clinical trials if possible evaluating research questions in a randomized fashion. However, children with rare diseases and a risk profile, which makes a controlled trial extremely difficult, do have the same right for a standardized treatment with the best available scientific evidence of activity.
The need for comprehensive data sets in AT/RT
To circumvent these obstacles German oncologists and other investigators around the world have chosen to recruit patients into “registry studies.” These competence structures offer a system of high‐quality reference diagnostics and expert counseling for diagnosis and therapy. The resulting data are inferior in quality to a controlled clinical trial, but superior to case series and individual reports. The reference system established within the GPOH has over the last decades achieved a near 99% coverage of affected children and is employed throughout German speaking countries.
Results of only one prospective clinical trial have been published in AT/RT 8. At the time of initiation of the EU‐RHAB registry in 2005 a standard of therapy had not been consented. As a European group we therefore chose the means of a registry. It has by now been joined by several European countries and has continued collecting data for almost 10 years. This modality was meant to bridge the gap between case series and the start of a clinical trial not to lose valuable data. The EU‐RHAB database allows conclusions on prognostic factors, course of disease and treatment modalities, onto which together with the diagnostic reference structure future phase I/II clinical trials will be built.
As expected there were deviations from to the protocol according to clinical conditions and individual preferences. The resulting groups with small numbers may not provide statistical significance and the results are complex to interpret. Despite these facts Rhabdoid 2007 therapy resulted in 6‐year OS and EFS rates of 46% (±0.10) and 45% (±0.09). This is an acceptable outcome when compared with published data and is based on a relevant follow‐up period of median of 6 years (5–8 years).
The prognostic variables determining outcome in AT/RT
Results from the registry study Rhabdoid 2007 provide information of prognostic factors in accordance with those previously described such as age at diagnosis 6, 7, 17. In our cohort seven of eight patients older than 3 years at diagnosis are long‐term survivors. Furthermore, Cox regression analysis for time‐dependent covariates demonstrated that a CR and the application of radiotherapy were positive prognostic factors. As described by Chi et al. CR by primary tumor therapy seems to be an important prognostic factor for survival 8, 18. Our analyses confirmed this finding. In the current cohort all surviving patients (n = 15) had achieved a CR. Nevertheless, of 23 who had achieved CR eight patients experienced late relapses and died.
Another negative prognostic factor appears to be the presence of GLM 19. Affected patients generally have a rather poor prognosis with a 2‐year survival of 29% 19. However, detailed data have recently been reported on four patients, including two of ours (no. 19, 25), with a predisposition to rhabdoid tumors and EFS of 5 and 7 years from diagnosis 14. In our cohort, no influence of GLM on OS was detected. However, only six patients had GLM, two of them are long‐term survivors (no. 19, 25). Two further cases in our cohort showed an unexpected long survival despite unfavorable prognostic factors. One patient with M1 is still alive more than 5 years from diagnosis (no. 27) and another patient survived recurrence for 22 months until death of disease (no. 11). These cases indicate that even in unfavorable situations a therapeutic attempt is justified and could result in an unexpected survival time.
In our cohort there was a high likelihood of survival once patients reached 3 years from diagnosis without relapse. The latest relapse occurred 37 months after diagnosis (no. 11) and the follow‐up in our cohort ranged from 60 up to 96 months. Still, the literature knows of a case of late intracranial recurrence 5.5 years following diagnosis 20. The majority of our patients experiencing treatment failure progressed on therapy, which indicates chemotherapy resistance. Especially those patients will most likely not benefit from further intensification of treatment but rather from agents targeting specific pathways. Since all relapsed patients in our cohort died, there is an evident need for relapse trials with innovative concepts in AT/RT.
The potential role of radiotherapy in the treatment of AT/RT
Radiotherapy has been deemed to be a significant positive prognostic factor on retrospective analyses 17. The NCI identified RT as an independent factor for survival in retrospective analysis of 144 children with AT/RT 21. It was concluded that RT could be of significant benefit particularly when looking at very young children <3 years. For our cohort the multivariate, time‐dependent statistical analysis demonstrated a significant beneficial effect of RT on OS and 60% of irradiated patients were <3 years. Assessment of the true value of RT in our cohort is complicated by the fact that most patients who did not receive RT were below 1 year of age at diagnosis and experienced rapid progressive disease. As a majority of progressions occurred on chemotherapy it deserves discussion whether RT as a rescue measure should be given earlier, for example, even to patients below 18 months of age 22. Late effects of RT in young children are, however, significant and have to be considered when tailoring treatment concepts 23. Increasingly, modern technologies like proton beam therapy are used and recent reports demonstrate both good feasibility and promising local control rates 24, 25.
The role of single therapeutic components in the concept of multimodal therapy such as HDCT, intraventricular MTX and MT remains to be clarified. In our cohort the analysis of these three modalities showed no beneficial effect on OS. Data from 19 patients with HDCT of the EU‐RHAB registry have been published 10 but due to the retrospective nature and a heterogeneously treated group with small patient numbers, no definitive conclusion about the effect of HDCT as primary therapy for AT/RT may be drawn.
Feasibility of intensive multimodal therapy in children with AT/RT
Despite a major proportion of >70% of patients below the age of 3 years in our cohort, including 35% infants, no toxic death due to chemotherapy or RT occurred. One treatment‐related death was reported following VP‐shunt insufficiency. In three cases (no. 7, 9, 22) early termination of chemotherapy was necessary due to complications of chemotherapy and intraventricular MTX. With respect to the young age of the patient population and treatment intensity, the recommended treatment of Rhabdoid 2007 showed feasibility and safety. Still, a potential for underreporting of adverse events due to the voluntary character in the framework of the registry has to be acknowledged.
Possible late sequelae which are recognized after intensive therapy for other malignancies in early childhood 26 will be investigated more detailed in AT/RT patients in the future.
Conclusion
Our results demonstrate that intensive multimodality treatment is feasible, safe and efficient even in very young patients. This supports the concept of EU‐RHAB as the basis for data analysis but also for future clinical trials in first line and relapse therapies. The implementation of a phase I/II study for AT/RT and other rhabdoid tumors in the frame of the established registry structures of EU‐RHAB is essential for further improvement of survival. Recently detected pathogenic mechanisms such as epigenetic modulation should give clues which compounds to choose for future innovative trials 27.
Conflict of Interest
None of the authors have any conflict of interest to declare.
Acknowledgments
Oral presentation of preliminary data of the Rhabdoid 2007 cohort at the 15th International Symposium on Pediatric Neuro‐Oncology (ISPNO), Toronto, Canada 2012. We thank P. Neumayer and I. Lechner for expert assistance in data acquisition, managing, and analyses.
Cancer Medicine 2016; 5(8):1765–1775
References
- 1. Judkins, A. R. , Mauger J., Ht A., Rorke L. B., and Biegel J. A.. 2004. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am. J. Surg. Pathol. 28:644–650. [DOI] [PubMed] [Google Scholar]
- 2. Hasselblatt, M. , Isken S., Linge A., Eikmeier K., Jeibmann A., Oyen F., et al. 2013. High‐resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosom. Cancer 52:185–190. [DOI] [PubMed] [Google Scholar]
- 3. Kieran, M. W. , Roberts C. W., Chi S. N., Ligon K. L., Rich B. E., Macconaill L. E., et al. 2012. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr. Blood Cancer 59:1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hasselblatt, M. , Nagel I., Oyen F., Bartelheim K., Russell R. B., Schüller U., et al. 2014. SMARCA4‐mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta. Neuropathol. 128:453–456. [DOI] [PubMed] [Google Scholar]
- 5. Reinhard, H. , Reinert J., Beier R., Furtwängler R., Alkasser M., Rutkowski S., et al. 2008. Rhabdoid tumors in children: prognostic factors in 70 patients diagnosed in Germany. Oncol. Rep. 19:819–823. [PubMed] [Google Scholar]
- 6. Tekautz, T. M. , Fuller C. E., Blaney S., Fouladi M., Broniscer A., Merchant T. E., et al. 2005. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high‐dose alkylator‐based chemotherapy. J. Clin. Oncol. 23:1491–1499. [DOI] [PubMed] [Google Scholar]
- 7. Dufour, C. , Beaugrand A., Le Deley M. C., Bourdeaut F., André N., Leblond P., et al. 2012. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118:3812–3821. [DOI] [PubMed] [Google Scholar]
- 8. Chi, S. N. , Zimmerman M. A., Yao X., Cohen K. J., Burger P., Biegel J. A., et al. 2009. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 27:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hilden, J. M. , Meerbaum S., Burger P., Finlay J., Janss A., Scheithauer B. W., et al. 2004. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J. Clin. Oncol. 22:2877–2884. [DOI] [PubMed] [Google Scholar]
- 10. Benesch, M. , Bartelheim K., Fleischhack G., Gruhn B., Schlegel P. G., Witt O., et al. 2014. High‐dose chemotherapy (HDCT) with auto‐SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU‐RHAB). Bone Marrow Transplant. 49:370–375. [DOI] [PubMed] [Google Scholar]
- 11. Chang, C. H. , Housepian E. M., and Herbert C. Jr. 1969. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93:1351–1359. [DOI] [PubMed] [Google Scholar]
- 12. Warmuth‐Metz, M. , Bison B., and Leykamm S.. 2009. Neuroradiologic review in pediatric brain tumor studies. Klin. Neuroradiol. 21:21. [DOI] [PubMed] [Google Scholar]
- 13. Kordes, U. , Gesk S., Frühwald M. C., Graf N., Leuschner I., Hasselblatt M., et al. 2010. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant tumor. Genes Chromosom. Cancer 49:176–181. [DOI] [PubMed] [Google Scholar]
- 14. Kordes, U. , Bartelheim K., Modena P., Massimino M., Biassoni V., Reinhard H., et al. 2014. Favorable outcome of patients affected by rhabdoid tumors due to rhabdoid tumor predisposition syndrome (RTPS). Pediatr. Blood Cancer 61:919–921. [DOI] [PubMed] [Google Scholar]
- 15. Slavc, I. , Chocholous M., Leiss U., Haberler C., Peyrl A., Azizi A. A., et al. 2014. Atypical teratoid rhabdoid tumor: improved long‐term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Med. 3:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lafay‐Cousin, L. , Hawkins C., Carret A. S., Johnston D., Zelcer S., Wilson B., et al. 2012. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 48:353–359. [DOI] [PubMed] [Google Scholar]
- 17. Sultan, I. , Qaddoumi I., Rodriguez‐Galindo C., Nassan A. A., Ghandour K., and Al‐Hussaini M.. 2010. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr. Blood Cancer 54:35–40. [DOI] [PubMed] [Google Scholar]
- 18. von Hoff, K. , Hinkes B., Dannenmann‐Stern E., von Bueren A. O., Warmuth‐Metz M., Soerensen N., et al. 2011. Frequency, risk‐factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr. Blood Cancer 57:978–985. [DOI] [PubMed] [Google Scholar]
- 19. Bourdeaut, F. , Lequin D., Brugieres L., Reynaud S., Dufour C., Doz F., et al. 2011. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin. Cancer Res. 17:31–38. [DOI] [PubMed] [Google Scholar]
- 20. Modena, P. , Sardi I., Brenca M., Giunti L., Buccoliero A. M., Pollo B., et al. 2013. Case report: long‐term survival of an infant syndromic patient affected by atypical teratoid‐rhabdoid tumor. BMC Cancer 13:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buscariollo, D. L. , Park H. S., Roberts K. B., and Yu J. B.. 2012. Survival outcomes in atypical teratoid rhabdoid tumors for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer 118:4212–4219. [DOI] [PubMed] [Google Scholar]
- 22. Seeringer, A. , Bartelheim K., Kerl K., Hasselblatt M., Leuschner I., Rutkowski S., et al. 2014. Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors ‐ experience of the EU‐RHAB registry. Klin. Padiatr. 226:143–148. [DOI] [PubMed] [Google Scholar]
- 23. Hasan, A. , Palumbo M., Atkinson J., Carret A. S., Farmer J. P., Montes J., et al. 2011. Treatment‐related morbidity in atypical teratoid/rhabdoid tumor: multifocal necrotizing leukoencephalopathy. Pediatr. Neurosurg. 47:7–14. [DOI] [PubMed] [Google Scholar]
- 24. Weber, D. C. , Ares C., Malyapa R., Albertini F., Calaminus G., Kliebsch U., et al. 2015. Tumor control and QoL outcomes of very young children with atypical teratoid/rhabdoid tumor treated with focal only chemo‐radiation therapy using pencil beam scanning proton therapy. J. Neurooncol. 121:389–397. [DOI] [PubMed] [Google Scholar]
- 25. Haskins, C. P. , Jyoti B., Hines M., Simoneaux V., and Buchsbaum J. C.. 2015. Single center results following proton beam therapy in children with atypical teratoid rhabdoid tumors of the central nervous system. Int. J. Particle Ther. 2:1–10. [Google Scholar]
- 26. Robinson, K. E. , Fraley C. E., Pearson M. M., Kuttesch J. F. Jr, and Compas B. E.. 2013. Neurocognitive late effects of pediatric brain tumors of the posterior fossa: a quantitative review. JINS 19:44–53. [DOI] [PubMed] [Google Scholar]
- 27. Kerl, K. , Holsten T., and Fruhwald M. C.. 2013. Rhabdoid tumors: clinical approaches and molecular targets for innovative therapy. Pediatr. Hematol. Oncol. 30:587–604. [DOI] [PubMed] [Google Scholar]