

Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia. Chronic kidney disease (CKD) is associated with a high prevalence of AF, and uremic toxins are an important risk factor for cardiovascular diseases associated with CKD. Uremic toxins can produce pro-fibrotic, pro-hypertrophic, and pro-inflammatory effects on cardiac tissues and enhance oxidative stress or neurohormonal phenomena of cardiovascular injury, which are recognized as arrhythmogenic factors of AF. This article reviews the clinical, molecular, and electrophysiological data of uremic toxins in CKD considered to induce AF through multiple mechanisms on structural and electrical remodeling of the cardiovascular system.
Keywords: Atrial fibrillation, Inflammation, Oxidative stress, Uremic toxins
INTRODUCTION
Atrial fibrillation (AF), the most common clinical arrhythmia, causes significant cardiovascular mortality and morbidity due to heart failure and stroke.1 AF is produced by enhanced trigger activity from ectopic foci or the genesis of reentry circuits in atrial substrates.2 Chronic kidney disease (CKD) is associated with a higher prevalence of AF.3 Uremic cardiomyopathy is a distinctive type of heart failure associated with CKD. Uremic cardiomyopathy can be produced by a reduced microvascular supply, enhanced cardiac fibrosis, progressive inflammation, and oxidative stress. In addition, anemia, hypertension, and activation of the renin-angiotensin-aldosterone and sympathetic nervous systems may contribute to the occurrences of uremic cardiomyopathy in CKD.4-7 The pathological effects of CKD are essential arrhythmogenic factors of AF.8-10 However, traditional cardiovascular risk factors are insufficient to accurately predict cardiovascular mortality or morbidity with CKD.11-16
A novel risk factor, uremic toxins, was proprosed as contributing to the cardiovascualr burden with CKD. Uremic toxins cannot be removed by conventional hemodialysis.13,15,16 Uremic toxins, such as indoxyl sulfate (IS), p-cresol (PC), and p-cresol sulfate (PCS), which originate from protein fermentation can increase oxidative stress and inflammation or activate the neurohormonal system which results in cardiovascular fibrosis and oxidative injury.17,18 On the other hand, uremic toxins produce pro-fibrotic, pro-hypertrophic, and pro-inflammatory phenomena in cardiomyocytes.19-21 In addition, IS was already proved to induce arrhythmogenesis in the pulmonary veins (PVs), sinoatrial node (SAN), and left atrium (LA) through oxidative stress in an animal study.22 Accordingly, uremic toxins may contribute to the genesis of AF through direct and indirect effects on structural and electrical remodeling.
CLINICAL STUDIES OF UREMIC TOXINS IN CARDIOVASCULAR SYSTEM
Uremic toxins play an important role in the progression of CKD and increase the risk of cardiovascular diseases (CVDs; Table 1). Higher level of IS increased all cause mortality and CV mortality in common or elderly hemodialysis patients, which was demonstrated with vascular disease in patients over 40 years of age.13,16 Serum total PCS and free PCS both were recognized as novel risk factor of CV events independent of age, diabetes, anemia, malnutrition, glomerular filtration rate, and calcium-phosphate imbalance in hemodialysis patients.11,15 Therefore, these results implied that uremic toxins may increase the potential risk of atrial arrhythmia during the progression of uremic toxins-induced CVDs.
Table 1. Effects of indoxyl sulfate (IS), p-cresol (PC), and p-cresol sulfate (PCS) on cardiovascular risks.
| Author | Study aim/patient characteristics | Cardiovascular risk |
| Barreto et al.13 | Relationships among IS, vascular calcification, vascular stiffness, and mortality in a cohort of 139 CKD patients during 2006~2008, including patients aged over 40 years who were all Caucasian. | IS in a multivariate analysis with increased pulse wave velocity: RR 2.165. |
| Meijers et al. (2002-2003)11 | A post-hoc analysis of the relationship between PC and cardiovascular (CV) disease with a set of novel and traditional CV risk factors in 175 hemodialysis patients within a mean follow-up of 56.2 months. | High free-PC: HR, 1.69; in a multivariate analysis, free-PC without diabetes mellitus: HR, 2.04. |
| Meijers et al. (2005-2006)15 | Multivariate association between the free PC serum concentration and CV events in 499 patients with mild to moderate kidney disease after a mean follow-up of 33 months. | Higher baseline concentrations of free PC: HR 1.79; in a multivariate analysis, independent of GFR and Framingham risk factors: HR 1.39. |
| Wu et al.16 | The association of serum IS and PCS with all-cause and CV-related mortality in 112 elderly hemodialysis patients within a mean follow-up of 33.2 months in a single medical center. | Serum total PCS with CV mortality: HR, 1.088; serum free PCS with CV mortality: HR, 1.027; the serum IS level was significantly associated with all-cause mortality after multivariate adjustment. |
CKD, chronic kidney disease; HR, hazard ratio; GFR, glomerular filtration rate.
UREMIC TOXINS ON CARDIAC OXIDATIVE STRESS AND INFLAMMATION
Oxidative stress is a substantial character in the pathophysiology of AF.23 Such oxidative stress modulates inflammation, ischemia, heart failure, and renin-angiotensin or adrenergic activation, which are known to increase the occurrence of AF.24,25 Oxidative stress produces structural remodeling through enhancing fibrosis, apoptosis, and fatty metamorphosis in the atrium.26-28 In addition, oxidative stress can change the atrial and thoracic venous electrophysiology.29 Fukunaga et al. demonstrated that oxidative stress can enhance AF vulner ability in an animal model of CKD.26
Oxidative stress was also demonstrated to shorten the atrial effective refractory period and induce automaticity of the atrium and thoracic veins by increasing calcium release through the opening of ryanodine receptors.24,30 Additionally, oxidative stress induces mitochondrial dysfunction in the atrium, which also plays a role in the pathophysiology of atrial arrythmogenesis.31,32 Uremic toxins, such as IS and PCS, can activate leukocytes in the endothelium, which generates an inflammatory reaction through reactive oxyen species (ROS), the nuclear factor-κB pathway, monocyte chemoattractant protein-1, and intercellular adhesion molesule-1 (Table 2).33,34 These data suggest that uremic toxins could have direct effects on atrial arrhythmogenecity and proinflammatory effects in CKD. Moreover, recent study has shown that the antioxidant supressed IS-induced PV brust firing.22 Additionally, slower beating rate and shorter action potential (AP) duration of SAN and LA cardiomyocytes, respectively, induced by IS also can be inhibited by the antioxidant. Hence, it demonstrated that IS can change electrical activities of PV, SAN, and LA cardiomyocytes through oxidative stress. Furthermore, oxidative stress can induce a Ca2+-overload, which interrupts the regulation protein of the calcium channel, and higher expression of the intracellular Ca2+ overload and Ca2+ release may increase the contractility of atrial cardiomyocytes.24,35
Table 2. The pathways of uremia toxins related to cardiovascular disease.
| Cytokine/transcription factor/ionic current | Target | Function/effect | Reference |
| TNF-α, IL-6, IL-1β, MAPK (p38, p42/44), NF-κB | Cardiac fibroblasts and myocytes in rats | Pro-fibrotic, pro-hypertrophic, and pro-inflammatory effects | [19] |
| Inhibit NO production; induce NADPH (Nox4) and superoxide | HUVECs | ROS generation | [33] |
| ICAM-1, MCP-1 | HUVECs | NF-κB | [34] |
| EMPs, calcium | in vivo and in vitro - HUVECs | Endothelial damage | [41] |
| TGF-β, CTGF, ANP, β-MHC, α-skeletal muscle actin | Cardiac tissues of rats | Fibrosis and hypertrophy | [21] |
| PKCα | Cardiac myoblasts of a cell line from rats | Disruption of cardiomyocyte adherent junctions | [20] |
ANP, atrial natriuretic peptide; CTGF, connective tissue growth factor; EMPs, endothelin microparticles; HUVECs, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molesule 1; IL-1β, interleukin-1β; IL-6, interleukin-6; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein 1; MHC, β-myosin heavy chain; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor-kappa B; ROS, reactive oxygen species; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; β-PKCα, protein kinase Cα.
UREMIC TOXINS ON STRUCTURAL REMODELING IN AF
Thoracic veins and the left atrium are critical AF triggers (AF initiation) and substrates (AF maintenance).36,37 Animals with CKD were associated with higher atrial arrhythmogenicity due to structural remodeling with enhanced atrial fibrosis, which can be produced by uremic toxins through their potential for pro-fibrotic and hypertrophic expression with activation of mitogen-activated protein kinase, transforming growth factor-β1, connective tissue growth factor, atrial natriuretic peptide, β-myosin heavy chain, and α-skeletal muscle actin (Table 2).
The dilated LA and thoracic veins are common findings of CKD patients, which could promote the occurrence of AF via structural remodeling with fibrotic molecular expression and mechano-electrical feedback.9,10,38,39 Furthermore, uremic toxins such as IS also activate tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β genes, which can cause cardiac fibrosis and hypertrophy and lead to the genesis of heart failure and atrial arrhythmias.19 Moreover, IS has been shown to modulate the PV vascular tone via increasing calcium leakage from increased ryanodine receptor open probability.22 Since mechanical stretch plays a critical role in the arrhythmogenesis of PV, IS may contribute to the occurrence of atrial arrhythmia in CKD.
UREMIC TOXINS ON ELECTRICAL REMODELING IN AF
Larger calcium leak in IS-treated PV cardiomyocytes was investigated to have more delayed after depolarizations (DADs) with increased PV beating rate in the rabbit study as shown in Figure 1.22 IS has been shown to increase calcium leaks in PV cardiomyocytes, which can facilitate the genesis of trigger activity due to diastolic calcium overload and DADs. In addition, IS-induced calcium leak may induce the increased PV diastolic tension shown in this cardiomyocyte study. However, IS did not change the calcium transient and sarcoplasmic reticulum calcium content of PV cardiomyocytes. Hence, abnormal calcium regulation plays an important role in electrical remodeling of IS-treated PV cardiomyocyte. Moreover, shorter AP duration of IS-treated LA cardiomyocytes had higher occurrence and longer duration of pacing-induced AF in the infusion of isoproterenol, which might indicate micro-reentry circuit with decreasing effective refractory period in the same study.
Figure 1.
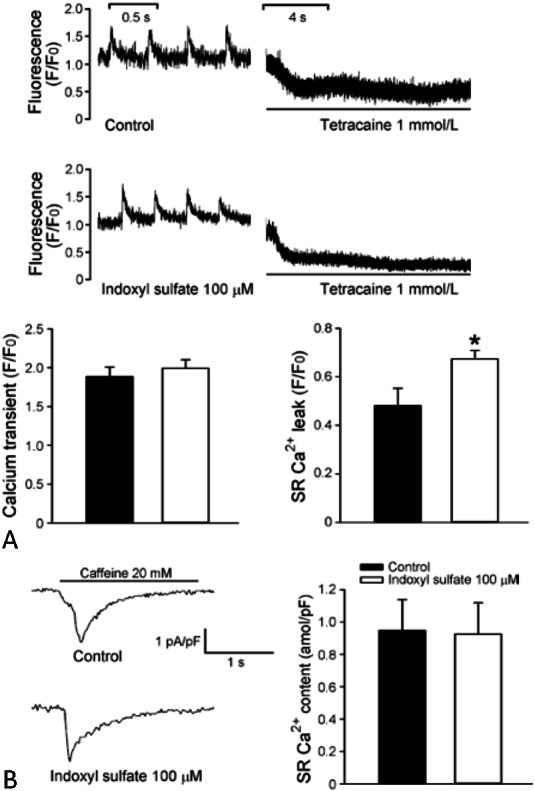
Intracellular calcium (Ca2+i) transient and sarcoplasmic reticulum (SR) stores in indoxyl sulfate (IS)-treated pulmonary vein (PV) cardiomyocytes and control. (A) SR Ca2+ leakage is measured during the shift down-ward in resting Ca2+i in the example of the tracings. The average data of calcium transient (left panel) and SR Ca2+ leakage (right panel). (B) The SR Ca2+ contest measured from sodium calcium exchanger currents in the example of tracings and average data. Reproduced with permission from Chen et al. Journal of Cardiovascular Electrophysiology 2015:26;203-210. © Wiley Materials
PC and PCS can disrupt nitric oxide signaling and induce shedding of endothelial microparticles with possibly higher cellular calcium concentration by blockage of the Rho-kinase pathway.15,40,41 Impairment of the endothelial system due to uremic toxins can modulate the arrhythmogenic activity of pulmonary veins through a vasoconstrictor effect and multiple electromechanical mechanisms.42,43 Moreover, activation of the renin-angiotensin-aldosterone system plays a critical role in the pathological processes of the AF trigger and substrate.44,45 Uremic toxins significantly activate angiotensin II type 1 receptor expression, which might shorten the action potential duration, modulate ionic currents, and disrupt calcium handling to cause AF.18,46,47 Further, uremic toxins increase intracellular calcium levels and activate protein kinase Cα that can damage adherent junctions of cardiomyocytes to increase irregular beats and electrical activity.20,48 Accordingly, the electrical effects of uremic toxins can enhance AF trigger arrhythmogenesis due to calcium dysregulation and facilitate AF maintenance with conduction block and shortening of refractoriness.18,26,41
CONCLUSIONS
Since there is a high incidence of CVDs and AF associated with CKD, researchers are dedicated to finding relationships among traditional and novel cardiovascular risk factors. Subsequently, uremic toxins are being emphasized as new targets for cardiovascular therapy, and even in dialysis patients. The AF trigger and AF substrate are modulated by structural and electrical remodeling to induce atrial arrhythmias with interactions among NADPH-oxidation, ROS, and the renin-angiotensin-aldosterone system (Figure 2). Correspondingly, uremic toxins produce pro-fibrotic, pro-hypertrophic, and pro-inflammatory effects in both renal and cardiovascular cells in response to oxidative stress and calcium handling. The mechano-electrical feedback directly increases trigger activity and maintains the reentry of the substrate as a result of atrial arrhythmogenicity. Therefore, the appropriate management of uremic toxins in CKD should attenuate the potential capacity for atrial arrhythmias and achieve better prognoses of CVDs.
Figure 2.
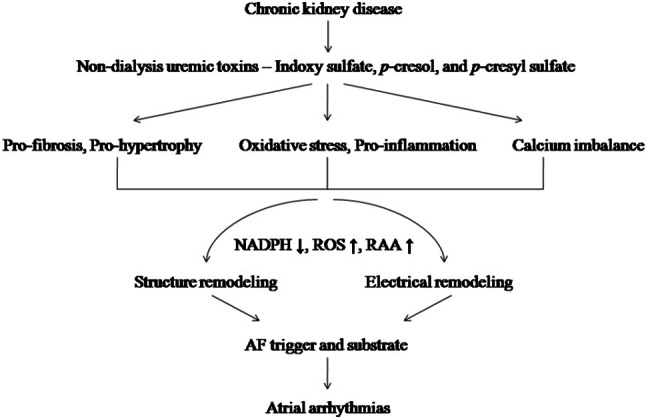
Proposed mechanism of uremic toxins on the genesis of atrial fibrillation (AF) in chronic kidney disease (CKD). Uremic toxins in CKD induce pro-fibrotic and pro-hypertrophic effects, cause interactions of oxidative stress and pro-inflammatory effects, and produce a calcium imbalance which enhance structural and electrical remodelings and produce atrial arrhythmias through increased reactive oxygen species (ROS) and renin-angiotensin-aldosterone (RAA) system with reduced nicotinamide adenine dinucleotide phosphate (NADPH).
CONFLICT OF INTEREST STATEMENT
None declared.
FINANCIAL SUPPORT
This work was supported by the Ministry of Science and Technology, Taiwan, (MOST 103-2314-B-038-041-MY2, MOST 103-2314-B-281-005-MY2, MOST 103-2314-B-281-006, MOST 103-2314-B-038-055), the Cathay General Hospital, Taiwan, (CGH-MR-10221, CGH-MR-10123), and the Taiwan Heart Rhythm Society (THRS-1501).
REFERENCES
- 1.Benjamin EJ, Wolf PA, D'Agostino RB, et al. Impact of atrial fibrillation on the risk of death:the Framingham Heart Study. Circulation. 1998;98:946–952. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 2.Chen SA, Hsieh MH, Tai CT, et al. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation. 1999;100:1879–1886. doi: 10.1161/01.cir.100.18.1879. [DOI] [PubMed] [Google Scholar]
- 3.Alonso A, Lopez FL, Matsushita K, et al. Chronic kidney disease is associated with the incidence of atrial fibrillation:the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2011;123:2946–2953. doi: 10.1161/CIRCULATIONAHA.111.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amann K, Buzello M, Simonaviciene A, et al. Capillary/myocyte mismatch in the heart in renal failure--a role for erythropoietin? Nephrol Dial Transplant. 2000;15:964–969. doi: 10.1093/ndt/15.7.964. [DOI] [PubMed] [Google Scholar]
- 5.Gross ML, Ritz E. Hypertrophy and fibrosis in the cardiomyopathy of uremia--beyond coronary heart disease. Semin Dial. 2008;21:308–318. doi: 10.1111/j.1525-139X.2008.00454.x. [DOI] [PubMed] [Google Scholar]
- 6.Vlahakos DV, Hahalis G, Vassilakos P, et al. Relationship between left ventricular hypertrophy and plasma renin activity in chronic hemodialysis patients. J Am Soc Nephrol. 1997;8:1764–1770. doi: 10.1681/ASN.V8111764. [DOI] [PubMed] [Google Scholar]
- 7.Zoccali C, Mallamaci F, Tripepi G, et al. Norepinephrine and concentric hypertrophy in patients with end-stage renal disease. Hypertension. 2002;40:41–46. doi: 10.1161/01.hyp.0000022063.50739.60. [DOI] [PubMed] [Google Scholar]
- 8.Tsai CT, Lai LP, Lin JL, et al. Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation. 2004;109:1640–1646. doi: 10.1161/01.CIR.0000124487.36586.26. [DOI] [PubMed] [Google Scholar]
- 9.Chang SL, Chen YC, Yeh YH, et al. Heart failure enhances arrhythmogenesis in pulmonary veins. Clin Exp Pharmacol Physiol. 2011;38:666–674. doi: 10.1111/j.1440-1681.2011.05553.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen SC, Chang JM, Liu WC, et al. Echocardiographic parameters are independently associated with increased cardiovascular events in patients with chronic kidney disease. Nephrol Dial Transplant. 2012;27:1064–1070. doi: 10.1093/ndt/gfr407. [DOI] [PubMed] [Google Scholar]
- 11.Meijers BK, Bammens B, De Moor B, et al. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008;73:1174–1180. doi: 10.1038/ki.2008.31. [DOI] [PubMed] [Google Scholar]
- 12.Foley RN, Parfrey PS, Sarnak MJ. Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol. 1998;9:S16–S23. [PubMed] [Google Scholar]
- 13.Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:1551–1558. doi: 10.2215/CJN.03980609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meijers BK, De Loor H, Bammens B, et al. p-Cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:1932–1938. doi: 10.2215/CJN.02940509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meijers BK, Claes K, Bammens B, et al. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin J Am Soc Nephrol. 2010;5:1182–1189. doi: 10.2215/CJN.07971109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu IW, Hsu KH, Hsu HJ, et al. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients--a prospective cohort study. Nephrol Dial Transplant. 2012;27:1169–1175. doi: 10.1093/ndt/gfr453. [DOI] [PubMed] [Google Scholar]
- 17.Raff AC, Meyer TW, Hostetter TH. New insights into uremic toxicity. Curr Opin Nephrol Hypertens. 2008;17:560–565. doi: 10.1097/MNH.0b013e32830f45b6. [DOI] [PubMed] [Google Scholar]
- 18.Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. 2012;7:e34026. doi: 10.1371/journal.pone.0034026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lekawanvijit S, Adrahtas A, Kelly DJ, et al. Does indoxyl sulfate,a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J. 2010;31:1771–1779. doi: 10.1093/eurheartj/ehp574. [DOI] [PubMed] [Google Scholar]
- 20.Peng YS, Lin YT, Wang SD, et al. p-Cresol induces disruption of cardiomyocyte adherens junctions. Toxicology. 2013;306:176–184. doi: 10.1016/j.tox.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 21.Lekawanvijit S, Kompa AR, Manabe M, et al. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin,indoxyl sulfate. PLoS One. 2012;7:e41281. doi: 10.1371/journal.pone.0041281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen WT, Chen YC, Hsieh MH, et al. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J Cardiovasc Electrophysiol. 2015;26:203–210. doi: 10.1111/jce.12554. [DOI] [PubMed] [Google Scholar]
- 23.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 24.Lin YK, Lin FZ, Chen YC, et al. Oxidative stress on pulmonary vein and left atrium arrhythmogenesis. Circ J. 2010;74:1547–1556. doi: 10.1253/circj.cj-09-0999. [DOI] [PubMed] [Google Scholar]
- 25.Van Wagoner DR. Redox modulation of cardiac electrical activity. J Cardiovasc Electrophysiol. 2001;12:183–184. doi: 10.1046/j.1540-8167.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- 26.Fukunaga N, Takahashi N, Hagiwara S, et al. Establishment of a model of atrial fibrillation associated with chronic kidney disease in rats and the role of oxidative stress. Heart Rhythm. 2012;9:2023–2031. doi: 10.1016/j.hrthm.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 27.Han W, Fu S, Wei N, et al. Nitric oxide overproduction derived from inducible nitric oxide synthase increases cardiomyocyte apoptosis in human atrial fibrillation. Int J Cardiol. 2008;130:165–173. doi: 10.1016/j.ijcard.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 28.Lin YK, Chen YC, Chen JH, et al. Adipocytes modulate the electrophysiology of atrial myocytes: implications in obesity-induced atrial fibrillation. Basic Res Cardiol. 2012;107:293. doi: 10.1007/s00395-012-0293-1. [DOI] [PubMed] [Google Scholar]
- 29.Suenari K, Chen YC, Kao YH, et al. Eicosapentaenoic acid reduces the pulmonary vein arrhythmias through nitric oxide. Life Sci. 2011;89:129–136. doi: 10.1016/j.lfs.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Shiroshita-Takeshita A, Brundel BJ, Lavoie J, Nattel S. Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc Res. 2006;69:865–875. doi: 10.1016/j.cardiores.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 31.Anderson EJ, Kypson AP, Rodriguez E, et al. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montaigne D, Marechal X, Lefebvre P, et al. Mitochondrial dysfunction as an arrhythmogenic substrate: a translational proof-of-concept study in patients with metabolic syndrome developping post-operative atrial fibrillation. J Am Coll Cardiol. 2013;62:1466–1473. doi: 10.1016/j.jacc.2013.03.061. [DOI] [PubMed] [Google Scholar]
- 33.Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;29:551–557. doi: 10.1159/000191468. [DOI] [PubMed] [Google Scholar]
- 34.Tumur Z, Shimizu H, Enomoto A, et al. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am J Nephrol. 2010;31:435–441. doi: 10.1159/000299798. [DOI] [PubMed] [Google Scholar]
- 35.Chen YJ, Chen YC, Wongcharoen W, et al. Effect of K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br J Pharmacol. 2008;153:915–925. doi: 10.1038/sj.bjp.0707564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haissaguerre M, Jais P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 37.Hsu LF, Jais P, Sanders P, et al. Catheter ablation for atrial fibrillation in congestive heart failure. N Engl J Med. 2004;351:2373–2383. doi: 10.1056/NEJMoa041018. [DOI] [PubMed] [Google Scholar]
- 38.Lu YY, Chen YC, Kao YH, et al. Extracellular matrix of collagen modulates arrhythmogenic activity of pulmonary veins through p38 MAPK activation. J Mol Cell Cardiol. 2013;59:159–166. doi: 10.1016/j.yjmcc.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Ko WC, Hong CY, Hou SM, et al. Elevated expression of connective tissue growth factor in human atrial fibrillation and angiotensin II-treated cardiomyocytes. Circ J. 2011;75:1592–1600. doi: 10.1253/circj.cj-10-0892. [DOI] [PubMed] [Google Scholar]
- 40.de Loor H, Bammens B, Evenepoel P, et al. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin Chem. 2005;51:1535–1538. doi: 10.1373/clinchem.2005.050781. [DOI] [PubMed] [Google Scholar]
- 41.Meijers BK, Van Kerckhoven S, Verbeke K, et al. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54:891–901. doi: 10.1053/j.ajkd.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 42.Udyavar AR, Chen YC, Chen YJ, et al. Endothelin-1 modulates the arrhythmogenic activity of pulmonary veins. J Cardiovasc Electrophysiol. 2008;19:285–292. doi: 10.1111/j.1540-8167.2007.01033.x. [DOI] [PubMed] [Google Scholar]
- 43.Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76:8–18. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Nakashima H, Kumagai K, Urata H, et al. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation. 2000;101:2612–2617. doi: 10.1161/01.cir.101.22.2612. [DOI] [PubMed] [Google Scholar]
- 45.Tsai CF, Chen YC, Lin YK, et al. Electromechanical effects of the direct renin inhibitor (aliskiren) on the pulmonary vein and atrium. Basic Res Cardiol. 2011;106:979–993. doi: 10.1007/s00395-011-0206-8. [DOI] [PubMed] [Google Scholar]
- 46.Wu J, Ding WG, Zhao J, et al. Irbesartan-mediated AT1 receptor blockade attenuates hyposmotic-induced enhancement of IKs current and prevents shortening of action potential duration in atrial myocytes. J Renin Angiotensin Aldosterone Syst. 2014;15:341–347. doi: 10.1177/1470320312474855. [DOI] [PubMed] [Google Scholar]
- 47.Lu YY, Chen YC, Kao YH, et al. Extracellular matrix of collagen modulates intracellular calcium handling and electrophysiological characteristics of HL-1 cardiomyocytes with activation of angiotensin II type 1 receptor. J Card Fail. 2011;17:82–90. doi: 10.1016/j.cardfail.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 48.Peng YS, Ding HC, Lin YT, et al. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology. 2012;302:11–17. doi: 10.1016/j.tox.2012.07.004. [DOI] [PubMed] [Google Scholar]