

Abstract
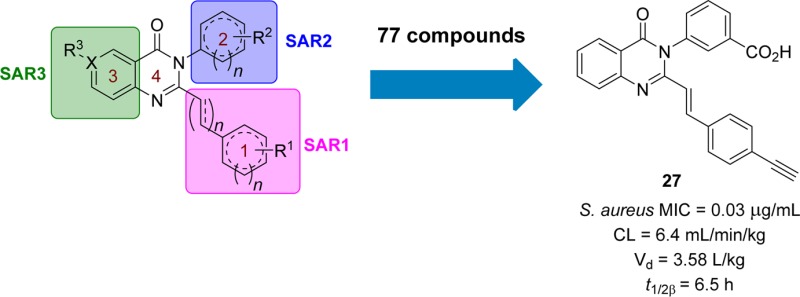
We recently reported on the discovery of a novel antibacterial (2) with a 4(3H)-quinazolinone core. This discovery was made by in silico screening of 1.2 million compounds for binding to a penicillin-binding protein and the subsequent demonstration of antibacterial activity against Staphylococcus aureus. The first structure–activity relationship for this antibacterial scaffold is explored in this report with evaluation of 77 variants of the structural class. Eleven promising compounds were further evaluated for in vitro toxicity, pharmacokinetics, and efficacy in a mouse peritonitis model of infection, which led to the discovery of compound 27. This new quinazolinone has potent activity against methicillin-resistant (MRSA) strains, low clearance, oral bioavailability and shows efficacy in a mouse neutropenic thigh infection model.
Introduction
There exists an urgent need for novel classes of antibiotics for treatment of infection by antibiotic-resistant bacteria.1 The ESKAPE panel of bacteria, comprising Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acetinobacter baumanii, Pseudomonas aeruginosa, and Enterobacteriaceae species (the underlined first letters for the names of the genera makes the acronym), account for the majority of these infections.2,3 Among these organisms, methicillin-resistant S. aureus (MRSA) alone accounts for nearly half of the deaths attributed to antibiotic-resistant infections.4 MRSA is essentially resistant to the entire class of β-lactam antibiotics, which used to be the mainstays of clinical treatment for S. aureus infections.5,6 Newer variants of β-lactams, antibiotics such as ceftaroline and ceftobiprole, exhibit anti-MRSA activities; however, they require intravenous infusions every 8–12 h.7
This broad resistance of MRSA to the β-lactam class is achieved through the acquisition of an additional penicillin-binding protein (PBP), designated PBP2a.8 This enzyme is able to evade inhibition by the β-lactams, as it maintains a closed active-site conformation that is regulated by allostery.9,10 We have shown that ceftaroline has a unique affinity for PBP2a due to its ability to bind noncovalently to the allosteric site at a distance of 60 Å from the active site.10 This binding leads to conformational changes that culminate in the opening of the active site, predisposing it to covalent inhibition by another molecule of ceftaroline.10 Resistance to this antibiotic has already emerged, which is linked to the critical role that this allostery makes to the mechanism of action.11−13 Whereas other antibiotics for treatment of MRSA have been introduced to the clinic,14 only the oxazolidinones, linezolid and tedizolid, are orally bioavailable.15 Resistance to all of these drugs has already emerged, and indeed it will to any other antibiotic.16−19 Hence, discovery of new antibiotics with novel mechanisms of action remains a high priority.20
We recently reported the discovery of the 4(3H)-quinazolinones from in silico screening of the PBP2a active site (1VQQ)21 using a 1.2-million ZINC compound library.22 High-ranking compounds were screened for antibacterial activity against the ESKAPE panel, which led to the discovery of compound 1, which displayed good activity against S. aureus (ATCC 29213) with a minimal-inhibitory concentration (MIC) of 2 μg/mL.22 Quinazolinone 2 was subsequently identified as a lead compound by synthesis of a focus library of analogs, which displayed in vivo efficacy in an MRSA mouse peritonitis model and showed good oral bioavailability.22 This compound works by inhibiting PBP1 and PBP2a in MRSA, consistent with the mechanism of action of β-lactams, which often inhibit more than one PBP.23 In an intriguing observation, we documented that compound 2 also binds to the allosteric site of PBP2a, as does ceftaroline.22 So, notwithstanding the distinct structures of ceftaroline and the lead quinazolinone, they mimic each other in their respective mechanisms of action. The quinazolinone class of antibacterials is of considerable interest, as they are non-β-lactam antibacterials that inhibit PBPs but also are orally bioavailable.
We report herein the first structure–activity relationship (SAR) of this class of antibacterials by systematically varying the quinazolinone structure in rings 1, 2, and 3 (Figure 1). Each derivative was screened for in vitro antibacterial activity by determining MICs against our panel of bacterial strains; however we specifically aimed to improve the activity for S. aureus. Several derivatives that displayed good activity in vitro were selected for testing for in vitro toxicity, pharmacokinetic (PK) properties in mice, and efficacy in mouse models of infection. A subsequent lead compound was identified from these select derivatives, and in vivo efficacy was further demonstrated in the neutropenic thigh infection model.
Figure 1.
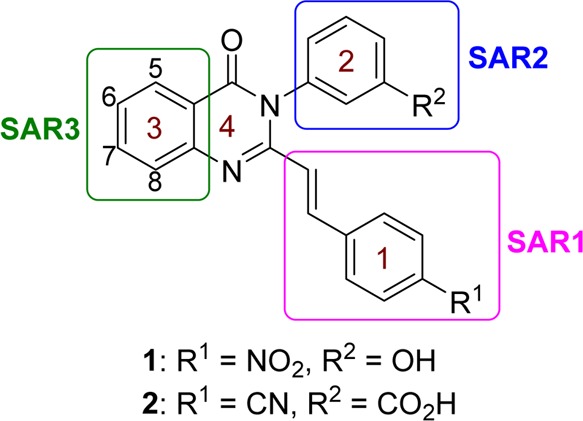
Structures of quinazolinones 1 and 2 are given. The sites for structure diversification under SAR1, SAR2, and SAR3 are highlighted by the colored boxes. Numbering of carbons on ring 3 is shown.
Results and Discussion
Synthesis
Variations of the structure of the hit quinazolinone were made on ring 1 (SAR1), ring 2 (SAR2), and ring 3 (SAR3), as shown in Figure 1. A general three-step synthetic route was used to synthesize 72 quinazolinone derivatives (Scheme 1), and an additional 5 compounds were purchased from ChemDiv. For syntheses, anthranilic acid (I) was cyclized by heating in triethyl orthoacetate, and the solution was cooled to −20 °C to crystallize intermediate II in high purity. Intermediate II was dissolved in glacial acetic acid by heating, a substituted aniline or amine was added, and the mixture was allowed to reflux for 4–6 h to afford intermediate III. To obtain the final compounds, various aldehydes were allowed to react with III in refluxing acetic acid. Further synthetic transformations of the final compounds could then be performed to expand the structural space. To obtain the amine or the functionalized amine (e.g., 17 and 41–43), the nitro analog was first prepared (e.g., 1 or 40), which was subsequently reduced in the presence of tin(II) chloride in ethanol. The free aniline was obtained (41) and acylated with acetic anhydride or mesyl chloride in pyridine to give compounds 42 and 43, respectively. To obtain O-acetylated compounds 37 and 38, the corresponding alcohol intermediate underwent reaction with the respective aldehyde in the presence of acetic anhydride. In order to reduce the double-bond linker of compound 19, hydrogenation on palladium on carbon was performed to yield compound 24.
Scheme 1. General Three-Step Synthetic Route for the Quinazolinones Prepared in This Report.

Structure–Activity Relationship
The SAR of the quinazolinone class of antibacterials was investigated by introducing variations on rings 1, 2, and 3 (Figure 1) and screening the final compounds for activity against the ESKAPE panel. These compounds displayed activity against the ESKAPE Gram-positive organism S. aureus and modest activity (MIC ≥ 8 μg/mL) against E. faecium but did not have activity (MIC ≥ 16 μg/mL) against the Gram-negative organisms. The MICs against S. aureus ATCC 29213, a quality control methicillin-susceptible strain, were used to determine the SAR. Thirty variations on ring 1 were prepared to generate SAR1, with ring 2 as either 3-hydroxyphenyl (series SAR1a) or 3-carboxyphenyl (series SAR1b), as shown in Figure 2. When ring 1 was a phenyl ring, para substitution (1) was found to be superior to meta (3) or ortho (4). In addition, small electron-withdrawing groups seemed to be favored; nitro (1), fluoro (5), chloro (8), nitrile (15), and alkynyl (16) groups all showed potent activities, with compounds 15 and 16 being the best with MICs of 0.03 μg/mL and 0.003 μg/mL, respectively. However, a trifluoromethyl group (9) was not active, while the electron-donating groups methyl (10) and ethyl (11) were well tolerated, but isopropyl (12) was not. Introduction of hydrogen-bonding moieties such as hydroxyl (13), amino (17), or carboxylic acid (18) was not tolerated. Substituting the hydroxyl group with a methoxyl (14) restored activity, indicating that hydrogen-bond acceptors might be allowed. In addition, an unsubstituted benzene ring was tolerated (19). A few heterocycles were explored, including a 1,3-benzodioxole (20) and 4-pyridine (21), both of which maintained activity (MIC 2 μg/mL). However, a furan ring (22) was not tolerated. Replacement of the benzene ring (19) with a cyclohexyl (23) resulted in an 8-fold reduction in the MIC. The necessity for the double-bond linker was also explored. The reduction of the double bond to give 24 resulted in an increase in activity; however shortening the linker length was not tolerated (25 and 26). A second series, SAR1b was prepared with 3-carboxyphenyl as ring 2. An alkyne (27) was found to be the most potent substituent in vitro with an MIC of 0.03 μg/mL versus 2 μg/mL for the corresponding nitrile (2), the lead compound previously disclosed. Substitution at the terminal alkyne was also tolerated (28 and 29). Introduction of halogens within the aromatic ring was tolerated (31 and 32); however a second halogen substituent only afforded a 2-fold improvement in the MIC compared to the parent (30). Finally, a mesyl group (33) was found to be inactive, consistent with the results observed for the corresponding carboxylic acid (18).
Figure 2.

Antibacterial activities of the 4(3H)-quinazolinones derivatives at ring 1. The MICs (μg/mL) were determined for S. aureus ATCC 29213. Red text is for inactive compounds (MIC ≥ 16 μg/mL) and blue for active compounds (MIC ≤ 8 μg/mL).
Three series were prepared for SAR2 while maintaining ring 1 as 4-nitrophenyl, 4-fluorophenyl, or 4-cyanophenyl (SAR2a, SAR2b, and SAR2c, respectively) with a total of 26 variations, as shown in Figure 3. With ring 2 as the phenyl group, meta (1) and ortho (35) substitutions were equally active but para (36) was generally not tolerated. We chose to move forward with meta substituents in order to be consistent with the SAR1 studies. Acetylation (37 and 38) or methylation (39) of the hydroxyl group resulted in loss of activity, suggesting that a hydrogen-bond donor might be favored at this position. This was further confirmed by the restoration of activity by reducing the nitroaromatic group (40) to the corresponding aniline (41). Acylation of the aniline to an N-acetyl (42) and N-mesyl group (43) improved activity, with the mesyl group being superior in vitro. The amide 44 was moderately active and the hydroxyethylamide 45 was 2-fold more active. Interestingly, adding a methylene group linker decreased activity for the carboxylic acid 46 but improved potency for O-acetyl derivative 47 and N-acetylated 53. Removal of the aromatic ring and replacement with the 3-acetoxylpropyl group in derivative 60 resulted in a complete loss of activity. However, replacing the benzene ring with a pyrazole (61) was well tolerated. Overall, hydrophilic groups were favored for R2 and the specific placement of hydrogen-bond donors and acceptors proved important for the potency. The most potent in vitro compound of SAR2 achieved an MIC of 0.004 μg/mL with an N-mesyl group (54), which contained a hydrogen-bond donor one atom away from the ring and two hydrogen-bond acceptors three atoms away. There is some flexibility in the placement of these groups, as shown by compound 53, which also had a very good MIC of 0.015 μg/mL. But the hydrogen-bond donors and acceptors were each shifted over by one atom. In addition, bulkier groups were tolerated, as demonstrated by compounds 51, 57, and 63.
Figure 3.
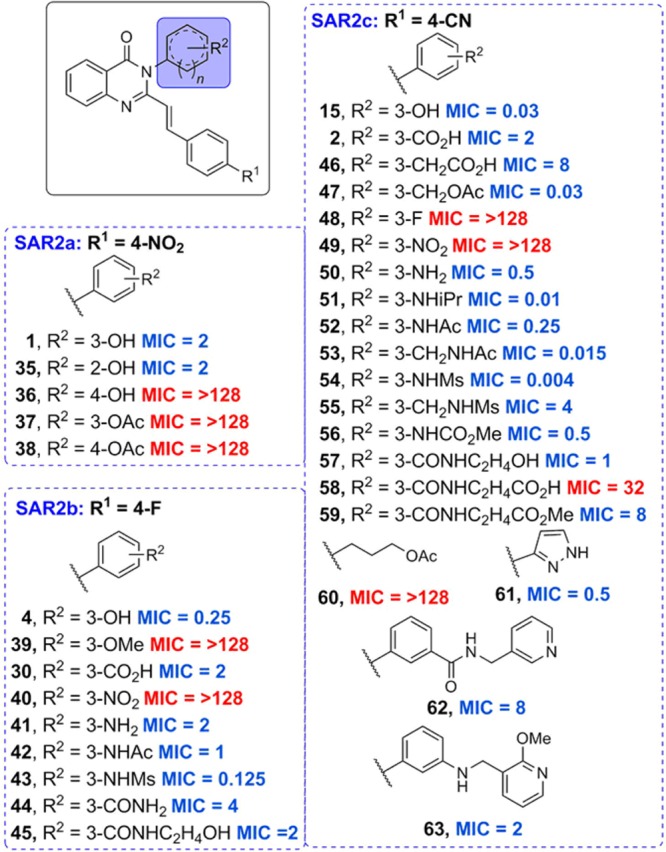
Antibacterial activities of the 4(3H)-quinazolinones derivatives at ring 2. As in Figure 2, the MICs (μg/mL) were determined for S. aureus ATCC 29213. Red text is for inactive compounds (MIC ≥ 16 μg/mL) and blue for active compounds (MIC ≤ 8 μg/mL).
Pyridine derivatives of ring 3, in which ring 2 was kept as 3-hydroxyphenyl, 3-carboxyphenyl, or 3-carbomylphenyl (series SAR3a, SAR3b, and SAR3c, respectively) and substitution at the C6 (SAR3d) and C7 (SAR3e) position, were explored for SAR3 (Figure 4). Replacing ring 3 with a pyridine was tolerated when ring 2 was 3-hydroxyphenyl (64–66); however this replacement was not tolerated when ring 2 was 3-carboxyphenyl (67–69) or 3-carbomylphenyl (70–72). Substitution at the C6 position of ring 3 with a bromo or hydroxyl group (73 and 74, respectively) was not tolerated. A similar trend was observed with substitution at the C7 position (75–77), with a loss of activity. In general, variations and substitutions on ring 3 resulted in reduced activity.
Figure 4.
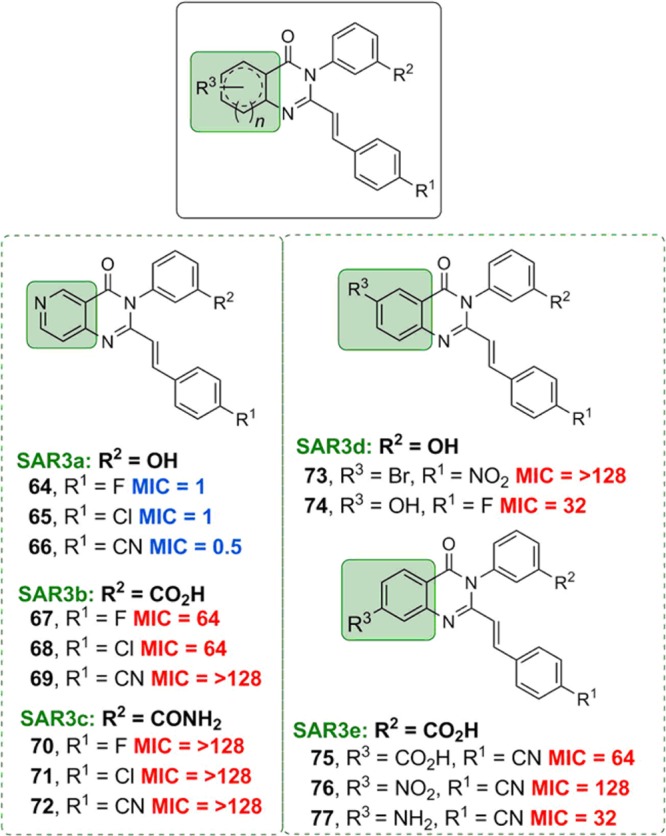
Antibacterial activities of the 4(3H)-quinazolinones derivatives at ring 3. As in Figure 2, the MICs (μg/mL) were determined for S. aureus ATCC 29213. Red text is for inactive compounds (MIC ≥ 16 μg/mL) and blue for active compounds (MIC ≤ 8 μg/mL).
Mouse Peritonitis Model
Selected compounds were evaluated for in vivo efficacy using the mouse peritonitis model of infection with MRSA (ATCC 27660). This model of infection was chosen because it is relatively easy to perform, the end points are simple (death or survival), and the model has been validated with marketed antibiotics.24,25 This model was used as the up-front animal model to identify compounds demonstrating efficacy in vivo, which could then be further evaluated. Two doses of the quinazolinones were given intravenously (iv) at 5 mg/kg. We evaluated the efficacy of compounds 5, 15, 23, 24, 27, 30, 42, 43, 50, 51, 52, 54, 57, 61, and 66 at 5 mg/kg to drive selection of compounds with better efficacy than compound 2, which showed an ED50 (effective dose that results in 50% of the mice surviving) of 9.4 mg/kg in the mouse peritonitis model of MRSA infection.22 Of the compounds tested, only compound 27 showed >50% efficacy at 5 mg/kg.
Fast Pharmacokinetics (PK)
In order to understand the lack of or diminished in vivo efficacy of quinazolinones that had exhibited potent in vitro activity against living bacteria, we assessed the PK properties of several of the compounds using fast PK studies, in which a limited number of animals (n = 2 per time point) and time points (5 time points) were utilized. This strategy significantly reduced the number of plasma samples for analysis by ultraperformance liquid chromatography (UPLC) and allowed for the estimation of systemic exposure (area under the curve, AUC) and clearance (CL). We carried out the fast PK studies after the mouse peritonitis model of infection, since the former required development of bioanalytical methods to determine the blood levels of the quinazolinones, while the latter was less resource intensive. Eleven compounds were selected in order to understand how various R1 and R2 groups affected the PK properties (Table 1). Compound 15 with a 4-CN in R1 and 3-OH in R2 had high CL of 186 mL min–1 kg–1, resulting in low systemic exposure of 26.8 μg·min/mL (Table 1). Substitution of the 3-OH in R2 for 3-CO2H in compound 2 reduced CL to 6.9 mL min–1 kg–1, with a concomitant increase in systemic exposure of 1180 μg·min/mL. The high CL of compound 15 was attributed to the propensity of phenols to undergo phase II metabolism and subsequent excretion. When R1 contained 4-F and R2 had a 3-OH (compound 5) or 3-CO2H (compound 30), no significant differences in CL (10.7 mL min–1 kg–1 vs 7.8 mL min–1 kg–1) or AUC (466 μg·min/mL at a dose of 5 mg/kg vs 2563 μg·min/mL at a dose of 20 mg/kg) were observed. Likewise, no differences in CL (6.9 vs 7.8 mL min–1 kg–1) or AUC (1180 μg·min/mL at a dose of 10 mg/kg vs 2560 μg·min/mL at a dose of 20 mg/kg) were observed when R2 contained 3-CO2H and R1 had 4-CN (compound 2) or 4-F (compound 30). Replacement of 3-CO2H in R2 with 3-NHAc or 3-NHMs increased CL (compare compound 30 vs 42 and 43; compound 2 vs 52 and 54). To reduce the CL of compound 52, 3-NHAc was replaced with 3-CH2NHAc (compound 53), which resulted in a 2-fold reduction of CL and 2-fold increase in AUC (after dose adjustment). These studies revealed that compound 27 had the best PK properties and potent in vitro and in vivo activities.
Table 1. In Vitro and in Vivo Evaluation of Select Quinazolinones.
| fast
PK parameters |
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 (R-benzyl) | R2 (R-benzyl) | MIC S. aureus (μg/mL) | HepG2 IC50 (μg/mL) | hemolysis (%)a | dose (mg/kg) | AUC0–lastb (μg·min/mL) | CLc (mL min–1 kg–1) |
| 2d | 4-CN | 3-CO2H | 2 | 62.9 | <1 | 10 | 1180 | 6.9 |
| 5 | 4-F | 3-OH | 0.25 | 16.4 | <1 | 5 | 466 | 11 |
| 15 | 4-CN | 3-OH | 0.03 | 53.6 | <1 | 5 | 27 | 186 |
| 27 | 4-C≡CH | 3-CO2H | 0.03 | 82.0 | <1 | 10 | 1540 | 6.4 |
| 30 | 4-F | 3-CO2H | 1 | 95.5 | <1 | 20 | 2560 | 7.8 |
| 42 | 4-F | 3-NHAc | 1 | 46.6 | 3.6 | 10 | 284 | 88 |
| 43 | 4-F | 3-NHMs | 0.125 | 66.0 | <1 | 10 | 819 | 28 |
| 50 | 4-CN | 3-NH2 | 0.5 | 77.2 | 3.3 | 10 | 204 | 25 |
| 52 | 4-CN | 3-NHAc | 0.25 | 51.0 | <1 | 5 | 38 | 131 |
| 53 | 4-CN | 3-CH2NHAc | 0.015 | 99.0 | <1 | 10 | 142 | 35 |
| 54 | 4-CN | 3-NHMs | 0.004 | 33.4 | <1 | 5 | 31 | 162 |
| 57 | 4-CN | 3-CONHC2H4OH | 1 | 70.2 | <1 | 10 | 425 | 29 |
The percent hemolysis was determined at a concentration of 64 μg/mL using Triton-X as positive control. PK parameters after a single iv dose.
AUC0–last = area under the curve from 0 min to the last quantifiable time point.
CL = clearance.
Data reproduced from Bouley et al. for comparison.
In Vitro Cytotoxicity
In order to evaluate the potential cytotoxicity of the quinazolinones, the XTT assay26 using HepG2 cells and hemolysis assays were performed. The resulting IC50 values from the XTT assay are one measure of cytotoxicity of the compounds to eukaryotic cells. The hemolysis of red blood cells was used as another measure of toxicity, primarily as a means for flagging of potential membrane-active compounds. Both assays were performed for each compound that was tested in mice (Table 1). Compound 5 displayed relatively high toxicity to HepG2 cells (IC50 = 16.4 μg/mL) but no significant hemolytic activity at 64 μg/mL (>120-fold MIC), which was >30-fold the HepG2 IC50. Although compound 50 displayed lower cytotoxicity to HepG2 cells (IC50 = 77.2 μg/mL), it caused some hemolysis of red blood cells at a concentration of 64 μg/mL.
Activity against Gram-Positive Panel of Bacteria
Six antibacterials from Table 1 (15, 27, 30, 50, 52, and 54) were tested for antibacterial activity against a panel of Gram-positive organisms and were compared to compound 2, vancomycin, and linezolid (Table 2). Quinazolinones mostly display activity against Staphylococcal species. Poor activity (MIC ≥ 16 μg/mL) was observed against a vancomycin-resistant E. faecalis strain. Importantly, compound 27 displayed MIC values of ≤0.5 μg/mL for all S. aureus strains tested, including vancomycin- and linezolid-resistant strains.
Table 2. Activities of Select Quinazolinones against a Gram-Positive Panel of Bacteria.
| MIC (μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| strain | 2a | 15 | 27 | 30 | 50 | 52 | 54 | vancomycin | linezolid |
| S. aureus ATCC 29213b | 2 | 0.03 | 0.03 | 1 | 0.5 | 0.25 | 0.004 | 1 | 2 |
| S. aureus ATCC 27660c | 8 | 0.5 | 0.06 | 2 | 0.5 | 1 | 0.25 | 1 | 2 |
| S. aureus NRS100 (COL)d | 16 | 0.25 | 0.25 | 8 | 4 | 1 | 0.5 | 2 | 2 |
| S. aureus NRS119d | 8 | 0.125 | 0.06 | 2 | 4 | 0.25 | 0.5 | 2 | 32 |
| S. aureus NRS120d | 8 | 0.125 | 0.5 | 8 | 4 | 0.25 | 0.25 | 2 | 32 |
| S. aureus VRS1e | 16 | 0.5 | 0.5 | 8 | 8 | 4 | 0.5 | 512 | 2 |
| S. aureus VRS2f | 2 | 0.06 | 0.03 | 1 | 4 | 0.125 | 0.125 | 64 | 2 |
| S. epidermis ATCC 35547 | 1 | 0.125 | 0.008 | 0.125 | 0.125 | 0.06 | 0.125 | 16 | 1 |
| S. haemolyticus ATCC 29970 | 1 | 0.125 | 0.008 | 0.25 | 1 | 0.25 | 0.125 | 2 | 2 |
| S. oralis ATCC 9811 | >128 | >128 | 128 | 128 | >128 | >128 | >128 | 0.5 | 1 |
| S. pyogenes ATCC 49399 | >128 | >128 | 128 | 128 | >128 | >128 | >128 | 0.5 | 1 |
| B. cereus ATCC 13061 | >128 | >128 | 128 | >128 | >128 | >128 | >128 | 1 | 1 |
| B. licheniformis ATCC 12759 | >128 | >128 | 128 | >128 | >128 | >128 | >128 | 0.5 | 1 |
| E. faecalis ATCC 29212b | >128 | 128 | >128 | >128 | >128 | 128 | >128 | 2 | 2 |
| E. faecalis 99g | >128 | 64 | 128 | >128 | 16 | 64 | 128 | 128 | 1 |
| E. faecium NCTC 7171 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | 0.5 | 2 |
Data reproduced from Bouley et al. for comparison.
Quality control strain for susceptibility testing.
mecA positive, resistant to methicillin, oxacillin, and tetracycline.
mecA positive, resistant to ciprofloxacin, gentamicin, oxacillin, penicillin, and linezolid.
Vancomycin-resistant MRSA (vanA) clinical isolate from Michigan.
Vancomycin-resistant MRSA (vanA) clinical isolate from Pennsylvania.
Vancomycin-resistant clinical isolate.
Minimal Bactericidal Concentration (MBC)
To determine whether compound 27 was bactericidal or bacteriostatic, its MBC was determined. The MBC was defined as the minimum concentration of compound required to achieve ≥99.9% reduction in the bacterial counts.27 Compound 2 was included as a comparator and linezolid as control. We used three strains of S. aureus: ATCC 29213 (a quality-control, methicillin-susceptible strain), ATCC 27660 (a methicillin-resistant strain used in the mouse peritonitis infection model), and NRS70 (a clinical MRSA strain used in the mouse neutropenic thigh model). Both compounds 2 and 27 show >4-fold difference between MIC and MBC, indicating that the quinazolinones are bacteriostatic, as is linezolid (Table 3).27 At higher concentrations, they exhibit bactericidal activity (lowered the initial inoculum of 1 × 106 CFU/mL to ≤500 CFU/mL). Since the MICs for antibacterial 27 were very low, the MBC values in the three cases that were evaluated in Table 3 were not prohibitively high (≥1 μg/mL). Hence, this compound will exhibit bactericidal activity at concentrations of ≥1 μg/mL.
Table 3. MBCs for Compounds 2 and 27.
| MBC (μg/mL) |
MIC (μg/mL) |
|||||
|---|---|---|---|---|---|---|
| strain | 2 | 27 | linezolid | 2 | 27 | linezolid |
| S. aureus ATCC 29213 | 8 | 0.125 | 16 | 2 | 0.03 | 2 |
| S. aureus ATCC 27660 | 64 | 1 | 16 | 8 | 0.06 | 2 |
| S. aureus NRS70 | 32 | 0.5 | 8 | 2 | 0.03 | 2 |
Full PK Studies
A full PK study in mice (n = 72 mice) was conducted with compound 27 after a single 10 mg/kg iv and oral (po) dose. This compound was converted to its sodium salt by the procedure published for 2,22 and this form was used for all mouse studies due to its superior water solubility (1.5 mg/mL). Concentrations were sustained above MIC (0.03 μg/mL) for 24 h (the last time point taken) when administered iv and for 30 h after po administration (Figure 5). This compound showed a similar rate of clearance compared to 2, 6.4 mL min–1 kg–1 versus 6.9 mL min–1 kg–1 (<10% of hepatic blood flow) and slightly higher systemic exposure (1540 μg·min/mL versus 1180 μg·min/mL). However, the volume of distribution (Vd) was 10-fold higher, 3.58 L/kg compared to 0.30 L/kg for 2 (Table 4), indicating that compound 27 had better distribution to tissues than compound 2. In addition, the peak concentration (Cmax) of compound 27 after po administration was 2.7 μg/mL and was achieved at 0.5 h, 2-fold higher than that of compound 2, which was attained at 1 h. The absolute oral bioavailability of compound 27 was 37%, while that of compound 2 was 50%. Given that the MIC values of compound 27 are 1–2 orders of magnitude smaller than those of compound 2 and had higher systemic exposure, we anticipated that compount 27 would be more efficacious in vivo than compound 2.
Figure 5.
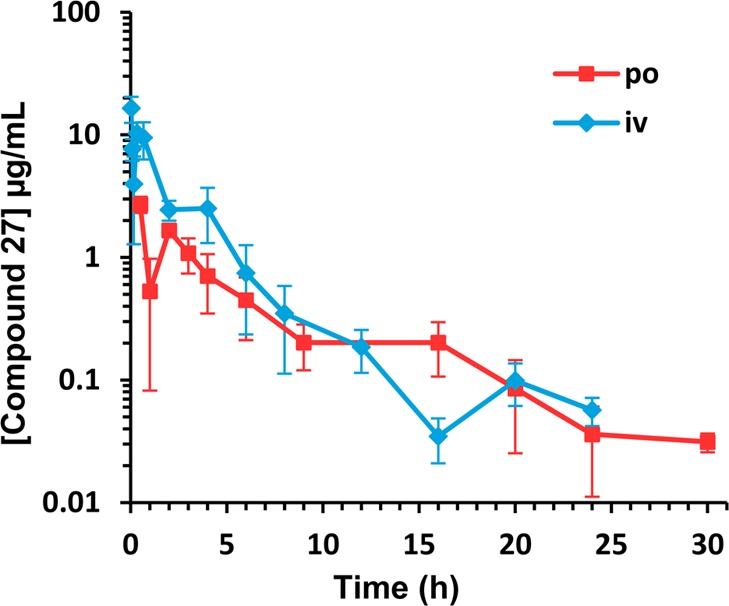
Pharmacokinetics of 27 in ICR mice after a single iv or po dose at 10 mg/kg (n = 3 per time point).
Table 4. Full Pharmacokinetic Parameters of Compound 27 Compared to Compound 2 at a Dose of 10 mg/kg.
| compd (route) | AUC0–lasta (μg·min/mL) | AUC0–∞b (μg·min/mL) | Cmaxc (μg/mL) | Tmaxd (h) | CLe (mL min–1 kg–1) | Vdf (L/kg) | t1/2g | Fh (%) |
|---|---|---|---|---|---|---|---|---|
| 27 (iv) | 1540 | 1570 | 6.4 | 3.58 | t1/2α = 3.6 min | |||
| t1/2β = 6.5 h | ||||||||
| 27 (po) | 552 | 582 | 2.7 | 0.5 | t1/2α = 2.4 h | 37 | ||
| t1/2β = 6.6 h | ||||||||
| 2 (iv)i | 1180 | 1460 | 6.9 | 0.30 | t1/2α = 35.2 min | |||
| t1/2β ≥ 20 h | ||||||||
| 2 (po)i | 408 | 738 | 1.3 | 1 | t1/2α = 1.9 h | 50 | ||
| t1/2β > 20 h |
AUC0–last = area under the curve from 0 min to the last quantifiable time point.
AUC0–-∞ = area under the curve from 0 min extrapolated to infinity.
Cmax = maximum concentration.
Tmax = time at which maximum concentration is achieved.
CL = clearance.
Vd = volume of distribution.
t1/2α = half-life of distribution. t1/2β = half-life of elimination.
F = oral bioavailbility.
Data for compound 2 reproduced from Bouley et al. for comparison.
Plasma Protein Binding
We ascertained protein binding in human plasma for compound 27 by equilibrium dialysis, as high plasma protein binding can decrease the in vivo efficacy of an otherwise potent in vitro compound.28 Compound 27 showed plasma protein binding of 99.6 ± 0.04%, which was higher than the plasma protein binding of the previous lead quinazolinone 2 (96.5 ± 0.7%).22
Mouse Neutropenic Thigh Infection Model
Compound 27 was evaluated in the mouse neutropenic thigh infection model. This model is more clinically relevant for S. aureus infections and allows us to be better able to quantitatively compare compound 27 to compound 2 and linezolid. Mice were rendered neutropenic by injection with cyclophosphamide, and then MRSA was administered intramuscularly to one thigh.29 Starting 1 h after infection, the compounds were dosed subcutaneously at 20 mg/kg three times a day (every 8 h) and the infected and uninfected thighs and plasma were harvested at 24 h. Linezolid (10 mg/kg) was used as a positive control and the vehicle as a negative control. The dosing regimen was chosen in order to maximize the time above MIC against the clinical MRSA strain used: NRS70. Compound 27 and linezolid showed statistically significant lowering of the bacterial load for NRS70 compared to the negative control (Figure 6), whereas compound 2 was not able to achieve statistically significant efficacy (p > 0.05) in this mouse model of infection. Additionally, the levels of 2, 27, or linezolid were quantified in plasma and uninfected thighs (n = 8 mice per group). At 24 h after infection (7 h after the last dose) levels in plasma were 2.43 ± 1.64 μg/mL for compound 27, 1.13 ± 1.07 μg/mL for compound 2, and not quantifiable for linezolid. The corresponding levels in the thighs were 0.22 ± 0.17 μg/mL for compound 27, 0.024 ± 0.023 μg/mL for compound 2, and not quantifiable for linezolid. Levels of compound 27 in thighs were 9-fold higher than those of compound 2, indicating better tissue distribution as reflected in its higher Vd.
Figure 6.
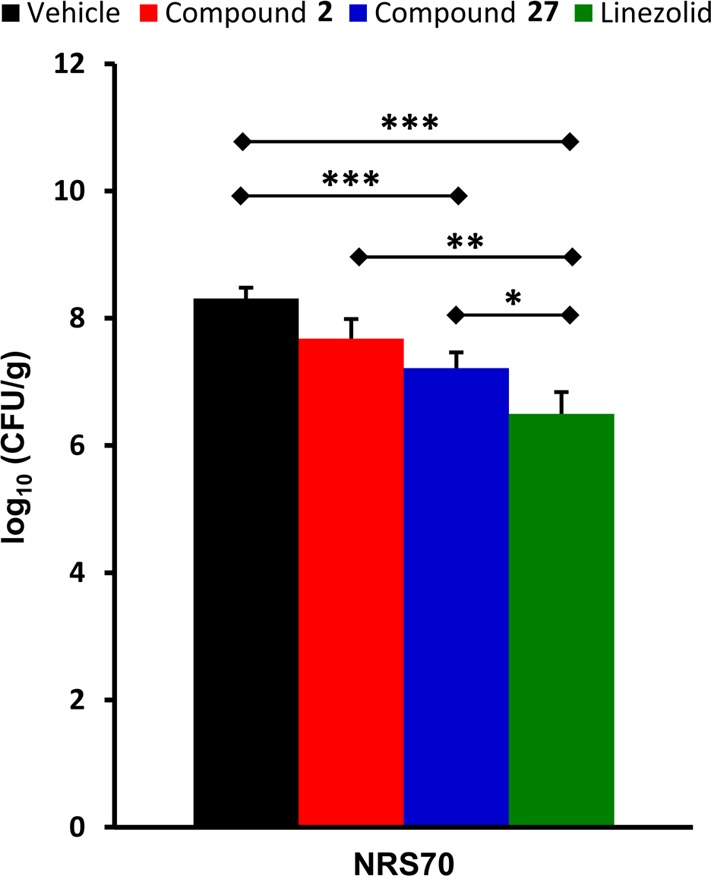
In vivo efficacy of compound 27 in a mouse neutropenic thigh infection model. The number of colony-forming units (CFU) per gram of thigh tissue was determined at 24 h after infection and averaged for each group (n = 8 mice). The initial inoculum corresponded to approximately 5.13 log10(CFU/g). Error is expressed as the standard error of the mean (SEM), and a Mann–Whitney U-test was used to determine statistical significance. Groups that were statistically significant from another are shown: (∗) p < 0.05, (∗∗) p < 0.01, or (∗∗∗) p < 0.001.
PBP Antisense-Mediated Sensitization
For further mechanistic support for select quinazolinones, an antisense technology developed by Roemer and colleagues was utilized.30 Four MRSA strains with expression of the genes for PBP1, PBP2, PBP2a, and PBP3 under antisense control were tested for susceptibility to the compounds. In the presence of xylose the antisense gene is transcribed and interferes with the translation of the corresponding protein, causing expression levels of the protein to be greatly reduced. Thus, the strain becomes increasingly sensitive to the action of an inhibitor of the corresponding protein as the xylose concentration increases. An increased susceptibility to compound 27 was observed under xylose induction for the PBP1 and PBP2a antisense strains but not for PBP2 or PBP3 (Figure S1), consistent with our earlier report with compound 2. Compound 27 was compared to ceftaroline as a positive control, in which an increase in susceptibility was observed for PBP1, PBP2, and PBP2a, as expected.
Conclusion
We have previously disclosed a lead 4(3H)-quinazolinone (2) that demonstrated good in vitro activity against MRSA strains, good PK properties and showed efficacy when dosed iv in the mouse peritonitis model of infection.22 Exploration of the SAR for the 4(3H)-quinazolinones is presented here with 77 analogs screened for antibacterial activity, of which 44 displayed activity (MIC ≤ 4 μg/mL) against S. aureus. Phenyl rings with para substituents were favored in ring 1; substituents such as fluoro (5), chloro (8), nitrile (15), and alkyne (16) had the best activity. Bulky groups such as isopropyl (12) and hydrogen-bond donors (13, 17) were not tolerated. Replacing the phenyl moiety with a pyridine (21) and cyclohexyl (23) was tolerated, but a furan (22) was inactive. Additionally, the double-bond linker was investigated; saturation (24) was tolerated, but shortening the linker (25 and 26) was not. For ring 2, hydrogen-bond donors in the meta position such as hydroxyl (15), amino (41), N-acetyl (42), N-mesyl (43) seemed to be favored. Replacing the hydroxyl with an O-acetyl (37) or methoxy (39) was not tolerated. Bulkier groups were also tolerated (51, 57–59, 62, 63) and replacing the phenyl moiety with a pyrazole (61) was allowed. A carboxyl substituent in ring 2 was less active than the parent hydroxyl in all cases; however these compounds demonstrated the best PK properties (2, 27, 30). A few variations of ring 3 were explored including a pyridine ring (64–72) and substitutions at the C6 (73 and 74) and C7 (75–77) positions; however in most cases these were not tolerated.
Eleven compounds (5, 15, 27, 30, 42, 43, 50, 52, 53, 54, and 57) were chosen based on their in vitro activity to be evaluated for in vitro toxicity, PK properties, in vivo efficacy, and activity against a panel of Gram-positive organisms. In the mouse peritonitis model of infection, compound 27 showed superior efficacy compared with compound 2. Compound 27 has low clearance, 10-fold higher volume of distribution than compound 2, and 2-fold higher Cmax after po administration than compound 2. Thus, compound 27 demonstrated better efficacy in the mouse neutropenic thigh MRSA infection model compared to 2.
Quinazolinones are non-β-lactam antibacterials targeting PBPs for inhibition. The lead quinazolinone of this study, compound 27, exhibits exquisite antibacterial activity (MIC = 0.03 μg/mL) and may have favorable pharmacodynamics. The excellent MIC values are reflective of avid binding of the critical PBPs in S. aureus. As the antisense methodology documented that more than one PBP is targeted for inhibition, as is the case with β-lactam antibiotics, the MIC values are a composite of the concentration that achieves this inhibition. These non-β-lactam antibacterials hold great promise in addressing the clinical need for infections by S. aureus.
Experimental Methods
Syntheses
The synthetic procedures for the 11 compounds evaluated in Table 1 are detailed below. These methods are representative for those used for the preparation of other derivatives. Purity of the final compounds was verified to be ≥95%, as determined by HPLC.
2-Methyl-4H-benzo[d][1,3]oxazin-4-one (II)
Anthranilic acid (I, 20 g, 146 mmol) was dissolved in triethyl orthoacetate (45 mL, 245 mmol) and refluxed for 2 h. The product was crystallized by cooling the reaction mixture on iced water for 4 h. The needle crystals were filtered from the reaction mixture and washed liberally with hexanes to give the product (17 g, 72% yield). 1H NMR (500 MHz, CDCl3) δ 2.47 (s, 3H), 7.50 (t, J = 7.38 Hz, 1H), 7.54 (d, J = 7.98 Hz, 1H), 7.80 (t, J = 7.18 Hz, 1H), 8.18 (d, J = 7.78 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 21.59, 116.84, 126.59, 128.42, 128.66, 136.77, 146.61, 159.89, 160.45. HRMS (m/z): [M + H]+, calcd for C9H8NO2, 162.0550; found, 162.0555.
3-(3-Hydroxyphenyl)-2-methylquinazolin-4(3H)-one (IIIa)
Compound II (2.1 g, 13.0 mmol) and 3-aminophenol (1.5 g, 13.7 mmol) were suspended in 5 mL of glacial acetic acid. The suspension dissolved completely upon heating. The reaction was refluxed for 4 h, cooled, and 10 mL of water was added to the reaction mixture. The resulting precipitate was filtered and was washed with water, cold ethanol, and hexanes to give the product (2.2 g, 67% yield). 1H NMR (300 MHz, CDCl3) δ 2.29 (s, 3H), 6.61 (s, 1H), 6.73 (d, J = 7.85 Hz, 1H), 6.85 (d, J = 8.37 Hz, 1H), 7.37 (t, J = 8.13 Hz, 1H), 7.50 (t, J = 7.89 Hz, 1H), 7.69 (d, J = 8.13 Hz, 1H), 7.80 (t, J = 8.37 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 24.18, 115.54, 117.57, 118.79, 120.57, 126.92, 127.17, 127.35, 131.22, 135.28, 138.01, 147.65, 154.76, 158.90, 163.15. HRMS (m/z): [M + H]+, calcd for C15H13N2O2, 253.0972; found, 253.0946.
3-(3-Carboxyphenyl)-2-methylquinazolin-4(3H)-one (IIIb)
The compound was prepared according to the procedure described for IIIa and as reported by Bouley et al.22
(E)-3-(3-Carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (2)
The compound was prepared according to the procedure described previously by Bouley et al.22
(E)-3-(3-Hydroxyphenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (5)
Compound IIIa (0.25 g, 1 mmol) was suspended in 5 mL of glacial acetic acid and dissolved upon heating, to which 4-fluorobenzaldehyde (0.11 mL, 1 mmol) was added. The reaction was refluxed overnight (18 h), and 10 mL of water was added to the cooled reaction mixture. The resulting precipitate was filtered and washed with water followed by cold ethanol and hexanes to give the product (0.14 g, 40% yield). 1H NMR (500 MHz, DMSO-d6) δ 6.32 (d, J = 15.55 Hz, 1H), 6.83 (s, 1H), 6.84 (d, J = 7.78 Hz, 1H), 6.96 (d, J = 8.17 Hz, 1H), 7.22 (m, 2H), 7.40 (t, J = 7.98 Hz, 1H), 7.44 (m, 2H), 7.53 (t, J = 8.17 Hz, 1H), 7.75 (d, J = 8.17 Hz, 1H), 7.87 (m, 2H), 8.12 (d, J = 7.78 Hz, 1H), 9.92 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 116.55, 116.75, 116.92, 117.00, 120.00, 120.57, 121.35, 127.18, 127.18, 127.85, 130.35, 130.41, 131.11, 132.25, 135.45, 138.15, 138.56, 148.05, 151.98, 159.03, 161.80, 162.52. HRMS (m/z): [M + H]+, calcd for C22H16FN2O2, 359.1190; found, 359.1186.
(E)-3-(3-Hydroxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (15)
The compound was prepared according to the procedure described for 5 (0.62 g, 84% yield). 1H NMR (500 MHz, DMSO-d6) δ 6.51 (d, J = 15.55 Hz, 1H), 6.85 (m, 2H), 6.97 (d, J = 7.98 Hz, 1H), 7.39 (t, J = 7.78 Hz, 1H), 7.55 (m, 3H), 7.77 (d, J = 7.78 Hz, 1H), 7.81 (d, J = 8.37 Hz, 2H), 7.88 (t, J = 7.18 Hz, 1H), 7.89 (d, J = 15.55 Hz, 1H), 8.12 (d, J = 7.98 Hz, 1H), 9.94 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 111.52, 115.87, 116.40, 118.61, 119.30, 120.81, 123.47, 126.50, 126.95, 127.29, 128.08, 130.42, 132.92, 134.80, 136.60, 137.65, 139.38, 147.17, 150.84, 158.32, 160.99. HRMS (m/z): [M + H]+, calcd for C23H16N3O2, 366.1237; found, 366.1254.
(E)-3-(3-Carboxyphenyl)-2-(4-ethynylstyryl)quinazolin-4(3H)-one (27)
The compound was prepared according to the procedure described for 2 and purified by silica gel column chromatography (0.377 g, 13% yield). The sodium salt form was prepared by dissolving the compound in 20% ethyl acetate/80% acetone and reacting with 1 equiv of sodium hexylethanoate at 0 °C for 30 min. The reaction mixture was concentrated in vacuo and the resulting yellow solid was thoroughly washed with ethyl acetate to obtain the final compound in 75% yield (0.260 g). 1H NMR (500 MHz, DMSO-d6) δ 4.32 (s, 1H), 6.34 (d, J = 15.55 Hz, 1H), 7.35 (d, J = 8.37 Hz, 2H), 7.44 (d, J = 8.37 Hz, 2H), 7.49 (d, J = 7.58 Hz, 1H), 7.55 (t, J = 7.98 Hz, 1H), 7.58 (t, J = 7.78 Hz, 1H), 7.78 (d, J = 8.17 Hz, 1H), 7.87 (m, 3H), 8.05 (d, J = 7.78 Hz, 1H), 8.13 (d, J = 7.98 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 82.70, 83.24, 120.66, 121.04, 122.84, 126.51, 126.81, 127.31, 127.83, 129.98, 130.12, 132.33, 132.39, 133.49, 134.90, 135.21, 137.21, 137.99, 147.36, 151.04, 161.37, 166.58. HRMS (m/z): [M + H]+, calcd for C25H17N2O3, 393.1234; found, 393.1250. HRMS (m/z): [M + Na]+, calcd for C25H16N2NaO3, 415.1053; found, 415.1054.
(E)-3-(3-Carboxyphenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (30)
The compound was prepared according to the procedure described for 2 (0.53 g, 76% yield). 1H NMR (500 MHz, DMSO-d6) δ 6.25 (d, J = 15.55 Hz, 1H), 7.19 (m, 2H), 7.46 (m. 2H), 7.55 (d, J = 8.17 Hz, 1H), 7.73 (m, 2H), 7.77 (d, J = 8.17 Hz, 1H), 7.87 (m, 2H), 8.02 (s, 1H), 8.13 (m, 2H). 13C NMR (126 MHz, DMSO- d6) δ 115.91, 116.20, 119.77, 120.59, 126.48, 126.66, 127.21,129.82, 129.93, 130.06, 131.39, 131.44, 132.34, 133.49, 134.84, 137.25, 137.79, 147.39, 151.15, 161.35, 166.55. HRMS (m/z): [M + H]+, calcd for C23H16N2O3, 387.1139; found, 387.1140.
3-(3-Nitrophenyl)-2-methylquinazolin-4(3H)-one (IIIc)
The compound was prepared according to the procedure described for IIIa and recrystallized by dissolving in hot ethanol and then allowing the solution to cool (2.4 g, 47% yield). 1H NMR (500 MHz, DMSO-d6) δ 2.14 (s, 3H), 7.54 (t, J = 7.18 Hz, 1H), 7.67 (d, J = 8.13 Hz, 1H), 7.88 (m, 2H), 7.99 (d, J = 7.89 Hz, 1H), 8.10 (d, J = 7.89 Hz, 1H), 8.38 (d, J = 8.13 Hz, 1H), 8.50 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 24.13, 120.41, 124.05, 124.25, 126.32, 126.56, 126.74, 130.99, 134.79, 135.66, 138.95, 147.34, 148.54, 161.43. HRMS (m/z): [M + H]+, calcd for C15H12N3O3, 282.0873; found, 282.0901.
(E)-3-(3-Nitrophenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (40)
The compound was prepared according to the procedure described for 5 (1.87 g, 68% yield). 1H NMR (500 MHz, DMSO-d6) δ 6.31 (d, J = 15.35 Hz, 1H), 7.18 (t, J = 8.97 Hz, 2H), 7.55 (m, 3H), 7.78 (d, J = 8.17 Hz, 1H), 7.89 (m, 3H), 7.95 (d, J = 7.78 Hz, 1H), 8.13 (d, J = 7.98 Hz, 1H), 8.43 (d, J = 8.18 Hz, 1H), 8.51 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 116.01, 116.18, 119.84, 120.70, 124.42, 124.89, 126.64, 126.86, 127.39, 130.32, 130.39, 131.15, 131.53, 131.56, 135.12, 136.11, 138.19, 138.34, 148.73, 151.16, 161.54, 162.06, 164.03. HRMS (m/z): [M + H]+, calcd for C22H15FN3O3, 388.1092; found, 388.1117.
(E)-3-(3-Aminophenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (41)
Compound 40 (1.80 g, 4.6 mmol) and tin(II) chloride dihydrate (5.24 g, 23.2 mmol) were suspended in 50 mL of ethanol to which 1 mL of concentrated HCl was added. The solution was refluxed for 3 h and then allowed to stir overnight at 25 °C. The solution was concentrated in vacuo, and ethyl acetate was added to completely dissolve the residue. The ethyl acetate solution was then neutralized with a NaOH solution and washed with saturated potassium fluoride. The mixture was then filtered through a bed of Celite to remove the tin precipitate. The resulting filtrate was washed three times with a saturated KF solution, and the organic layer was dried with MgSO4 and concentrated in vacuo. The residue was then purified by silica gel chromatography to give the product (0.54 g, 32% yield). 1H NMR (500 MHz, DMSO-d6) δ 5.46 (s, 2H), 6.38 (d, J = 15.55 Hz, 1H), 6.50 (d, J = 7.58 Hz, 1H), 6.54 (s, 1H), 6.73 (d, J = 8.17 Hz, 1H), 7.23 (m, 3H), 7.43 (m, 2H), 7.52 (t, J = 7.18 Hz, 1H), 7.74 (d, J = 7.78 Hz, 1H), 7.78 (m, 2H), 8.12 (d, J = 7.98 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 113.50, 114.48, 115.42, 116.05, 116.23, 119.95, 120.62, 126.45, 126.55, 127.12, 129.57, 129.64, 129.99, 131.58, 131.60, 134.69, 137.33, 137.53, 147.32, 150.05, 151.35, 161.03, 163.77. HRMS (m/z): [M + H]+, calcd for C22H17FN3O, 358.1350; found, 358.1360.
(E)-3-(3-Acetamidophenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (42)
Compound 41 (0.15 g, 0.42 mmol) was dissolved in 4 mL of pyridine, to which acetic anhydride was added (0.21 g, 2.1 mmol). The reaction was stirred for 3 h at 25 °C and then refluxed for 1 h. The reaction was then poured over ice in 2 M HCl and the resulting precipitate was filtered out and washed with cold water to give the product (0.15 g, 83% yield). 1H NMR (500 MHz, DMSO-d6) δ 2.06 (s, 3H), 6.30 (d, J = 15.55 Hz, 1H), 7.12 (d, J = 7.38 Hz, 1H), 7.22 (t, J = 8.77 Hz, 1H), 7.46 (m, 2H), 7.55 (m, 2H), 7.73 (d, J = 6.58 Hz, 2H), 7.82 (d, J = 7.98 Hz, 1H), 7.90 (t, J = 6.78 Hz, 1H), 7.93 (d, J = 15.75 Hz, 1H), 8.13 (d, J = 7.98 Hz, 1H), 10.33 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 24.09, 111.17, 116.12, 116.30, 118.45, 118.87, 119.70, 120.31, 123.21, 126.64, 127.10, 127.41, 130.03, 135.13, 136.70, 140.61, 141.61, 158.28, 160.81, 168.78. HRMS (m/z): [M + H]+, calcd for C24H19FN3O2, 400.1456; found, 400.1440.
(E)-3-(3-Methylsulfonamidophenyl)-2-(4-fluorostyryl)quinazolin-4(3H)-one (42)
Compound 41 (0.15 g, 0.42 mmol) was dissolved in 4 mL of pyridine and cooled to 0 °C, to which mesyl chloride (0.24 g, 2.1 mmol) was added. The reaction was stirred at 25 °C for 3 h and then poured over ice in 2 M HCl. The resulting precipitate was filtered out and washed with cold 2 M HCl followed by cold water to give the product (0.19 g, 99% yield). 1H NMR (500 MHz, DMSO-d6) δ 3.05 (s, 3H), 6.31 (d, J = 15.75 Hz, 1H), 7.22 (m, 3H), 7.33 (s, 1H), 7.42 (d, J = 8.17 Hz, 2H), 7.48 (m, 2H), 7.58 (t, J = 7.98 Hz, 2H), 7.92 (d, J = 3.59 Hz, 2H), 7.99 (d, J = 15.75 Hz, 1H), 78.12 (m, 2H), 10.20 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 54.50, 115.94, 116.11, 118.14, 119.37, 120.08, 120.27, 123.88, 126.52, 127.01, 127.27, 129.92, 129.99, 130.43, 135.05, 139.56, 141.42, 146.46, 160.54, 161.94. HRMS (m/z): [M + H]+, calcd for C23H19FN3O3S, 436.1126; found, 436.1136.
(E)-3-(3-Nitrophenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (49)
The compound was prepared according to the procedure described for 5 (4.8 g, 75% yield). 1H NMR (500 MHz, DMSO-d6) δ 6.54 (d, J = 15.35 Hz, 1H), 7.58 (t, J = 7.78 Hz, 1H), 7.68 (d, J = 8.17 Hz, 2H), 7.78 (m, 3H), 7.91 (m, 4H), 8.14 (d, J = 7.98 Hz, 1H), 8.43 (d, J = 8.17 Hz, 1H), 8.52 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 111.77, 118.80, 120.88, 123.48, 124.50, 124.94, 126.68, 127.24, 127.54, 128.73, 131.16, 132.89, 135.18, 136.10, 137.54, 137.97, 139.35, 147.37, 148.72, 150.76, 161.45. HRMS (m/z): [M + H]+, calcd for C23H14N4NaO3, 417.0958; found, 417.0941.
(E)-3-(3-Aminophenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (50)
The compound was prepared according to the procedure described for 41 (2.2 g, 37% yield). 1H NMR (500 MHz, DMSO-d6) δ 5.47 (s, 2H), 6.51 (d, J = 7.78 Hz, 1H), 6.56 (m, 2H), 6.73 (d, J = 8.17 Hz, 1H), 7.21 (t, J = 7.98 Hz, 1H), 7.53 (m, 3H), 7.76 (d, J = 8.17 Hz, 1H), 7.82 (d, J = 8.37 Hz, 2H), 7.87 (t, J = 6.98 Hz, 1H), 7.90 (d, J = 15.55 Hz, 1H), 8.12 (d, J = 7.98 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 111.50, 113.50, 114.57, 115.42, 118.61, 120.79, 123.52, 126.48, 126.92, 127.27, 128.00, 130.00, 132.92, 134.73, 136.48, 137.34, 139.43, 147.14, 150.04, 150.89, 160.91. HRMS (m/z): [M + H]+, calcd for C23H17N4O, 365.1397; found, 365.1367.
(E)-3-(3-Acetamidophenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (52)
The compound was prepared according to the procedure described for 42 (0.19 g, 85% yield). 1H NMR (500 MHz, DMSO-d6) δ 3.16 (s, 3H), 6.50 (d, J = 15.55 Hz, 1H), 7.13 (d, J = 7.38 Hz, 1H), 7.56 (m, 4H), 7.71 (d, J = 7.98 Hz, 2H), 7.80 (m, 3H), 7.91 (m, 2H), 8.14 (d, J = 7.98 Hz, 1H), 10.22 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 24.23, 111.73, 118.77, 119.17, 119.75, 120.90, 123.50, 126.67, 127.22, 127.52, 128.34, 130.14, 133.06, 135.06, 137.03, 137.07, 139.50, 140.64, 147.33, 150.92, 161.24, 168.86. HRMS (m/z): [M + Na]+, calcd for C25H18N4NaO2, 429.1322; found, 429.1291.
(E)-3-(3-(Acetamidomethyl)phenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (53)
This compound was prepared according to the procedure for 42 in 21% yield (0.17 g). 1H NMR (500 MHz, DMSO-d6) δ 1.83 (s, 3H), 4.32 (m, 2H), 6.45 (d, J = 15.5 Hz, 1H), 7.33 (d, J = 5.5 Hz, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.52 (m, 4H), 7.77 (m, 3H), 7.86 (m, 2H), 8.12 (dd, J1 = 1.0 Hz, J2 = 8.0 Hz, 1H), 8.43 (t, J = 11.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 23.3, 42.4, 112.2, 119.2, 121.4, 124.0, 127.1, 127.6, 127.9, 128.0, 128.1, 128.7, 128.8, 130.2, 133.5, 135.5, 137.4, 137.5, 140.0, 142.3, 147.8, 151.4, 161.8, 170.0. HRMS (m/z): [M + H]+, calcd for C26H21N4O2, 421.1658; found, 421.1675.
(E)-3-(3-Methylsulfonamidophenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (54)
The compound was prepared according to the procedure described for 42 (0.33 g, 77% yield). 1H NMR (500 MHz, DMSO-d6) δ 3.04 (s, 3H), 6.52 (d, J = 15.75 Hz, 1H), 7.21 (d, J = 7.58 Hz, 1H), 7.27 (s, 1H), 7.37 (d, J = 8.57 Hz, 1H), 7.59 (m, 4H), 7.81 (m, 3H), 7.90 (m, 2H), 8.15 (d, J = 7.98 Hz, 1H), 10.08 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 111.60, 118.65, 119.73, 120.32, 120.76, 123.51, 125.24, 126.56, 127.12, 127.39, 128.22, 130.60, 132.90, 134.98, 136.89, 137.45, 139.39, 139.62, 147.19, 150.78, 161.10. HRMS (m/z): [M + Na]+, calcd for C24H18N4NaO3S, 465.0992; found, 465.0973.
(E)-3-(3-(2-Hydroxyethyl)carbomylphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one (57)
Compound 2 (0.19 g, 0.5 mmol) was dissolved in 12 mL of THF, to which 2-aminoethanol (0.037 g, 0.6 mmol) and DIC (0.069 g, 0.55 mmol) were added. The mixture was stirred at room temperature for 8 h, and then 25 mL ethyl acetate was added and washed with a saturated NH4Cl solution. The organic phase was separated and dried over Na2SO4, concentrated in vacuo, and purified by silica gel column chromatography to give the product in 63% yield (0.14 g). 1H NMR (400 MHz, DMSO-d6) δ 3.28 (m, 2H), 3.51 (q, J = 6.0 Hz, 2H), 4.75 (t, J = 5.6 Hz, 1H), 6.50 (d, J = 15.6 Hz, 1H), 7.57 (m, 3H), 7.63 (m, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.89 (m, 3H), 8.04 (m, 1H), 8.14 (m, 1H), 8.59 (t, J = 5.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 42.3, 59.6, 111.6, 118.6, 120.6, 126.5, 127.1, 127.4, 127.9, 128.1, 128.2, 129.7, 131.6, 132.8, 135.0, 136.0, 136.7, 137.0, 139.2, 147.2, 150.8, 161.2, 165.2. HRMS (m/z): [M + H]+, calcd for C26H21N4O3, 437.1608; found, 437.1635.
HPLC Analysis
Compounds were analyzed for purity using a PerkinElmer series 200 chromatography system 524 (PerkinElmer, Waltham, MA) equipped with a LC pump, UV/vis detector, with an autosampler. The samples were diluted in acetonitrile (20 μM) and analyzed on a Zorbax RX-C8 5.0 μm, 4.6 mm × 250 mm analytical column (Agilent Technologies, Santa Clara, CA). Isocratic elution was at 1.0 mL/min of 20% water containing 0.1% TFA and 20% acetonitrile containing 0.1% TFA for 6 min with UV detection from 349 to 351 nm.
Screening for Interference Compounds
Compounds were screened for presence of known pan assay interference compounds (PAINS) using the PAINS-remover.31 Compounds were input as a SMILES strings, and all 77 compounds passed the filter.
Bacterial Strains
E. faecium NCTC 7171 (ATCC 19734), S. aureus ATCC 29213, K. pneumonia ATCC 700603, A. baumannii ATCC 17961, P. aeruginosa ATCC 17853, E. aerogenes ATCC 35029, E. coli ATCC 25922, S. aureus ATCC 27660, S. epidermis ATCC 35547, S. haemolyticus ATCC 29970, S. oralis ATCC 9811, S. pyogenes ATCC 49399, B. cereus ATCC 13061, B. licheniformis ATCC 12759, and E. faecalis ATCC 29212 were purchased from American Type Culture Collection (Manassas, VA). S. aureus strains NRS70, NRS100, NRS119, NRS120, VRS1, and VRS2 were obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (Chantilly, VA). E. faecalis 99 was collected from Wayne State University School of Medicine.
MIC Determination
MICs were determined following the CLSI microdilution method.32 2-Fold serial dilutions of each compound in cation-adjusted Mueller–Hinton broth (supplemented with 5% lysed horse blood for Streptococcus strains) were prepared in triplicate in 96-well plates and inoculated with 5 × 105 CFU/mL bacteria. The plates were incubated at 36 °C for 16–20 h, and the MIC was recorded as the lowest concentration that visibly inhibited growth.
MBC Determination
Compounds 2 and 27 were serially diluted 2-fold in 96-well plates and inoculated with 5 × 105 CFU/mL S. aureus ATCC 29213, ATCC 27660, or NRS70 according to the procedure outlined for MIC determination. Following incubation, 25 μL aliquots were spotted onto agar plates in triplicate for 1× MIC to 128 μg/mL. The MBC was determined as the concentration of compound that reduced the initial inoculum by 99.9% (≤500 CFU/mL).
XTT Assay
Cytoxicity assays were performed in triplicate against HepG2 cells (ATCC HB-8065), as previously described.22 The IC50 values were calculated by nonlinear regression with GraphPad Prism 5 (San Diego, CA).
Hemolysis
The percent hemolysis was determined for each compound at a fixed concentration of 64 μg/mL using a 10% red-blood cell suspension from heparinized human blood, as previously described.22 This concentration was selected, as it was the highest concentration in which all tested compounds retained solubility and was several-fold above the respective MICs.
Animals
Outbred mice (ICR female, 6–8 weeks old, ∼20 g body weight, Harlan Laboratories, Indianapolis, IN), providing a heterogeneous population, were used for PK and mouse models of infection. Mice were maintained on a 12:12 light/dark cycle at 72 ± 2 °F in polycarbonate cages with a mixture of corncob and Alpha-dri bedding (Shepherd Specialty Papers Inc., Richard, MI). Mice were provided with Teklad 2818 extruded rodent diet (Harlan Laboratories, Indianapolis, IN) and water ad libitum. All procedures were approved and performed in accordance with the University of Notre Dame Institutional Animal Care and Use Committee.
Mouse Peritonitis Studies
Mice (n = 6 per group) were infected intraperitoneally with 0.5 mL of a S. aureus ATCC 27600, as previously described.22 The bacterial inoculum was adjusted to approximately 1 × 108 CFU/mL and mixed 50:50 with 10% mucin (Sigma-Aldrich, St. Louis, MO) to obtain the final inocula. Mice were given two iv doses of the compound, vehicle, or positive control (5 mg/kg vancomycin) at 30 min and 4.5 h after infection by tail-vein injection. Mice were monitored for 48 h, and the final number of surviving mice per group was recorded.
Fast PK Studies
Mice (n = 2 per time point) were given a single 50 μL iv injection of the test compound (equivalent to either 5 mg/kg, 10 mg/kg, or 20 mg/kg) via the tail vein, and terminal blood was collected in heparinized syringes by cardiac puncture at 5 min, 40 min, 2 h, 4 h, and 8 h. Compounds were dissolved in 25% DMSO/65% propylene glycol/10% water at a concentration of 2.5 mg/mL, 5 mg/mL, or 10 mg/mL, respectively, and filtered through a Acrodisc syringe filter (Pall Life Sciences, Ann Arbor, MI) prior to dosing.
Full PK Study
Mice (n = 3 per time point) were given a 50 μL tail vein iv injection of compound 27 (10 mg/kg), and terminal blood was collected at 2, 5, 10, 20, and 40 min and at 1, 2, 3, 4, 8, 12, 18, 20, and 24 h. A separate group of mice were given 100 μL of compound 27 (10 mg/kg) by oral gavage (po), and terminal blood was collected at 0.5, 1, 2, 3, 4, 6, 9, 16, 20, 24, and 30 h after po dosing. Compound 27 was dissolved in 25% DMSO/55% propylene glycol/20% PBS at a concentration of 5 mg/mL or 2.5 mg/mL, respectively, and filtered through an Acrodisc syringe filter prior to dosing.
PK Sample Analysis
Blood was centrifuged at 1200g for 10 min at 4 °C to obtain plasma, and samples were stored at −80 °C until analysis. A 50 μL aliquot of plasma was quenched with 100 μL of acetonitrile containing 10 μM internal standard (compound 73). Samples were centrifuged at 21 000g for 15 min at 4 °C to precipitate proteins, and the supernatant was analyzed by UPLC with UV/vis detection or mass spectrometry detection using multiple-reaction monitoring (MRM). Calibration curves for each compound from 0 to 50 μg/mL were prepared in control mouse plasma (50 μL) quenched with two volumes of acetonitrile containing internal standard. Compound concentrations were plotted against the peak area ratio relative to internal standard, and a linear regression was performed. Concentrations in plasma samples were determined from the linear regression parameters.
UPLC Analysis
Analysis was performed using a Waters Acquity UPLC system (Waters Corporation, Milford, MA) equipped with a binary solvent manager, column heater, photodiode array detector, and autosampler. An Acquity UPLC HSS C18 1.8 μm, 2.1 mm × 100 mm column was used. The following chromatographic conditions were used: 10% acetonitrile/90% water for 2 min, a 10 min linear gradient to 80% acetonitrile/20% water, and a 3 min linear gradient to 100% acetonitrile with a flow rate of 0.4 mL/min and UV/vis detection at 350 nm. Mass spectrometry analysis was performed using a Waters TQD tandem quadrupole detector (Waters Corporation, Milford, MA) and an Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 50 mm column. The mobile phases were supplemented with 0.1% formic acid, and the following chromatographic conditions were used: 30% acetonitrile/70% water for 0.1 min, a 2.9 min linear gradient to 97% acetonitrile/3% water, 97% acetonitrile/3% water for 0.5 min, and a 0.5 min linear gradient to 30% acetonitrile/70% water with a flow rate of 0.4 mL/min. Mass spectrometry acquisition was performed in the positive electrospray ionization mode with MRM. The capillary, cone, extractor, and RF lens voltages were set at 3.2 kV, 22 V, 2 V, and 0.2 V, respectively. The desolvation and cone gas (nitrogen) flow rates were 650 L/h and 50 L/h, respectively. The source temperature was set at 150 °C, and the desolvation temperature was 350 °C. The MRM transitions used were 393.4 → 274.4 for compound 27, 394.1 → 273.9 for compound 2, 338.2 → 296.1 for linezolid, and 464.1 → 418.4 for the internal standard.
PK Parameters
The area under the curve (AUC), clearance (CL), volume of distribution (Vd), and terminal half-life were calculated by noncompartmental analysis using uniform weighing with Phoenix WinNonlin 6.3 (Certara LP, St. Louis, MO). The remaining half-lives were estimated from the linear portion of the initial or terminal phase of the concentration–time curve by linear regression, where the slope of the line was the rate constant (k) and t1/2 = ln 2/k.
Plasma Protein Binding
The percent of plasma protein binding of compound 27 was determined in triplicate (with duplicate measurements of each replicate) in human plasma using a rapid equilibrium dialysis device as previously described.22 Briefly, compound 27 was added to the sample chamber to a final concentration of 20 μM and dialyzed in an orbital shaker for 2 h at 37 °C. Samples were quenched with acetonitrile containing internal standard and analyzed by UPLC with MRM detection.
Mouse Neutropenic Thigh Infection Model
Mice (n = 8 per group) were rendered neutropenic by ip dosing with cyclophosphamide (200 mg/kg) 4 days and 1 day prior to infection. The bacterial inoculum of NRS70 was prepared by suspending colonies into brain–heart infusion and adjusting to approximately 1 × 106 CFU/mL. A 100 μL aliquot of the bacterial inocula was injected intramuscularly into the right thigh. Mice were then administered three subcutaneous doses of 2 or 27, vehicle (negative control), or linezolid (positive control) every 8 h (1, 9, and 17 h after infection). Compounds 2 and 27 were administered at 20 mg/kg and linezolid at 10 mg/kg. After 24 h, the mice were euthanized by CO2 asphyxiation, and then terminal blood was collected by cardiac puncture in heparinized syringes and both thighs were surgically removed and transferred to sterile tubes. The thighs were weighed and an equal volume of sterile PBS was added. The infected thighs were homogenized using a bullet blender (Next Advance Inc., Averill Park, NY), and dilutions of the homogenate were prepared in triplicate and plated onto LB agar and incubated overnight. The colonies were then counted and averaged for each group and reported as log10(CFU/g tissue). Uninfected thighs were homogenized with a bullet blender and centrifuged at 15 000g for 5 min, and a 50 μL aliquot of the supernatant was mixed with 100 μL of acetonitrile containing internal standard and centrifuged at 21 000g for 15 min and the supernatant analyzed by UPLC with MRM detection. Plasma was analyzed following the procedure for PK samples.
MRSA Antisense-Mediated Sensitization
Compounds 27 and 30 were tested against MRSA COL strains with pbpA, pbp2, mecA, or pbp3 under antisense control using a previously described method.22 Briefly, overnight cultures were grown on agar supplemented with 34 μg/mL chloramphenicol. The next day, molten agar containing chloramphenicol and supplemented with either 0 mM or 50 mM xylose was cooled to 48 °C and seeded with 1 × 107 CFU/mL and allowed to set and dry. Compounds were spotted onto the agar (10 μL) and the plates incubated at 36 °C for 18–20 h.
Acknowledgments
This project is supported by Grant AI116548 from the National Institutes of Health (to M.C.). R.B. was supported by Training Grant T32GM075762 and by an individual Ruth L. Kirschstein National Research Service Award F31AI115851 from the National Institutes of Health and by an American Chemical Society Division of Medicinal Chemistry Predoctoral Fellowship. The antisense strains were generous gifts from Dr. Terry Roemer of Merck.
Glossary
Abbreviations Used
- AUC
area under the curve
- CL
clearance
- CFU
colony-forming unit
- iv
intravenous
- MBC
minimal bactericidal concentration
- MIC
minimal inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- MRM
multiple-reaction monitoring
- PBP
penicillin-binding protein
- PK
pharmacokinetics
- po
per os, oral administration
- UPLC
ultraperformance liquid chromatography
- UV
ultraviolet
- Vd
volume of distribution
- XTT
2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00372.
The authors declare no competing financial interest.
Supplementary Material
References
- Bassetti M.; Merelli M.; Temperoni C.; Astilean A. New antibiotics for bad bugs: where are we?. Ann. Clin. Microbiol. Antimicrob. 2013, 12, 22. 10.1186/1476-0711-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control (CDC). Antibiotic Resistance Threats in the United States, 2013; CDC: Atlanta, GA, 2013. [Google Scholar]
- Kong K. F.; Schneper L.; Mathee K. Beta-lactam antibiotics: from antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. 10.1111/j.1600-0463.2009.02563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitout J. D.; Sanders C. C.; Sanders W. E. Jr. Antimicrobial resistance with focus on beta-lactam resistance in gram-negative bacilli. Am. J. Med. 1997, 103, 51–59. 10.1016/S0002-9343(97)00044-2. [DOI] [PubMed] [Google Scholar]
- Biek D.; Critchley I. A.; Riccobene T. A.; Thye D. A. Ceftaroline fosamil: a novel broad-spectrum cephalosporin with expanded anti-Gram-positive activity. J. Antimicrob. Chemother. 2010, 65 (Suppl. 4), iv9–iv16. 10.1093/jac/dkq251. [DOI] [PubMed] [Google Scholar]
- Hartman B. J.; Tomasz A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 1984, 158, 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda C.; Hesek D.; Lee M.; Morio K.; Nowak T.; Mobashery S. Activation for Catalysis of Penicillin-Binding Protein 2a from Methicillin-Resistant Staphylococcus aureus by Bacterial Cell Wall. J. Am. Chem. Soc. 2005, 127, 2056–2057. 10.1021/ja0434376. [DOI] [PubMed] [Google Scholar]
- Otero L. H.; Rojas-Altuve A.; Llarrull L. I.; Carrasco-Lopez C.; Kumarasiri M.; Lastochkin E.; Fishovitz J.; Dawley M.; Hesek D.; Lee M.; Johnson J. W.; Fisher J. F.; Chang M.; Mobashery S.; Hermoso J. A. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16808–16813. 10.1073/pnas.1300118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes R. E.; Tsakris A.; Sader H. S.; Jones R. N.; Biek D.; McGhee P.; Appelbaum P. C.; Koswska-Shick K. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J. Antimicrob. Chemother. 2012, 67, 1321–1324. 10.1093/jac/dks069. [DOI] [PubMed] [Google Scholar]
- Long S. W.; Olsen R. J.; Mehta S. C.; Palzkill T.; Cernoch P. L.; Perez K. K.; Musick W. L.; Rosato A. E.; Musser J. M. PBP2a mutations causing high-level Ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2014, 58, 6668–6674. 10.1128/AAC.03622-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishovitz J.; Rojas-Altuve A.; Otero L. H.; Dawley M.; Carrasco-Lopez C.; Chang M.; Hermoso J. A.; Mobashery S. Disruption of allosteric response as an unprecedented mechanism of resistance to antibiotics. J. Am. Chem. Soc. 2014, 136, 9814–9817. 10.1021/ja5030657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodvold K. A.; McConeghy K. W. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin. Infect. Dis. 2014, 58 (Suppl. 1), S20–S27. 10.1093/cid/cit614. [DOI] [PubMed] [Google Scholar]
- Sahre M.; Sabarinath S.; Grant M.; Seubert C.; Deanda C.; Prokocimer P.; Derendorf H. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int. J. Antimicrob. Agents 2012, 40, 51–54. 10.1016/j.ijantimicag.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. L.; Pearson M. L.; Wilcox K. R.; Cruz C.; Lancaster M. V.; Robinson-Dunn B.; Tenover F. C.; Zervos M. J.; Band J. D.; White E.; Jarvis W. R. Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-Intermediate Staphylococcus aureus Working Group. N. Engl. J. Med. 1999, 340, 493–501. 10.1056/NEJM199902183400701. [DOI] [PubMed] [Google Scholar]
- Jones T.; Yeaman M. R.; Sakoulas G.; Yang S. J.; Proctor R. A.; Sahl H. G.; Schrenzel J.; Xiong Y. Q.; Bayer A. S. Failures in clinical treatment of Staphylococcus aureus Infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother. 2008, 52, 269–278. 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales G.; Picazo J. J.; Baos E.; Candel F. J.; Arribi A.; Pelaez B.; Andrade R.; de la Torre M. A.; Fereres J.; Sanchez-Garcia M. Resistance to linezolid is mediated by the cfr gene in the first report of an outbreak of linezolid-resistant Staphylococcus aureus. Clin. Infect. Dis. 2010, 50, 821–825. 10.1086/650574. [DOI] [PubMed] [Google Scholar]
- Sahm D. F.; Deane J.; Bien P. A.; Locke J. B.; Zuill D. E.; Shaw K. J.; Bartizal K. F. Results of the surveillance of Tedizolid activity and resistance program: in vitro susceptibility of gram-positive pathogens collected in 2011 and 2012 from the United States and Europe. Diagn. Microbiol. Infect. Dis. 2015, 81, 112–118. 10.1016/j.diagmicrobio.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Talbot G. H.; Bradley J.; Edwards J. E. Jr.; Gilbert D.; Scheld M.; Bartlett J. G. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 2006, 42, 657–668. 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- Lim D.; Strynadka N. C. J. Structural basis for the β-lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]
- Bouley R.; Kumarasiri M.; Peng Z.; Otero L. H.; Song W.; Suckow M. A.; Schroeder V. A.; Wolter W. R.; Lastochkin E.; Antunes N. T.; Pi H.; Vakulenko S.; Hermoso J. A.; Chang M.; Mobashery S. Discovery of antibiotic (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one. J. Am. Chem. Soc. 2015, 137, 1738–1741. 10.1021/jacs.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E.; Kerff F.; Terrak M.; Ayala J. A.; Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Ford C. W.; Hamel J. C.; Wilson D. M.; Moerman J. K.; Stapert D.; Yancey R. J. Jr.; Hutchinson D. K.; Barbachyn M. R.; Brickner S. J. In vivo activities of U-100592 and U-100766, novel oxazolidinone antimicrobial agents, against experimental bacterial infections. Antimicrob. Agents Chemother. 1996, 40, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebeisen P.; Heinze-Krauss I.; Angehrn P.; Hohl P.; Page M. G.; Then R. L. In vitro and in vivo properties of Ro 63–9141, a novel broad-spectrum cephalosporin with activity against methicillin-resistant staphylococci. Antimicrob. Agents Chemother. 2001, 45, 825–836. 10.1128/AAC.45.3.825-836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Pankey G. A.; Sabath L. D. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin. Infect. Dis. 2004, 38, 864–870. 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- Trainor G. L. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discovery 2007, 2, 51–64. 10.1517/17460441.2.1.51. [DOI] [PubMed] [Google Scholar]
- Hegde S. S.; Okusanya O. O.; Skinner R.; Shaw J.; Obedencio G.; Ambrose P. G.; Blais J.; Bhavnani S. M. Pharmacodynamics of TD-1792, a Novel Glycopeptide-Cephalosporin Heterodimer Antibiotic Used against Gram-Positive Bacteria, in a Nuetropenic Murine Thigh Model. Antimicrob. Agents Chemother. 2012, 56, 1578–1583. 10.1128/AAC.05382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Jarantow L. W.; Wang H.; Sillaots S.; Cheng H.; Meredith T. C.; Thompson J.; Roemer T. Antagonism of chemical genetic interaction networks resensitize MRSA to beta-lactam antibiotics. Chem. Biol. 2011, 18, 1379–1389. 10.1016/j.chembiol.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Wikler M. A.; Cockerill F. R.; Craig W. A.; Dudley W. A.; Eliopoulos G. M.; Hecht D. W.; Hindler J. F.; Low D. E.; Sheehan D. J.; Tenover F. C.; Turnidge J. D.; Weinstein M. P.; Zimmer B. L.; Ferraro M. J.; Swenson J. M.. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard; Document M7-A7; Clinical Laboratory Standards Institute, 2009; Vol. 29. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.