

Abstract
Background
Genome-wide association studies have identified several common susceptibility alleles for epithelial ovarian cancer (EOC). To further understand EOC susceptibility, we examined previously ungenotyped candidate variants, including uncommon variants and those residing within known susceptibility loci.
Results
At nine of eleven previously published EOC susceptibility regions (2q31, 3q25, 5p15, 8q21, 8q24, 10p12, 17q12, 17q21.31, and 19p13), novel variants were identified that were more strongly associated with risk than previously reported variants. Beyond known susceptibility regions, no variants were found to be associated with EOC risk at genome-wide statistical significance (p <5×10−8), nor were any significant after Bonferroni correction for 17,000 variants (p< 3×10-6).
Methods
A customized genotyping array was used to assess over 17,000 variants in coding, non-coding, regulatory, and known susceptibility regions in 4,973 EOC cases and 5,640 controls from 13 independent studies. Susceptibility for EOC overall and for select histotypes was evaluated using logistic regression adjusted for age, study site, and population substructure.
Conclusion
Given the novel variants identified within the 2q31, 3q25, 5p15, 8q21, 8q24, 10p12, 17q12, 17q21.31, and 19p13 regions, larger follow-up genotyping studies, using imputation where necessary, are needed for fine-mapping and confirmation of low frequency variants that fall below statistical significance.
Keywords: ovarian cancer, high-grade serous carcinoma, genetic association, susceptibility loci, NF-κB
INTRODUCTION
Epithelial ovarian cancer (EOC) is the second most common gynecologic cancer in the US, but it leads in deaths owing to its tendency to be diagnosed in the late stages of disease [1]. EOC is composed of five major histologic types [2]: high-grade serous carcinoma (HGSC), accounting for most cases (∼70%); and the rarer clear cell, endometrioid, mucinous, and low-grade serous carcinomas (LGSC). Known rare mutations in DNA repair and mismatch repair genes are thought to account for 10%-15% of all EOCs [3-9]. Common alleles identified by genome-wide association studies (GWAS) are thought to account for an additional 3%-4% of EOC risk [10-17]. Still, much of about the heritability of EOC remains unaccounted for. Here, we sought to identify additional EOC susceptibility variants through direct genotyping and analysis of EOC cases and controls from 13 independent studies. We targeted variants based on innovative pilot studies, hypothesizing that previously ungenotyped variants may be responsible for a proportion of the unexplained EOC susceptibility.
RESULTS
Known EOC susceptibility regions
One goal of this project was to compare the relative strength of the associations between known and novel variants within the first eleven published EOC risk loci [10-12, 14, 18] (Supplemental Table 1). The variants most strongly associated with EOC risk in this study (all histology or HGSC) are given in Table 1 and plotted regionally in Figure 1, Figure 2, and Supplemental Figure 1. Compared to published variants, novel variants were more strongly associated with susceptibility of all histologies of EOC at nine loci (2q31, 3q25, 5p15, 8q21, 8q24, 10p12, 17q12, 17q21.31, and 19p13) (Table 1); all but three (8q21, 17q21.31, and 19p13) are in moderate LD (r2> 0.4) with known variants (Supplemental Figure 1). At the 3q25 locus variant rs62273902 (pall-histology= 2 × 10−8) was the most strongly associated variant (Figure 1), and at the 17q21.31 locus variant rs2532240 (pall-histology= 3 × 10−7) was the most strongly associated variant (Figure 2). In the HGSC only analysis, novel variants were more strongly associated with susceptibility at seven loci (2q31, 3q25, 8q24, 10p12, 17q12, 17q21.31, and 19p13; Table 1); all but two (2q31, 17q21.31) are in moderate LD (r2> 0.4) with known variants (Supplemental Figure 1). With two exceptions (noted below), novel variants were common (minor allele frequency (MAF)>0.05), in the intron of genes or intergenic, in moderate-to-strong LD with known variants, and conferred modest effects on susceptibility. One exception was the association of rare intergenic variant rs74955251 at 8q21 (MAFoverall= 0.00028, OR= 3.9 × 10−6, 95% confidence interval [CI]: 3.4 × 10−118-4.6 × 10106, pall-histology= 4 × 10−3). Given its rarity, rs74955251 requires assessment in a much larger sample of cases and controls. A second exception was the association of common (MAFoverall= 0.50) missense variant rs2363956 in the gene ANKLE1 at 19p13 (OR= 0.91, 95% CI: 0.87-0.97, pall-histology = 2 × 10−3, protein change Leu184Trp).
Table 1. Most significant associations within eleven known EOC susceptibility regions.
| All histologies (4,973 cases, 5,640 controls) | High grade serous (3,573 cases, 5,640 controls) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | Variant | Position (hg19) | Nearest Gene | MAF overall | OR (95% CI) | P value | Variant | Position (hg19) | Nearest Gene | MAF overall | OR (95% CI) | P value |
| 2q31 | rs711830 | 177037311 | 3′ UTR of HOXD3 | 0.33 | 1.12 (1.05-1.18) | 2.05 × 10−4 | rs1374325 | 177043971 | Intron of HOXD1-AS | 0.32 | 0.89 (0.83-0.96) | 1.37 × 10−3 |
| 3q25 | rs62273902 | 156544488 | 5′ UTR of LEKR1 | 0.06 | 1.40 (1.25-1.57) | 1.52 × 10−8 | rs62275810 | 156647851 | Intron of LEKR1 | 0.05 | 1.46 (1.27-1.68) | 2.58 × 10−7 |
| 5p15 | rs4975538 | 1280830 | Intron of TERT | 0.35 | 1.13 (1.06-1.20) | 8.24 × 10−5 | rs10069690 | 1279790 | Intron of TERT | 0.26 | 1.14 (1.06-1.22) | 6.28 × 10−4 |
| 8q21 | rs74955251 | 82230643 | 34kb 3′ of FABP5 | 2.8 × 10−4 | 3.94 × 10−6 (3.4 × 10−118-4.6 × 10106) | 5.71 × 10−3 | rs11782652 | 82653644 | Intron of CHMP4C | 0.07 | 1.23 (1.09-1.39) | 6.49 × 10−4 |
| 8q24 | rs1400482 | 129541931 | Intron of LINC00824 | 0.12 | 0.82 (0.75-0.89) | 2.57 × 10−6 | rs1400482 | 129541931 | Intron of LINC00824 | 0.13 | 0.80 (0.73-0.89) | 1.71 × 10−5 |
| 9p22 | rs3814113 | 16915021 | 44kb 5′ of BNC2 | 0.32 | 0.81 (0.76-0.86) | 7.96 × 10−13 | rs3814113 | 16915021 | 44kb 5′ of BNC2 | 0.32 | 0.75 (0.70-0.81) | 3.20 × 10−5 |
| 10p12 | rs4364959 | 22285371 | Intron of DNAJC1 | 0.30 | 1.15 (1.08-1.22) | 9.66 × 10−6 | rs9971210 | 21879084 | Intron of MLLT10 | 0.49 | 1.11 (1.04-1.19) | 1.21 × 10−3 |
| 17q12 | rs7405776 | 36093022 | Intron of HNF1B | 0.38 | 1.18 (1.11-1.24) | 1.12 × 10−8 | rs7405776 | 36093022 | Inton of HNF1B | 0.37 | 1.25 (1.17-1.34) | 2.23 × 10−11 |
| 17q21.31 | rs2532240 | 44265839 | Intron of KANSL1 | 0.41 | 1.16 (1.10-1.23) | 2.99 × 10−7 | rs3785880 | 43993376 | Intron of MAPT | 0.45 | 0.87 (0.82-0.93) | 4.95 × 10−5 |
| 17q21.32 | rs9303542 | 46411500 | Intron of SKAP1 | 0.29 | 1.15 (1.08-1.23) | 8.39 × 10−6 | rs9303542 | 46411500 | Intron of SKAP1 | 0.27 | 1.16 (1.08-1.25) | 4.88 × 10−5 |
| 19p13 | rs2363956 | 17394124 | Missense mutation (L184W) in ANKLE1 | 0.50 | 0.91 (0.87-0.97) | 1.57 × 10−3 | rs56069439 | 17393925 | Intron of ANKLE1 | 0.30 | 1.15 (1.07-1.23) | 1.82 × 10−4 |
Bolded variants were previously reported as the most strongly associated variants in these susceptibility regions (Supplemental Table 1). MAF, minor allele frequency; OR, odds ratio; CI, confidence interval; UTR, untranslated region. Associations adjust for age, site, and three European principal components.
Figure 1. Novel variant rs62273902 in the 5′-untranslated region of LEKR1 has the strongest association signal at 3q25.
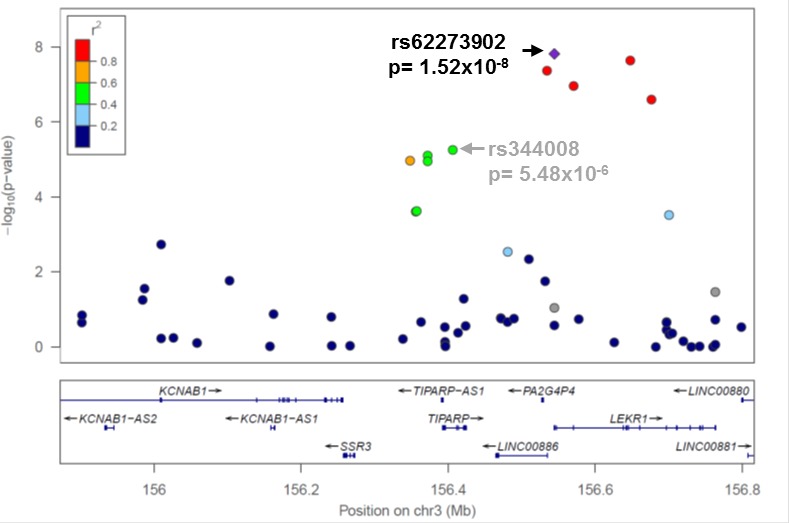
Regional association plot for variants genotyped at 3q25 in all EOC histologies cases (N = 4,973) and controls (N = 5,640). Linkage disequilibrium between rs62273902 and each variant is estimated using data from 5,640 controls and indicated by the color scheme. The previously reported risk variant rs2665390 in this region {Goode, 2010 #23} was not genotyped; rs344008 (p = 5×10−6) is indicated in its place to allow comparison of the novel (rs62273902) and known (rs2665390) most associated variants (r2 = 1 for rs344008 and rs2665390 in 1000 Genomes Project phase 1 European data).
Figure 2. Novel variant rs2532240 has the strongest association signal at 17q21.31.
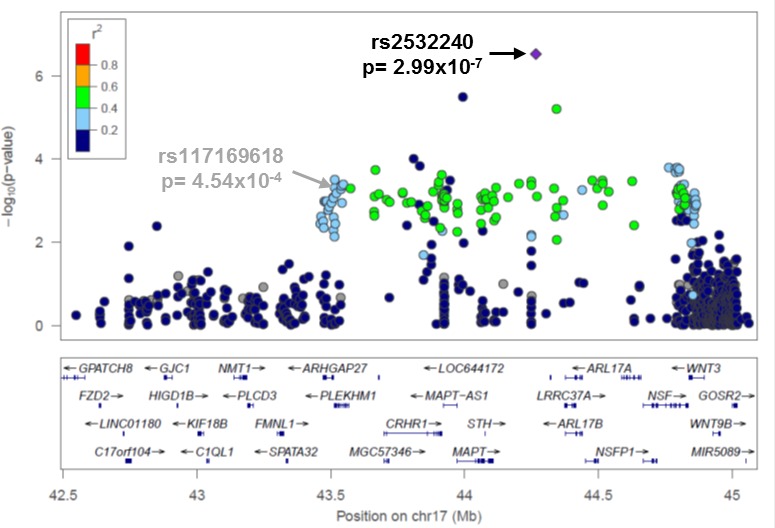
Regional association plot for variants genotyped at 17q21.31 in all EOC histologies cases (N = 4,973) and controls (N = 5,640). The most associated variant was rs2532240 (p = 3 × 10−7). Linkage disequilibrium between rs2532240 and each variant is estimated using data from 5,640 controls and indicated by the color scheme. The previously reported risk variant rs12942666 in this region {Permuth-Wey, 2013 #28} was not genotyped, but rs117169618 (p = 5 × 10−4) is indicated in its place to allow comparison of the novel (rs2532240) and known (rs12942666) most variants (r2 = 0.8 for rs117169618 and rs12942666 in 1000 Genomes Project phase 1 European data).
Beyond known EOC susceptibility regions
We targeted 5,320 variants which showed suggestive association with susceptibility in a pilot-scale whole genome sequence analysis that compared germline sequence of EOC patients (N= 19) to 1000 GP participants (N= 174). No novel variants reached genome-wide significance for association with EOC risk overall or HGSC (p≤ 5×10−8), nor were significant after Bonferroni correction (p≤ 9×10−6). Nonetheless, the risk estimates generally were in the expected direction based on pilot data, and several variants merit investigation in larger case-control collections (Table 2). For example, among variants targeted because they were present only in EOC germline sequence data (WGS EOC+ in Table 2), the most strongly associated risk variant was rs138643956 (OR= 3.68; pHGSC= 2 × 10−4). For variants selected because they were absent from whole genome sequenced EOC cases (WGS EOC- in Table 2), the most associated variant was rs9380516 (OR= 0.83; pHGSC= 6 × 10−5); in the current study this variant showed a case MAF of 0.15, suggesting that this was a missed variant in the pilot sequencing study. For variants targeted which were present in whole genome sequenced EOC cases and in 1000 GP data, but differed in MAF (WGS EOC↑ and WGS EOC↓ in Table 2), the current analyses were generally consistent, including rs117841616 on chromosome 20 (pall histology= 2 × 10−4) and rs240783 (pHGSC= 8 × 10−4) on chromosome 6. In general, very few variants targeted based on suggestive association in pilot sequence study had appreciable MAF differences (case vs. control) in the current genotyping study. As expected due to small sample size, we observed that case MAF estimates in our pilot whole genome sequencing study were both inflated and deflated compared to the current study.
Table 2. Most significant EOC risk associations by selection criteria outside of eleven known susceptibility regions.
| Pilot WGS Study | Case-control study data (13 sites) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Selection Criteria | Variant | Chr. | Position (hg19) | Location | EOC MAF |
1000 GP MAF |
Histology | Case MAF |
Control MAF |
N case, N control |
OR (95%CI) | P value |
| WGS EOC+ | rs138643956 | 10 | 79367857 | Intron of KCNMA1 | 0.053 | 0.000 | HGSC | 0.004 | 0.001 | 3035, 5637 | 3.68 (1.79 - 7.55) | 1.85 × 10−4 |
| WGS EOC↑ | rs117841616 | 20 | 57855211 | 20kb 5′ of EDN3 | 0.079 | 0.006 | All | 0.008 | 0.005 | 4973, 5634 | 1.93 (1.36 - 2.74) | 2.06 × 10−4 |
| WGS EOC↓ | rs240783 | 6 | 100968737 | Intron of ASCC3 | 0.184 | 0.494 | HGSC | 0.400 | 0.428 | 2956, 5518 | 0.89 (0.84 - 0.95) | 7.90 × 10−4 |
| WGS EOC− | rs9380516 | 6 | 35502202 | 10kb 5′ of RP3-340B19.3 | 0.000 | 0.155 | HGSC | 0.147 | 0.171 | 3027, 5633 | 0.83 (0.76 - 0.91) | 6.44 × 10−5 |
| NF-κB | rs10143322 | 14 | 91556577 | 24kb 5′ of C14orf159 | n.a. | n.a. | All | 0.220 | 0.246 | 4562, 5634 | 0.87 (0.82 - 0.93) | 2.99 × 10−5 |
| NF-κB | rs6092485 | 20 | 56045014 | 26kb 3′ of CTCFL | n.a. | n.a. | HGSC | 0.335 | 0.311 | 3019, 5625 | 1.13 (1.05 - 1.21) | 6.77 × 10−4 |
| Endometrioid GWAS | rs9264042 | 6 | 31196801 | 25kb 3′ of HCG27 | n.a. | n.a. | All | 0.125 | 0.107 | 4440, 5505 | 1.17 (1.07 - 1.28) | 4.71 × 10−4 |
| Endometrioid GWAS | rs2638653 | 8 | 18666210 | Intron of PSD3 | n.a. | n.a. | EC | 0.409 | 0.362 | 832, 5619 | 1.23 (1.11 - 1.37) | 1.28 × 10−4 |
WGS, whole-genome sequencing; EOC, epithelial ovarian cancer; 1000 GP, 1000 Genomes Project; GWAS, genome wide association study; Chr, chromosome; MAF, minor allele frequency; OR, odds ratio; CI, confidence interval; HGSC, high grade serous carcinoma; n.a., not applicable. WGS EOC+ variant selection criteria: MAF> 0% in serous EOC cases, monomorphic in 1000 GP European individuals; WGS EOC↑ variant selection criteria: polymorphic in cases and 1000 GP, MAF greater in WGS patients; WGS EOC↓ variant selection criteria: polymorphic in cases and 1000 GP, MAF greater in 1000 GP; WGS 1000 EOC− variant selection criteria: monomorphic in cases, MAF> 0% in 1000 GP.
Finally, among NF-κB-related variants and those hypothesized to associate with endometrioid EOC risk, the most suggestive results for variants which disrupt NF-κB binding [19, 20] were intergenic variants rs10143322 on chromosome 14 (pall-histology= 3 × 10−5) and rs6092485 on chromosome 20 (pHGSC= 7×10−4) (Table 2). If Bonferroni correction for the number NF-κB binding site variants tested is applied, the threshold for statistical significance is p<4 × 10−5 (p= 0.05/1,302), and this single variant, rs10143322, is declared significant; using experiment-wide and certainly genome-wide multiple testing corrections, it is not significant. Among variants previously identified in a pilot GWAS of endometrioid EOC, the most significant variants were intronic variant rs2638653 (pendometrioid= 1 × 10−4) on chromosome 8 (Table 2), and intergenic rs9264042 on chromosome 6 (pall-histology= 5 × 10−4). These modest associations may also warrant follow-up in larger studies.
DISCUSSION
The objective of this study was to test whether novel variants identified through a combination of approaches were associated with EOC susceptibility. We took an innovative approach to the selection of variants, including the use of whole genome sequencing data to target novel variants correlated with known GWAS risk variants, comparison of sequencing EOC cases to 1000 GP participants beyond these regions, NF-κB functional data, and GWAS analysis of EOC cases with endometrioid histology.
In nine of the eleven susceptibility regions investigated, novel variants were more highly associated with all histology EOC risk than previously reported variants, and, in the HGSC only analysis, novel variants were more strongly associated with susceptibility at seven loci. Further work on thesevariant may provide more biological insight. For example, at the 3q25 locus, the novel variant rs62273902 (all histology) coincides with a genomic sequence that appears functionally active in a range of cell lines and tissues relevant to EOC, including ovary, as assayed by the Roadmap Epigenomics Mapping Consortium (REMC), http://www.epigenomebrowser.org/. rs62273902 resides within a DNase peak, an active transcription start site (TSS), and multiple proteins across diverse tissues bind the sequence spanning this variant. rs62273902 is therefore a good functional candidate variant at this locus. As well, at 17q21.31, the novel variants (rs2532240 in all histologies and rs3785880 in HGSC-only analysis) are separated by 272 kb and not correlated with each other or the previously reported variant rs1294266. rs2532240 coincides with a chromatin region marked as a weak/poised enhancer in several tissues, including ovary (REMC [21]); however, it does not overlap transcription factor binding sites (TFBS) or DNase peaks. rs12942666 does not coincide with promoter or enhancer regions in tissues relevant to EOC in the REMC data, suggesting it is unlikely to be functionally relevant. The 17q21.31 variants are located in a large region of strong LD previously identified as the “17q21.31 inversion” (∼900kb long), which exists either as a direct (H1) or inverted (H2) haplotype in the European population [14, 15, 22]. Further investigation of how these variants might impact EOC risk is needed.
Of critical note, a large EOC meta-GWAS with imputation to revised Phase I 1000 GP data was recently completed with over 23,000 cases and 35,000 controls of the Ovarian Cancer Association Consortium, including many of the participants in the current analysis. We inspected our most associated variants from the 11 known susceptibility regions in in an online look-up of results based on these data (http://apps.ccge.medschl.cam.ac.uk/consortia/ocac/contact/contact.html). In general, the variants reported here were highly ranked in the EOC meta-GWAS data (i.e., in the top 50 most associated variants in the regions we defined). At 8q24, the novel directly genotyped variant presented here (rs1400482) was the most associated variant in the larger imputation-based study. At 3q25, 10p12, 17q21.31, and 17q21.32, the most significant variant in the current study was not among the most significant in the imputation-based study. Nonetheless, novel variants at 3q25 and 17q21.32 remained highly significant (rs62273902 at 3q25 pmeta= 2 × 10−28, and rs9303542 at 17q21.32 pmeta= 3 × 10−12). Although novel variants at 3q25 and 17q21.32 remained highly significant (rs62273902 at 3q25 pmeta= 2 × 10−28, and rs9303542 at 17q21.32 pmeta= 3 × 10−12), at these two regions as well as at 10p12 and 17q21.31, the imputation-based study revealed stronger associations with other variants.
Among variants genotyped based on our pilot study comparing whole genomes of EOC cases and 1000 GP participants, none were significant after multiple testing correction for 5,320 variants. Despite our sample size (4,973 cases and 5,640 controls), power to detect associations with low MAF variants was limited. Variants in NF-κB binding sites were also not associated with EOC risk at genome-wide significance. Noting the debate regarding the use of p<5 × 10−8 as the threshold for statistical significance when evaluating potentially functional variants with presumed higher prior probability [23], Bonferroni correction for the number of NF-κB binding site variants yields one statistically significant variant (rs10143322, p= 3×10−5). rs2638653, a variant selected based on an unpublished GWAS of endometrioid EOC, and found here to be the variant most associated with endometrioid EOC risk (p= 1 × 10−4), coincides with chromatin marked as being an active promoter in ovary tissues (of PSD3), but not an enhancer or DNase site. Interestingly, loss of heterozygosity on chromosome 8p22, where this variant is located, is common in EOC tumors, and reduced expression of genes in this region has been found to negatively impact survival in EOC [24].
In summary, we developed a diverse panel of previously ungenotyped variants to directly test for association with EOC susceptibility in 4,973 EOC cases and 5,640 controls from 13 independent studies. Our innovative approach to variant selection included the first use of whole-genome sequencing data from EOC cases in novel variant discovery. At several EOC susceptibility regions, we report novel risk variants for further association and functional investigation. Beyond known regions, this first pass at using whole genome sequencing pilot analyses, although underpowered, also yielded variants of potential interest (rs138643956 and rs117841616). The key strength of this report is the use of direct genotyping of novel variants, some rare, while its key limitation is an inability to more comprehensively examine rare variation. Larger scale genotyping and/or improved genotype imputation accuracy will facilitate further scrutiny of the variants highlighted here.
MATERIALS AND METHODS
Study participants
Study participants were drawn from 13 independent EOC case-control studies of the Ovarian Cancer Association Consortium and were restricted to women of European ancestry. Characteristics of the contributing studies are given in Supplemental Table 2 and have been described previously [18]. Cases (N=4,973) consisted of women aged 18 and older with a pathologically confirmed primary invasive EOC, fallopian tube cancer, or primary peritoneal cancer; controls (N=5,640) were matched by age and region.
Genotyping array
A total of 17,439 germline DNA variants were genotyped using a customized Affymetrix Axiom Exome array (Affymetrix Corporation, Santa Clara, CA). These variants were drawn from four discovery categories: 1) from eleven known EOC susceptibility regions (N =6,948; Supplemental Table 3) [10-14, 18], identified by in silico fine-mapping and a small germline whole genome sequencing study of EOC cases, 2) variants outside these eleven regions which showed suggestive association in pilot whole genome sequencing of serous EOC cases (compared to 1000 Genomes Project [GP] data)(N = 7,189), 3) variants with a hypothesized role in disrupted binding of NF-κB transcription factors, which are known to have central roles in immune and inflammatory responses and cancer [19, 20, 25, 26](N = 1,302), and 4) the top associated variants from a pilot GWAS of endometrioid EOC (N = 2,000). See the Supplemental Methods for more detail on the selection of these variants.
Quality control
Germline DNA was genotyped at the Affymetrix Research Services Laboratory (Santa Clara, CA) using default quality control (QC) and genotype calling criteria. Variants failed QC if: (1) the call rate was < 95%; (2) p-values of Hardy-Weinberg equilibrium in controls were < 10−7; or (3) there was > 2% discordance in duplicate pairs. Further, monomorphic variants were removed. Of 6,948 variants genotyped within 11 known EOC risk regions, 4,919 (71%) met these QC criteria and were polymorphic. Outside of these regions, of 7,189 variants selected based on whole-genome sequencing data, 5,286 (74%) met QC criteria and were polymorphic. Of 1,302 variants associated with NF-κB binding, 980 (75%) met QC criteria and were polymorphic, and, of 2,000 variants selected from a GWAS of endometrioid EOC, 1,826 (91%) met QC criteria and were polymorphic. Most variants were excluded for being monomorphic. Thus, a total of 13,011 genotyped variants remained for analysis.
Association analysis
All cases were included in the overall EOC risk association analyses (N=4,973). Subset analyses were performed on histologic subsets based on a priori selection; HGSC (N=3,573) and endometrioid EOC (N=835). For each analysis, 5,640 controls were used. Associations were estimated using logistic regression assuming an additive genetic model, adjusting for age, study site, and population substructure by including the first three eigenvalues from principal components analysis [18]. All analyses were conducted in R version 3.0.2 (http://www.R-project.org/).
SUPPLEMENTARY MATERIAL METHODS FIGURE AND TABLES
Acknowledgments
The NHS/NHSII studies thank the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, and WY.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
GRANT SUPPORT
Funding support for the Follow-up of Ovarian Cancer Genetic Association and Interaction Studies (FOCI) was provided through the National Cancer Institute's Cancer Post-GWAS Initiative, Genetic Associations and Mechanisms in Oncology (GAME-ON) (U19-CA148112). In addition, we acknowledge the following: DOV: National Institutes of Health R01-CA112523 and R01-CA87538; HAW: National Institutes of Health (R01-CA58598, N01-CN-55424 and N01-PC-67001); HOP: DOD DAMD17-02-1-0669 and NCI K07-CA080668, R01-CA95023, P50-CA159981; NIH/National Center for Research Resources/General Clinical Research Center grant M01-RR000056; MAY: National Institutes of Health (R01-CA122443, P30-CA15083-41, P50-CA136393); NCO: National Institutes of Health (R01-CA76016) and the Department of Defense (DAMD17-02-1-0666); NEC: National Institutes of Health R01-CA54419 and P50-CA105009 and Department of Defense W81XWH-10-1-02802; NHS: NIH (P01-CA87696 and R01-CA49449): NJO: National Cancer Institute (K07-CA095666, R01-CA83918, K22-CA138563, and P30-CA072720) and the Cancer Institute of New Jersey; NCI CCSG award (P30-CA008748); OVA: This work was supported by Canadian Institutes of Health Research grant (MOP-86727); LEK is supported by a Canadian Institutes of Health Research New Investigator award (MSH-87734); POL: Intramural Research Program of the National Cancer Institute; UCI: R01-CA058860, R01-CA092044, US Public Health Service PSA-042205, and the Lon V Smith Foundation grant LVS-39420; USC: P01-CA17054, P30-CA14089, R01-CA61132, N01-PC67010, R03-CA113148, R03-CA115195, N01-CN025403, and California Cancer Research Program (00-01389V-20170, 2II0200). CMP is supported by National Institutes of Health R01-CA-149429.
REFERENCES
- 1.Center M, Siegel R, Jemal A. Global Cancer Facts & Figures. Atlanta: American Cancer Society; 2011. [Google Scholar]
- 2.Kobel M, Kalloger SE, Lee S, Duggan MA, Kelemen LE, Prentice L, Kalli KR, Fridley BL, Visscher DW, Keeney GL, Vierkant RA, Cunningham JM, Chow C, Ness RB, Moysich K, Edwards R, et al. Biomarker-based ovarian carcinoma typing: a histologic investigation in the ovarian tumor tissue analysis consortium. Cancer Epidemiol Biomarkers Prev. 2013;22:1677–1686. doi: 10.1158/1055-9965.EPI-13-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Schuelke GS, Kimberling WJ, Albano WA, Lynch JF, Biscone KA, Lipkin ML, Deschner EE, Mikol YB, Sandberg AA, et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). II. Biomarker studies. Cancer. 1985;56:939–951. doi: 10.1002/1097-0142(19850815)56:4<939::aid-cncr2820560440>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 5.Lynch HT, Conway T, Lynch J. Hereditary ovarian cancer. Pedigree studies, Part II. Cancer Genet Cytogenet. 1991;53:161–183. doi: 10.1016/0165-4608(91)90094-b. [DOI] [PubMed] [Google Scholar]
- 6.Boyd J, Rubin SC. Hereditary ovarian cancer: molecular genetics and clinical implications. Gynecol Oncol. 1997;64:196–206. doi: 10.1006/gyno.1996.4572. [DOI] [PubMed] [Google Scholar]
- 7.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, King MC, Swisher EM. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narod SA, Madlensky L, Bradley L, Cole D, Tonin P, Rosen B, Risch HA. Hereditary and familial ovarian cancer in southern Ontario. Cancer. 1994;74:2341–2346. doi: 10.1002/1097-0142(19941015)74:8<2341::aid-cncr2820740819>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 9.Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B, Narod SA. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68:700–710. doi: 10.1086/318787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, Anton-Culver H, Chang-Claude J, Cramer DW, DiCioccio R, Dork T, Goode EL, Goodman MT, Schildkraut JM, Sellers T, Baglietto L, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22. 2. Nat Genet. 2009;41:996–1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, Widschwendter M, Vierkant RA, Larson MC, Kjaer SK, Birrer MJ, Berchuck A, Schildkraut J, Tomlinson I, Kiemeney LA, Cook LS, et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874–879. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, Sher T, Gentry-Maharaj A, Wozniak E, Tsai YY, Weidhaas J, Paik D, Van Den Berg DJ, Stram DO, Pearce CL, Wu AH, et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 2010;42:880–884. doi: 10.1038/ng.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA, Shen HC, Smart CE, Hillman KM, Mai PL, Lawrenson K, Stutz MD, Lu Y, Karevan R, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. doi: 10.1038/ng.2566. 384e371-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Permuth-Wey J, Lawrenson K, Shen HC, Velkova A, Tyrer JP, Chen Z, Lin HY, Chen YA, Tsai YY, Qu X, Ramus SJ, Karevan R, Lee J, Lee N, Larson MC, Aben KK, et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21. 31. Nat Commun. 2013;4:1627. doi: 10.1038/ncomms2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, Barrowdale D, Dennis J, Gaudet MM, Dicks E, Kosel M, Healey S, Sinilnikova OM, Bacot F, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013;9:e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen K, Ma H, Li L, Zang R, Wang C, Song F, Shi T, Yu D, Yang M, Xue W, Dai J, Li S, Zheng H, Wu C, Zhang Y, Wu X, et al. Genome-wide association study identifies new susceptibility loci for epithelial ovarian cancer in Han Chinese women. Nat Commun. 2014;5:4682. doi: 10.1038/ncomms5682. [DOI] [PubMed] [Google Scholar]
- 17.Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, Lawrenson K, McGuffog L, Healey S, Lee JM, Spindler TJ, Lin YG, Pejovic T, Bean Y, Li Q, Coetzee S, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015 doi: 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, Buckley M, Fridley BL, Tyrer JP, Shen H, Weber R, Karevan R, Larson MC, Song H, Tessier DC, Bacot F, et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362–370. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, et al. Variation in transcription factor binding among humans. Science. 2010;328:232–235. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karczewski KJ, Tatonetti NP, Landt SG, Yang X, Slifer T, Altman RB, Snyder M. Cooperative transcription factor associations discovered using regulatory variation. Proc Natl Acad Sci U S A. 2011;108:13353–13358. doi: 10.1073/pnas.1103105108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, Lema G, Nyambo TB, Omar SA, Bodo JM, Froment A, Donnelly MP, et al. Structural diversity and African origin of the 17q21. 31 inversion polymorphism. Nat Genet. 2012;44:872–880. doi: 10.1038/ng.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broer L, Lill CM, Schuur M, Amin N, Roehr JT, Bertram L, Ioannidis JP, van Duijn CM. Distinguishing true from false positives in genomic studies: p values. Eur J Epidemiol. 2013;28(2):131–138. doi: 10.1007/s10654-012-9755-x. [DOI] [PubMed] [Google Scholar]
- 24.Pils D, Horak P, Gleiss A, Sax C, Fabjani G, Moebus VJ, Zielinski C, Reinthaller A, Zeillinger R, Krainer M. Five genes from chromosomal band 8p22 are significantly down-regulated in ovarian carcinoma: N33 and EFA6R have a potential impact on overall survival. Cancer. 2005;104:2417–2429. doi: 10.1002/cncr.21538. [DOI] [PubMed] [Google Scholar]
- 25.Charbonneau B, Block MS, Bamlet WR, Vierkant RA, Kalli KR, Fogarty Z, Rider DN, Sellers TA, Tworoger SS, Poole E, Risch HA, Salvesen HB, Kiemeney LA, Baglietto L, Giles GG, Severi G, et al. Risk of ovarian cancer and the NF-kappaB pathway: genetic association with IL1A and TNFSF10. Cancer Res. 2014;74:852–861. doi: 10.1158/0008-5472.CAN-13-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Block MS, Charbonneau B, Vierkant RA, Fogarty Z, Bamlet WR, Pharoah PD, Rossing MA, Cramer D, Pearce CL, Schildkraut J, Menon U, Kjaer SK, Levine DA, Gronwald J, Culver HA, Whittemore AS, et al. Variation in NF-kappaB signaling pathways and survival in invasive epithelial ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2014;23:1421–1427. doi: 10.1158/1055-9965.EPI-13-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.