

Abstract
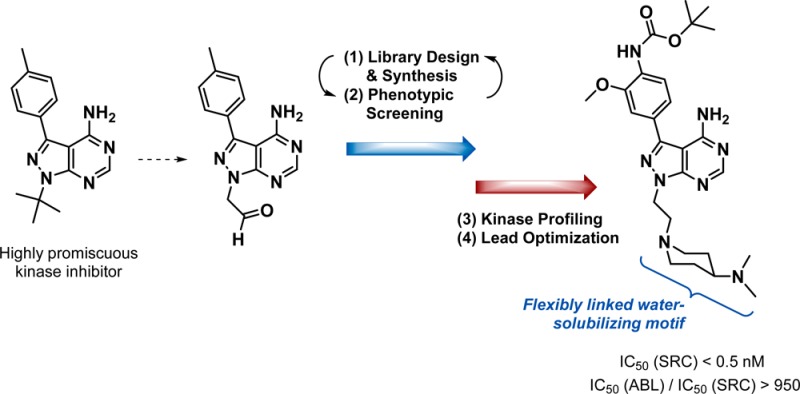
Novel pyrazolopyrimidines displaying high potency and selectivity toward SRC family kinases have been developed by combining ligand-based design and phenotypic screening in an iterative manner. Compounds were derived from the promiscuous kinase inhibitor PP1 to search for analogs that could potentially target a broad spectrum of kinases involved in cancer. Phenotypic screening against MCF7 mammary adenocarcinoma cells generated target-agnostic structure–activity relationships that biased subsequent designs toward breast cancer treatment rather than to a particular target. This strategy led to the discovery of two potent antiproliferative leads with phenotypically distinct anticancer mode of actions. Kinase profiling and further optimization resulted in eCF506, the first small molecule with subnanomolar IC50 for SRC that requires 3 orders of magnitude greater concentration to inhibit ABL. eCF506 exhibits excellent water solubility, an optimal DMPK profile and oral bioavailability, halts SRC-associated neuromast migration in zebrafish embryos without inducing life-threatening heart defects, and inhibits SRC phosphorylation in tumor xenografts in mice.
Introduction
Modern drug discovery programs typically begin with a screening campaign (e.g., biochemical, virtual, or biophysical) for agonists, antagonists, or inhibitors of a nominated target associated with a particular disease.1−4 After hit identification, subsequent chemical optimization is fundamentally based upon “on-target” potency.1 The generation of high-affinity ligands (so-called lead compounds) is followed by chemical refinement into derivatives of superior potency, selectivity, and desirable pharmacokinetic properties.1,5 Selected drug candidates are then validated in vivo and, upon verification of safety and efficacy, progressed to human trials.5 While the merits of this well-defined process are undeniable, including several major breakthroughs in anticancer therapy,6 this strategy is also associated with declining productivity in the pharmaceutical industry and limited success to tackle the most aggressive cancers of unmet therapeutic need.7,8 High attrition rates at late stages of drug development underlines that cancer heterogeneity across patients and adaptive drug resistance mechanisms are major obstacles for the development of effective and long-lasting anticancer targeted therapies.9−12 These challenges have stimulated out-of-the-box thinking in pharmacotherapy research (e.g., targeted polypharmacology,10 antibody-drug conjugates,13 innovative prodrug approaches,14−17 etc.) and the re-examination of the core principles of drug discovery in complex diseases.18−20 The rise of modern phenotypic drug discovery18,19 together with the use of more clinically relevant disease models to guide early drug development20 are representative examples of the paradigm shift initiated in the field to trigger a positive inflection point.
Protein kinases are integral components of signal transduction cascades. They govern a wide range of basic intracellular functions and coordinate cell-to-cell and extracellular matrix-to-cell communication to modulate cell and tissue physiology. Consequently, their malfunctioning is directly linked to progressive diseases including cancer and inflammation.21 The success in the clinic of several anticancer kinase inhibitors has validated a number of kinases as oncotargets,22 while the increasing understanding of cancer cell biology has demonstrated the essential role of different kinases in tumor suppressor pathways (antitargets).23−26
The vast majority of kinase inhibitors bind to the kinase adenosine triphosphate (ATP) pocket. Since all kinases (>500) necessarily possess this relatively well-conserved catalytic site, there is a great potential for cross-reactivity.10 In fact, even though most kinase inhibitors are developed from single target hypotheses, they typically display broad selectivity profiles which, in some cases, have resulted in unanticipated clinical applications (e.g., sorafenib).26 Inhibitor promiscuity may also be advantageous for anticancer therapy when off-target activities assist to address bioactivity issues related to pathway redundancies, molecular heterogeneity, or resistance mechanisms.9,10,26 However, if these activities result in the inhibition of antioncogenic pathways or lead to severe side effects, drug promiscuity becomes a major drawback.27,28 Paradoxically, some kinases may behave as a target or an antitarget depending on the cancer context. The expression of the activated fusion oncoprotein BCR-ABL is a genetic abnormality associated with chronic myeloid leukemia (CML), and ABL inhibitors (imatinib, dasatinib) are clinically used in chronic phase CML treatment.29 Also, ABL family kinases are abnormally activated in various solid tumors, supporting their involvement in oncogenesis.29 However, ABL (ABL1) and ARG (ABL2) have been found to negatively modulate breast cancer progression in vivo,30−32 indicating that ABL inhibition could be counterproductive for breast cancer treatment (= antitarget). This example serves to delineate the complexity of cancer etiology and highlights the necessity of developing kinase inhibitors with tailor-made pharmacodynamic profiles for the effective targeting of each cancer subtype.33 Unfortunately, despite significant investments in the development of kinase inhibitors and the biomedical knowledge compiled over several decades, our still limited understanding of cancer biology prevents us from anticipating and optimally targeting the complex orchestrated actions that generate, maintain, and progress most neoplastic processes.
Acknowledging these limitations, many research groups including ours are frontloading the collection of robust empirical data to progress anticancer drug development programs away from classical black-and-white anticancer target hypotheses to more unbiased and evidence-led strategies for hit selection and lead generation. Following that principle, in this manuscript we show that cooperative ligand-based design and phenotypic screening, complemented with biochemical assays and the use of published data (literature, patents, etc.), can be effectively applied to accelerate the generation of preclinical drug candidates. Our strategy builds on three wide-ranging hypotheses: (i) targeting the kinase ATP pocket with compounds derived from promiscuous kinase inhibitors can enable “rationally-biased” serendipitous discoveries; (ii) early optimization of drug-likeness can be concurrently applied to explore pharmacodynamic diversity; and (iii) phenotypic screening of chemically related compounds in designated models of cancer can generate target-agnostic structure–bioactivity relationships and tailor ligand optimization to particular cancer types/subtypes. By means of this pragmatic approach to anticancer kinase inhibitor discovery, target deconvolution of identified hits and leads is largely simplified, thereby assisting the mechanistic elucidation of the molecular targets and antitargets involved in the observed phenotype.
In this work we describe how the implementation of such an approach led to the discovery of a kinase inhibitor with potent activity against breast cancer cells and a unique selectivity profile: the first small molecule able to inhibit SRC at subnanomolar levels with a 1000-fold selectivity over ABL.
Results and Discussion
Design, Synthesis, and Screening of Compounds 7a–l
PP1 is a promiscuous inhibitor that indiscriminately targets protein tyrosine kinases, many of which are involved in oncogenesis such as the SRC family kinases (SFK), RET, KIT, and ABL.34−37 Moreover, related derivatives developed thereafter38−40 have shown strong inhibition of a variety of kinases with relevance in cancer including IGF-1R, EGFR, BTK, VEGFR, PDGFR, PI3K, and mTOR. According to the co-crystal structure of PP1 with HCK41 and RET42 kinases, this small molecule is an archetypical type I kinase inhibitor, with its N5 and 4-NH2 groups forming multiple H-bonds with the hinge region of the kinase ATP site (Figure 1). The C3 p-tolyl group is positioned toward a hydrophobic region well-conserved across tyrosine kinases, thus being responsible for the partial selectivity of PP1 over other kinase families. Although PP1’s potent inhibition of disease-associated kinases make it a valuable biological tool, its use is limited by low solubility in water and poor selectivity, major limiting factors for the clinical translation of many drug candidates.
Figure 1.
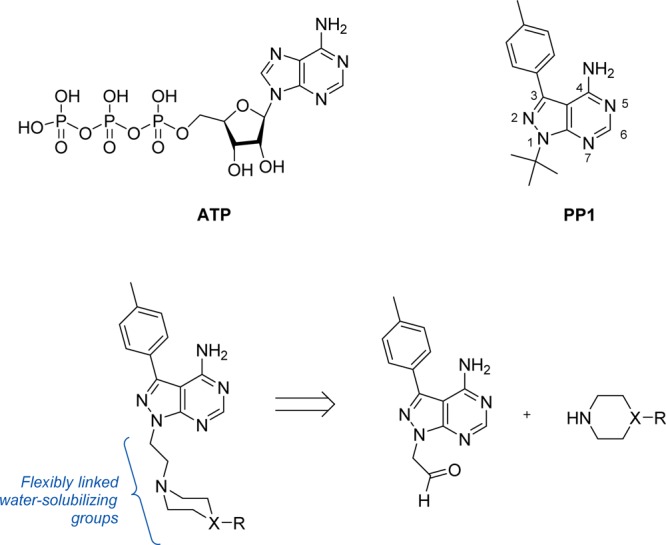
Upper panel: ATP and PP1 (neutral forms). Lower panel: General structure of the novel compounds and retrosynthetic analysis.
We envisaged that the substitution of PP1’s tert-butyl group at the N1 position with flexible water-solubilizing groups could be used to improve drug-like properties and, at the same time, explore the accessible sugar/phosphate regions occupied by the natural ligand ATP in the search for novel binding affinity profiles. As shown in Figure 1, compounds were designed to display a cyclic tertiary amine connected to the N1 position of the pyrazolopyrimidine ring through an ethylene linker. Following the route described in Scheme 1 (see full details in the Supporting Information), a highly focused 12-member library was generated by coupling a selection of cyclic secondary amines to an aldehyde-modified derivative43 of PP1 (6) via reductive amination.
Scheme 1. Five-Step Synthesis of Compounds 7a–l from Commercially Available 5-amino-1H-pyrazole-4-carbonitrile 1.

Compound 6 was employed as the common intermediate to generate compounds 7a–l by reductive amination using 12 commercially available cyclic secondary amines (piperidines, morpholine, and piperazines).
The 12-member library was then tested against human mammary adenocarcinoma MCF7 cells, using the inhibition of cell growth as the primary output of the screening. Live-cell high-content image-based phenotypic assays were carried out to kinetically profile the antiproliferative response of MCF7 breast cancer cells following treatment with a single dose (100 μM) of compounds 7a–l for 5 d. Experiments were performed in triplicate, using cells treated with PP1 (100 μM) or DMSO as a positive and negative control, respectively. Cell proliferation was monitored by time-lapse imaging using an IncuCyte ZOOM microscope and analyzed by its software, enabling determination of cell density (% confluence) over time after compound addition. As shown in Figure 2a, compounds 7d and 7i–l (colored lines) led to a strong inhibition of cell proliferation, maintaining confluence levels equal or below those imaged at time zero (point of compound addition). Interestingly, all these hits presented at least two tertiary amines in their N1 motifs, providing the first insights into structure/antiproliferative activity relationships.
Figure 2.

(a) Time-lapse imaging analysis of breast cancer MCF7 cell proliferation over 5 d. Cells were imaged every 3 h using an IncuCyte ZOOM microscope and growth measured by its integrated software. Curves represent % confluence of cells treated with compounds 7a–l (100 μM, 0.1% DMSO) over 5 d of incubation. DMSO (0.1%, v/v) was used as untreated cell control (in black). Error bars: ± SD from n = 3. (b) Dose–response curves and calculated EC50 values determined by PrestoBlue cell viability assay after incubation of MCF7 cells with compounds 7d and 7i–l and the positive control PP1 (dose range: 0.03–100 μM). Error bars: ± SD from n = 3.
Half-maximal effective concentration (EC50) values were then calculated for the five hits and PP1 in MCF7 cells using an 8-point half-log dose–response study (0.03 to 100 μM). Based on the MCF7 growth kinetics data provided by the IncuCyte assay, cell viability was determined at day 5 using the PrestoBlue reagent. Figure 2b shows that derivatives 7j and 7k were the most potent among the novel compounds, with EC50 values of 12 μM. Due to its potency and lower molecular weight, the dimethylamino-containing piperidinyl group of compound 7j was chosen as the N1 motif for the preparation of a second library of pyrazolopyrimidines.
Design, Synthesis, and Screening of Compounds 8a–f and 9a–f
Several investigations have reported that substitutions of the p-tolyl group at the C3 position of PP1 by different aryl moieties (even closely related ones) have a major impact on protein–ligand binding. Medicinal chemistry at that position has generated inhibitors for a variety of kinases, including receptor and nonreceptor tyrosine kinases (e.g., SRC, ABL, RET, EGFR, BTK, PDGFRs, VEGFRs, KIT)34−38,40 and nontyrosine kinases (e.g., PI3Ks, mTOR).39,44 To exploit this feature as an opportunity to expand the prospective pharmacodynamic scope of 7j, a selection of five arylboronic acids and phenylacetylene were employed to functionalize the C3 position of the heterocyclic ring by palladium-catalyzed cross-coupling chemistry to synthesize derivatives 9a–f (Scheme 2). Together with the piperidine-containing compounds 9a–f, the corresponding acetal intermediates 8a–f were selected for testing against breast cancer cells to increase the chemical diversity of the screening.
Scheme 2. Second Library of Pyrazolopyrimidines: Compounds 8a–f and 9a–f.

Antiproliferative properties of compounds 8a–f and 9a–f were evaluated in MCF7 cells (dose–response studies), using compound 5 (acetal intermediate of library 1), 7j (most potent derivative of library 1), and PP1 as controls. As shown in Figure 3, compounds 8d and 9d (both having a 7-azaindol-5-yl group at C3) exhibited superior antiproliferative properties (EC50 < 2 μM). Remarkably, time-lapse imaging of compounds 8d and 9d (1–100 μM) revealed patently distinct antiproliferative mode of actions (Figure 3b and Movies S1 and S2). Compound 9d—containing the piperidinyl moiety of 7j—induced cell death, while its acetal precursor 8d halted cell division.
Figure 3.

(a) EC50 values calculated after treatment of MCF7 cells with acetal-functionalized compounds 5 and 8a–f (top panel) and piperidine-functionalized derivatives 7j and 9a–f (bottom panel). Dose range: 0.03–300 μM. DMSO and PP1 were used as negative and positive controls, respectively. Cell viability was determined using the PrestoBlue reagent. (b) IncuCyte ZOOM images of MCF7 cells treated with 8d, 9d, and DMSO at 0, 48, and 120 h after compound addition. Inhibitor dose: 100 μM.
To identify the target/s that could be responsible of the phenotype induced by hits 8d and 9d in MCF7 cells and thereby shed some light over further optimization campaigns, inhibition activities were tested for PP1, 7i–k, 8d, and 9d against a selection of kinases involved in human cancer. Proteins were chosen in accordance with the profile of related pyrazolopyrimidines.34−39 Kinase profiling (Reaction Biology Corp., USA) was performed by measuring 33P incorporation on the substrate (poly [Glu, Tyr] 4:1) relative to DMSO. Dose–response curves and calculated half-maximal inhibitory concentration (IC50) values are shown in Figure S1 and Table 1, respectively. Derivatives 7i–k and 9d strongly inhibited SFK members, whereas acetal-functionalized compound 8d preferentially inhibited mTOR (>28-fold more potent than PP1). The most potent inhibitor was compound 9d, which displayed similar potency to PP1 against SRC but higher selectivity over RET, PDGFRα, and KIT. Interestingly, according to the inhibition profile of compounds 7i–k (only varying on the spacer between the dimethylamino group and the piperidine), potency and selectivity for SRC were enhanced by the proximity of the dimethylamino to the ring (IC50(ABL)/IC50(SRC) for 7i = 50).
Table 1. IC50 Values (in nM) Calculated for PP1, 7i–k, 8d, 9d, 11a, and Dasatinib in a Selection of Recombinant Kinases.
| kinase \ hit | PP1 | 7i | 7j | 7k | 8d | 9d | 11a | dasatinib |
|---|---|---|---|---|---|---|---|---|
| ABL | 147 | 6323 | 7525 | 7249 | 1207 | 116 | 479 | <0.5 |
| FYNa | 27 | 311 | 913 | 1964 | 1226 | 38 | 2.1 | <0.5 |
| KIT | 1318 | >104 | >104 | >104 | >104 | >104 | >104 | 39 |
| mTOR | 9318 | >104 | >104 | >104 | 328 | 8565 | >104 | >104 |
| PDGFRα | 657 | >104 | >104 | >104 | >104 | >104 | >104 | 9.9 |
| RET | 11 | 6584 | >104 | >104 | 598 | 289 | >104 | 433 |
| SRCa | 17 | 126 | 303 | 1040 | 2453 | 27 | <0.5 | <0.5 |
| YESa | 36 | 71 | 344 | 696 | 566 | 12 | <0.5 | <0.5 |
| IC50(ABL) /IC50(SRC) | 8 | 50 | 25 | 7 | 0.5 | 4 | >950 | 1 |
Member of SFK.
Using the information provided by the phenotypic screening and the kinase inhibition profiling of leads 8d and 9d, two independent optimization campaigns were implemented toward the generation of anticancer drug candidates inhibiting mTOR and SFK, respectively. Optimization of lead 8d into the highly selective mTOR inhibitor eCF309 has been reported elsewhere.45 Herein is described the generation and structure–activity relationships (SAR) of pyrazolopyrimidine derivatives with high potency and selectivity for SFK.
Design, Synthesis, and Phenotypic Screening of Compounds 11a,b
The data collected from the kinase profiling study indicated that the 2-[4-(dimethylamino)-1-piperidyl]ethyl group at the N1 position was optimal to generate SFK inhibitors of high selectivity, while the superior potency of 9d against SRC suggested that on-target potency could be further enhanced by optimization of the substituent at the C3 position. A literature survey on SRC inhibitors was thus performed to assist in the design of derivatives with improved SRC binding. We focused our attention on the C3 substituent of PP20, a potent dual SRC/ABL inhibitor developed by Apsel et al. (Figure 4a).36 Novel derivatives were designed by substituting the 7-azaindol-5-yl group at C3 of 9d with the functionalized phenyl ring found in PP20, whereas the piperidines used for 7i and 7j were introduced at the N1 position. Acetal deprotection of compound 4 in TFA/water (1:1) followed by reductive amination with either 4-(dimethylamino) or 4-(dimethylaminomethyl)piperidine generated iodo intermediates 10a,b, respectively, which afforded derivatives 11a (also known as eCF506) and 11b (Figure 4a) by Suzuki cross-coupling with 4-(N-Boc-amino)-3-methoxyphenylboronic acid (see synthesis in the Supporting Information).
Figure 4.

(a) Structures of PP20 and compounds 11a and 11b. (b) EC50 values calculated after treating MCF7 and MDA-MB-231 cells with compounds 11a, 11b, PP20, and dasatinib (dose range: 1–10,000 nM) for 5 d. Cell viability was determined using the PrestoBlue reagent. Error bars: ± SD from n = 3. (c) Western blot analysis of SRC and FAK activity in MDA-MB-231 cells treated with 11a and dasatinib for 1.5 h. (d) Scratch-wound migration assay. MDA-MB-231 cells were treated with 11a or dasatinib (10 nM), and cell migration compared with untreated cell control (DMSO, 0.1%, v/v) at 6, 12, and 24 h. Cells were imaged and analyzed using an IncuCyte-ZOOM microscope with integrated scratch-wound migration software module. Error bars: ± SD from n = 3; p < 0.001, ***; p < 0.01, **. P value calculated from t-test. (e) Snapshots of the scratch-wound area of MDA-MB-231 cells at 0 and 24 h. Yellow lines highlight the gap formed by the scratch. Blue lines indicate the advance of cells into the wound after 24 h incubation in the absence and presence of 11a (10 nM).
Antiproliferative properties of compounds 11a,b were then tested in cells using PP20 and dasatinib (a clinically approved dual SRC/ABL inhibitor)22 as positive controls. Along with MCF7 cells, breast adenocarcinoma MDA-MB-231 cells were tested as a model of triple-negative breast cancer known to be particularly sensitive to SRC inhibitors.46 As shown in Figure 4b, compound 11a induced a very potent antiproliferative effect in both MCF7 and MDA-MB-231 cells, significantly superior to the activity displayed by derivatives 9d, 11b, and PP20. Notably, 11a also outperformed the gold-standard SRC inhibitor dasatinib.
Compound 11a was then selected for further screening. Inhibition of SRC kinase activity in cells was studied by Western blot. Upon activation, SRC undergoes autophosphorylation of the Y416 residue within its kinase domain.47 Study of phospho-SRCY416 levels can then serve both as evidence for the presence of active SRC and to evaluate direct inhibition of SRC kinase activity. Phosphorylation of the focal adhesion kinase (FAK), a downstream substrate of SRC with high relevance in cancer progression,48 was also studied in both MDA-MB-231 (Figure 4c) and MCF7 (Figure S2) cells. Dasatinib was used as a positive control. Cell lysates were prepared following 24 h of serum starvation and 1.5 h exposure to each inhibitor and serum stimulation for 1 h. Analyses demonstrated that 11a inhibits phosphorylation of SRC and FAK at low nanomolar levels, with complete inhibition observed at 100 nM. Dasatinib activity was close to that of 11a, although it also induced a dose-dependent increment of total SRC (Figure 4c).
Having demonstrated that 11a targets SRC in cells, we investigated FAK activity and cell viability in cells that lack expression of SRC. SYF murine embryonic fibroblasts (SRC–/–, YES–/–, FYN–/−) were treated with either 11a or dasatinib and levels of total/phospho-FAK analyzed by protein immunoblot. As expected (Figure S3), FAK activity was found to be unaffected by drug treatments, indicating that the inhibition of phospho-FAKY861 in the breast cancer cell lines was a consequence of SRC inhibition. Compounds 11a and 11b displayed significantly less antiproliferative activity in SYF cells than dasatinib, suggesting increased selectivity against SRC family kinases (Figure S3).
An automated 96-well scratch-wound cell migration assay (analyzed in real-time by the IncuCyte ZOOM system) was then set up to determine whether compound 11a could halt migration of MDA-MB-231 cells, as would be expected for a SRC inhibitor.49 Cells treated with 11a were monitored for 24 h at a dose range (1–10,000 nM) and compared with untreated cells (0.1% v/v DMSO) and dasatinib treatment. Compound 11a significantly reduced cell motility at 10 nM as early as 6 h into the study, with equivalent efficacy to dasatinib (Figure 4d,e). 11a treatments at higher concentrations induced greater inhibition of cell migration (Figure S4), although cell viability was also significantly reduced. Image-based measurement of caspase 3/7 activity in MCF7 breast cancer cells demonstrated significant levels of apoptotic cell death after 5 d treatment with 11a at concentrations ≥100 nM (Figure S4), in accordance with the EC50 values observed for this cell line.
Kinome Profiling of SRC Inhibitor 11a
IC50 values were determined for 11a and dasatinib against the panel of recombinant kinases used in previous screenings (see IC50 values in Table 1 and dose–response curves in Figure S5). As expected, dasatinib induced potent inhibition of nonreceptor tyrosine kinases, such as ABL and SRC,22 but also high potency against receptor tyrosine kinases (e.g., PDGFRα, KIT). In contrast, 11a exclusively inhibited SFK, with subnanomolar IC50 values against SRC and YES (activities for the rest of the SFK members are shown in Table S1). It is important to highlight that 11a displayed a vast difference in activity (>950-fold difference) between ABL and its primary target SRC. While small molecule inhibitors with reverse binding capabilities have been developed (e.g., imatinib strongly targets ABL without inhibiting SRC),49 this is, to the best of our knowledge, the first case of a small molecule with subnanomolar IC50 for SRC that requires a concentration 3 orders of magnitude greater to reach the same level of inhibition in ABL. Such properties have only been previously reported in peptide-based bisubstrate inhibitors of SRC.50
Design and Synthesis of Compounds 12a–x and SAR Analysis
To shed light over the structural features responsible for the high antiproliferative properties and unique selectivity profile of 11a, a library of 24 closely related analogs was developed (see syntheses in the Supporting Information) by introducing small changes on key functional groups of 11a (see Figure 5). The activity of compounds 12a–x was then tested in cells. MDA-MB-231 was the cell line chosen for the screening due to its superior sensitivity to 11a treatment. Cell viability assays were performed as previously described, and EC50 values calculated for each compound. IC50 values were subsequently determined against SRC and ABL for those compounds exhibiting high antiproliferative activity (12d, 12e, 12m, 12w, and 12x) and for selected compounds with low activity (12a, 12i, 12j, 12t, and 12v).
Figure 5.

Compounds 12a–x and their biological activities. Structural motifs modified on 11a for the generation of compounds 12a–x are highlighted as colored circles. EC50 values against MDA-MB-231 breast cancer cells are shown below each structure. SRC and ABL inhibition values (as IC50) are reported for selected compounds.
Analysis of the properties of compounds 12a–e (Figure 5) indicates that the presence of a tertiary amino group at the position 4 of the pyperidinyl ring is essential for the activity. Substitution of the methyl groups by larger aliphatic rings was tolerated, however, with compounds 12d and 12e being among the most potent SRC inhibitors of the series. Removal of the pyrimidine ring (see compound 12f) resulted in >3000-fold decrease in antiproliferative activity. Screening of compounds 12g–x evidenced the limited structural variations permitted at the top of the molecule to achieve high bioactivity. Most modifications of the Boc and the methoxy groups of the phenyl moiety at the C3 position led to a significant reduction in bioactivity. Even minor chemical modifications such as the substitution of the carbamate group by urea (12i) or amide (12j), or the change of the methoxy by OH (12s), resulted in >200-fold decrease in both antiproliferative properties and SRC inhibitory activity. Remarkably, introduction of a benzylamino group instead of the Boc was tolerated well, as observed from the bioactivity exhibited by compound 12m. However, introduction of endocyclic nitrogen atoms in the ring (see 12n and 12o) dramatically reduced compounds’ activity, particularly in the ortho position. Interestingly, while the methoxy group is required for maintaining high levels of activity when the Boc group is present in the structure (see 12s–u), substitution of the Boc group by an ester tolerated the elimination of the methoxy group (see 12w and 12x).
Biochemical and in Silico Studies of 11a Binding to the SRC Kinase
Most kinase inhibitors, especially adenine analogues, bind to the catalytic domain of the enzyme in its active conformation,22 thus competing with the natural substrate ATP. To determine whether this was the mode of binding of 11a to its primary target SRC, competitive inhibition studies with ATP were performed (Reaction Biology Corp., USA). Experiments were designed to obtain Km values for ATP in the absence and presence of different concentrations of inhibitor 11a. The reactions were monitored every 10 min to obtain progress curves with time course (Figure S6). These were found to be linear regardless of the compound’s concentration, suggesting that SRC inhibition by 11a is not time-dependent (= reversible). The slopes (μM/min) were represented against ATP concentration for a Michaelis–Menten plot and a subsequent Lineweaver–Burk plot (double-reciprocal plot), using GraphPad Prism software (Figure S6). Apparent Km increased when inhibitor concentration increased and all lines converged on the Y-axis in the double-reciprocal plot, thus suggesting that 11a is competitive with respect to ATP against SRC. Dose–response curves of SRC inhibition by 11a at different concentrations of ATP are plotted in Figure S7. Calculated IC50 values increased in direct proportion to ATP concentration, further confirming that 11a is a type I kinase inhibitor and therefore binds to the SRC ATP site in its active conformation. Global fit analysis using GraFit software provided a Ki of 0.8 nM (Figure S7).
To gain insight on the selective binding affinity of 11a for SRC over ABL, in silico docking studies were performed with both the dual ABL/SRC inhibitor PP20 and the SRC-selective inhibitor 11a. Docking of PP20 into SRC and ABL produced a plausible predicted binding mode in both cases (Figure 6a,b), with the adenine-mimicking pyrazolopyrimidine making H-bonds to the hinge region of the proteins. The tert-butyl group of the carbamate occupies a hydrophobic pocket formed by several lipophilic residues in SRC and ABL, including the Phe of their aspartate-phenylalanine-glycine (DFG) motif. Free energy of binding was predicted by Autodock to be approximately the same for both proteins (−8.5 and −8.9 kcal/mol, respectively). Docking of 11a into SRC also produced a plausible predicted binding mode (Figure 6c) and a higher predicted affinity than PP20 (calculated free energy of binding of −10.6 kcal/mol). In SRC, the dimethylamino group of the ethylpiperidinyl moiety on N1 interacts with Asp404, one of the protein’s phosphate binding residues (see Figure 6c,e).21,22 In contrast, the large N1 group of 11a (compared to the isopropyl on PP20) clashes with Tyr253 in the catalytic site of ABL. This resulted in Autodock being unable to find a position in the ATP binding site of ABL where 11a could mimic the interactions made by the adenine moiety of ATP (Figure 6d). This study indicates that the flexible polyamine linker on N1 is responsible for the low affinity of compound 11a for ABL.
Figure 6.

(a-d) Predicted binding modes of PP20 and 11a in SRC and ABL kinases. (a) PP20 in SRC, (b) PP20 in ABL, (c) 11a in SRC, and (d) 11a in ABL. The P-loop, αC helix, and DFG motif are in red, orange, and magenta, respectively. PP20 and 11a are represented by sticks, where carbon atoms are in black, nitrogen atoms are in blue, and oxygen atoms are in red. (e) 11a in SRC active site compared to adenosine monophosphate (AMP) (from PDB 3DQX). AMP is represented by sticks, where carbon atoms are in green, nitrogen atoms are in blue, oxygen atoms are in red, and the phosphorus atom in orange. Predicted binding mode indicates that the dimethylamino group at the piperidine ring is optimally placed to interact with the carboxylic group of Asp404 (DFG motif, in pink).
Phenotypic Screening of SRC Inhibitor 11a in Zebrafish
Developing zebrafish provides a rapid phenotypic assay to simultaneously test safety and efficacy of novel compounds in a living vertebrate.51 Small molecule phenotypic-based screens in zebrafish have recently implicated SRC kinase in the migration of the posterior lateral line primordium,52 a cohesive cluster of cells that migrates horizontally under the skin along the myoseptum to the end of the tail, periodically depositing neuromasts. To determine the effects of 11a on cell migration in vivo, we treated Tg(brn3c:mGFP) transgenic zebrafish53 that express green fluorescent protein (GFP) in the mechanosensory hair cells of the lateral line (which form part of the neuromasts) with 11a for 2 d and measured the distance of the last neuromast to the tip of the tail (marked by the end of the notochord and the presence of black melanocytes, Figure 7a, in red). 11a significantly reduced neuromast migration (>100 μm in average) with minimal effect on the development of the embryos (Figure 7a–c). In contrast, dasatinib treatment at >10 μM resulted in severe cardiotoxicity and death of most embryos. At concentrations that were compatible with embryo survival (1–10 μM), dasatinib did not inhibit the migration of neuromasts, whereas it did still induce a patent cardiotoxic phenotype (note heart enlargement in Figure 7c). Further safety studies (see Figure S8) showed that dual ABL/SRC inhibitor PP20 also induces severe cardiotoxicity in zebrafish even after short treatment. These results, which correlate with the essential role of ABL in heart development and healing,54,55 suggests that the selectivity of 11a over ABL might be advantageous for therapy when ABL inhibition is not required.
Figure 7.

(a,b) Neuromast migration assay. Fresh E3 media with DMSO or 11a (500 μM) was added to zebrafish embryos at 20 hpf, 36 hpf, and 48 hpf and imaged at 72 hpf. (a) Representative images of the tail of a 3 dpf zebrafish without (top) and with 11a treatment (bottom). Neuromasts are identified by GFP expression (green) and the tip of the notochord as a red line. Yellow arrow indicates shortest distance from tail tip to a neuromast. (b) Imaging analysis of the distance between the last neuromast and the tip of the tail (n = 3) under treatment with DMSO (negative control) or 11a (500 μM). P value calculated from t-test. (c) Study of zebrafish heart development under short treatment with 11a (500 μM) and dasatinib (10 μM). Compounds were added to 2 dpf zebrafish embryos and incubated for 4 h (n = 3). Subsequently, fresh media was added, and the fish imaged after 48 h incubation.
Physicochemical and Drug Metabolism and Pharmacokinetics (DMPK) Profiling of SRC Inhibitor 11a in Vitro and in Vivo
Compound 11a displays very high solubility in water (>100 mg/mL, pH = 7.4), a significant advantage over dasatinib, whose solubility in water ranges from moderate (18 mg/mL at pH = 2.6) to extremely low (<0.001 mg/mL at pH = 7).56 Notably, analysis of cLogP with the OSIRIS Property Explorer57 provided an estimated value of 2.2 for 11a, while experimental log D at pH = 7.4 was determined to be −0.04.
Inhibition of hERG channel and cytochrome P450 (CYP) enzymes was determined for 11a to assess off-target liabilities associated with cardiotoxicity (QT interval prolongation) and potential drug–drug metabolic interactions. Results demonstrated weak hERG inhibition (50.1% inhibition at 25 μM) and marginal inhibition of CYP enzymes at 10 μM (Figure S9).
A plasma protein binding assay was performed with compound 11a to test to what extent the compound can bind to proteins in the blood, an important factor to predict free drug levels in the body. Warfarin (positive control, 99–100% bound) and 11a were incubated with human or rat plasma in duplicates at 37 °C for 24 h and the free (unbound) fraction of compound determined by LC-MS/MS. Percentages of unbound 11a were 9.4 and 19.1% in rat and human plasma, respectively.
In vitro metabolic stability of 11a was systematically assessed in human liver microsomes (HLM), blood plasma, and primary hepatocytes (Cyprotex, UK). LC-MS/MS analysis of 11a showed excellent compound stability (94%) after 30 min incubation with HLM at 37 °C. 11a was subsequently incubated (1 μM, 2 h, 37 °C) with plasma from either human, mouse, or rat and analyzed by LC-MS/MS at different time points. Regardless of the species, analyses showed full plasma stability (Figure S10). Lastly, compound 11a (3 μM) was incubated with mouse and human primary hepatocytes for 60 min and samples analyzed by LC-MS/MS at various time points. Verapamil and umbelliferone were used as control compounds. In agreement with the HLM study, compound 11a showed high stability in human hepatocytes with a half-life of 751 min (Figure S10). As expected, faster clearance was observed in mice hepatocytes (7-fold decrease in half-life).
Encouraged by the in vitro DMPK profile of 11a, an in vivo study was performed in mice to determine oral bioavailability and half-life (Cyprotex, U.K.). Female CD1 mice were given a single dose of 10 mg/kg (0.25 mg/25 g mouse) of compound 11a orally (as a solution in nanopure water) or via IV injection (as a saline solution). Blood/plasma samples were taken at different time points (final sample taken after 8 h), and compound levels measured by LC-MS/MS (Figure S11). The calculated half-life value for 11a was 2.9 h, substantially higher than that reported for dasatinib in mice (0.9 h).58 Compound 11a showed moderate oral bioavailability (25.3%), although it is important to note that no excipients (just pure water) were used for its oral formulation.
In Vivo SRC Inhibition Study in a Tumor Xenograft Model
The presence of active (phosphorylated) SRC in human colorectal cancer HCT116 cells and its inhibition under 11a treatment was verified by Western blot (Figure S12). Subsequently, an in vivo PD study was performed in a xenograft model of HCT116 cells in mice.59 HCT116 cells were injected subcutaneously, and tumors were allowed to grow up to 3–4 mm in diameter. Subsequently, mice were dosed daily for 3 d with 11a (50 mg/kg, in nanopure water) or vehicle (nanopure water) by oral gavage and culled 3 h after the last dose (n = 4). Tumors were excised, fixed, and sections labeled for phospho-SRCY416 and stained with hematoxylin. As shown in Figure 8, microscopy analysis demonstrated significant reduction of phospho-SRCY416 in the xenograft sections from mice treated with 11a relative to the untreated animal controls.
Figure 8.

Immunohistochemical analysis of phospho-SRCY416 in human tumor xenografts. (a) Images of representative sections (low and high resolution) of HCT116 xenografts from (left) untreated mice and (right) mice treated with 11a (n = 4). (b) Histoscore analysis (6–7 sections analyzed per experiment). Quantification of immunohistochemistry across tumor tissue sections from untreated animals (water) and 11a treated groups performed in blinded fashion. P value calculated from t-test.
Conclusions
To move away from orthodox target-centric strategies, a pragmatic approach for rapid discovery of novel high-quality anticancer kinase inhibitors was implemented through the generation and phenotypic screening of analogues of PP1, a pyrazolopyrimidine known to inhibit multiple oncogenic pathways.32−34 Derivatives of this promiscuous kinase inhibitor were designed to improve physicochemical properties and explore favorable pharmacological features through small chemical modifications. To accelerate the advance from hits to leads to drug candidates, compounds were screened by phenotypic assays in the search for derivatives with potent antiproliferative properties. Chemical design was biased toward human breast cancer treatment (rather than to a particular target) by using the breast adenocarcinoma cell line MCF7 as a discriminating cell model. Such a pseudotarget-agnostic strategy identified compounds that inhibited pathways involved in breast cancer survival and also disregarded compounds with low cell penetrability (= deficient drug-likeness). After two rounds of design, synthesis, and screening of highly focused libraries, novel candidates with low micromolar activity in MCF7 cells and high solubility in water were identified (compounds 8d and 9d). Elucidation of the potential target/s linked with their bioactivity was followed by a literature survey to explore chemical motifs that could further improve these phenotypic leads. From that point, two independent medicinal chemistry campaigns were initiated. This manuscript has focused on the development of SRC inhibitor 11a, a novel pyrazolopyrimidine analogue that exhibits potent antiproliferative properties against breast cancer cells, halts MDA-MB-231 cell migration, and inhibits intracellular SRC signaling at low nanomolar concentrations. Remarkably, kinase profiling revealed that 11a is the first small molecule that inhibits SRC at subnanomolar concentration (IC50 < 0.5 nM) with a 1000-fold selectivity over ABL. This is of relevance because the manifestation of cardiac events, especially in elderly patients, is a well-established adverse effect of ABL inhibition.51,52 Synthesis and phenotypic screening of 24 closely related derivatives of 11a were carried out to gain further insight into the optimal chemical space for the generation of potent, selective, and cell-active SRC inhibitors. Analysis of SAR provided evidence of the strict structural requirements of the pyrazolopyrimidine scaffold required to create such a class of antiproliferative SRC/non-ABL inhibitors. The relatively low molecular weight of 11a (510 Da) and its good physicochemical properties, including excellent water solubility and cLogP of 2.2, prompted us to investigate further the biological properties of this compound. Biochemical and in silico studies confirm that 11a is a Type I kinase inhibitor of SRC and provided rationale for its selective affinity for SRC over ABL. Screening of hERG channel and CYP induction, together with in vitro and in vivo PK/PD assays (including zebrafish PD/safety studies) support the nomination of 11a as a SRC-targeting drug candidate that could potentially offer a superior therapeutic window than dual SRC/ABL inhibitors.60,61 This novel SRC-targeting inhibitor belongs to a second generation of SRC inhibitors that do not target the ABL kinase,50,62 being the first one to demonstrate suppression of SRC phosphorylation in vivo. Finally, in concordance with the primary role of SRC in driving tumor invasion and drug resistance mechanisms,63,64 more in-depth preclinical proof-of-concept studies using appropriate in vivo models of disease progression and rational drug combination studies will be required to demonstrate the efficacy and therapeutic value of SRC/no-ABL inhibitors in cancer treatment.
Experimental Section
Synthetic Procedures
General Methods
Microwave-irradiated reactions were carried out in a Biotage Initiator microwave synthesizer. Nonmicrowave reactions were performed under an inert atmosphere of nitrogen using anhydrous solvents. All commercially available chemicals were obtained from either Fisher Scientific, Matrix Scientific, Sigma-Aldrich, or VWR International Ltd. NMR spectra were recorded at ambient temperature on a 500 MHz Bruker Avance III spectrometer. Samples were dissolved in deuterated solvents commercially available from Sigma-Aldrich. Chemical shifts are reported in parts per million (ppm) relative to the peak of the solvent. The data are presented as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (in Hertz, Hz) and interpretation. TLCs were ran on Merck TLC Silica gel 60 F254 plates, typically 5 × 10 cm, and monitored using a 254 nm UV source or permanganate staining. Purifications were carried out using flash column chromatography with commercially available silica gel and solvents. All compounds used in the biological screenings were determined to be >95% pure by analytical HPLC with evaporative light scattering detection (Agilent).
Synthesis of Compound 11a
1H-Pyrazolo[3,4-d]pyrimidin-4-amine (2)
5-amino-1H-pyrazole-4-carbonitrile, 1 (3 g, 27.77 mmol), and formamide (15 mL) were added to a 20 mL microwave vial, and the mixture heated at 180 °C for 2 h using microwave radiation. The precipitate formed on cooling was filtered off and washed with water (50 mL) and allowed to dry giving the product as a pale brown solid (3.5 g, 25.9 mmol, 93%). The experiment was repeated to give a second batch of product (3.44 g, 25.5 mmol, 92%). 1H NMR (500 MHz, DMSO) δ 13.34 (s, 1H), 8.13 (s, 1H), 8.07 (s, 1H), 7.69 (br. m, 2H); 13C NMR (126 MHz, DMSO) δ 158.2, 156.0, 155.0, 132.8 (CH), 99.8; MS (ES +ve) (M + H)+: 136.0, 157.9 (+Na), (ES −ve) (M – H)−: 133.9.
3-Iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3)
Compound 2 (1.5 g, 11.11 mmol) was suspended in 15 mL of DMF, and N-iodosuccinimide (1.2 equiv, 3.0 g, 13.3 mmol) added. The mixture was heated at 180 °C in the microwave for 40 min. EtOH (80 mL) was added to the reaction, and a precipitate began to form, which was aided by sonication. The precipitate was filtered and washed with EtOH (×3, 20 mL) and allowed to dry in an oven at 40 °C overnight to give a sand colored solid (2.115 g, 8.1 mmol, 73%). 1H NMR (500 MHz, DMSO) δ 13.80 (s, 1H), 8.16 (s, 1H), 7.79–6.44 (m, 2H); 13C NMR (126 MHz, DMSO) δ 157.6, 156.1, 155.0, 102.5, 89.8; MS (ES +ve) (M + H)+: 283.9 (+Na), (ES −ve) (M – H)−: 259.9, 287.8 (Na+).
1-(2,2-Diethoxyethyl)-3-iodo-pyrazolo[3,4-d]pyrimidin-4-amine (4)
To a solution of 3 (500 mg, 1.9 mmol) in DMF (15 mL) was added sodium hydride (1.5 equiv, 2.9 mmol, 60% dispersion in mineral oil, 115.2 mg), and the solution allowed to stir for 30 min until the gas evolution stopped. Bromoacetaldehyde diethyl acetal (1.5 eq 2.9 mmol, 0.435 mL) was then added dropwise, and the mixture heated at 150 °C in the microwave for 40 min. EtOAc and water (50 mL) were added to the mixture, and the organics separated. The aqueous layer was washed with EtOAc (50 mL, ×3), and the organics combined and washed with water (×3, 30 mL), dried over anhydrous MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography MeOH/DCM (0–5%) to give a light orange solid (461 mg, 1.2 mmol, 64%). 1H NMR (500 MHz, DMSO) δ 8.21 (s, 1H), 7.90–6.30 (m, 2H), 4.93 (t, J = 5.7, 1H), 4.33 (d, J = 5.8, 2H), 3.62 (dq, J = 9.4, 6.9, 2H), 3.40 (dq, J = 9.6, 7.0, 2H), 0.98 (t, J = 7.0, 6H); 13C NMR (126 MHz, DMSO) δ 157.9, 156.3 (CH), 154.0, 103.2, 99.5, 89.5, 61.4 (CH2), 48.8 (CH2), 15.39 (CH3); MS (ES +ve) (M + H)+: 377.8, 400.0 (Na+), (ES −ve) (M – H)−: 376.0.
1-(2,2-Diethoxyethyl)-3-(1H-pyrrolo[2,3-b]pyridin-5-yl)pyrazolo[3,4-d]pyrimidin-4-amine (8d)
To a solution of 4 (1.135 g, 3.0 mmol) in dioxane/water (10 mL/1 mL) were added 7-azaindole-5-boronic acid pinacol ester (1.5 equiv, 614 mg, 4.5 mmol), potassium carbonate (1.5 equiv, 624.7 mg, 4.5 mmol), and followed by palladium acetate (5 mol %, 33.8 mg), and the mixture heated in the microwave at 120 °C for 1 h. EtOAc and water (50 mL) were added to the mixture, and the organic layer separated, dried over anhydrous MgSO4, and concentrated in vacuo. The crude products were purified by column chromatography. Purified by column chromatography, MeOH/DCM (0–6%), to give a white solid (93 mg, 0.253 mmol, 96%). 1H NMR (500 MHz, CDCl3) δ 9.53 (s, 1H), 8.62 (d, J = 1.9, 1H), 8.41 (s, 1H), 8.24 (d, J = 2.0, 1H), 7.45 (d, J = 3.4, 1H), 6.62 (d, J = 3.5, 1H), 6.29–5.86 (br. s, 2H), 5.14 (t, J = 5.7, 1H), 4.62 (d, J = 5.7, 2H), 3.79 (dq, J = 9.4, 7.0, 2H), 3.55 (dq, J = 9.4, 7.0, 2H), 1.14 (t, J = 7.0, 6H); 13C NMR (126 MHz, CDCl3) δ 156.8, 154.6, 153.4 (CH), 148.9, 143.8, 142.8 (CH), 128.8 (CH), 126.8 (CH), 121.3, 120.5, 101.8 (CH), 99.9 (CH), 98.5, 62.2 (CH2), 49.3 (CH2), 15.3 (CH3); MS (ES +ve) [M + H]+: 368.2, 390.2 (Na+), (ES −ve) [M – H]−: 366.2; HRMS (ES +ve), C18H22N7O2 (M + H)+: calculated 368.18295, found 368.18090.
Synthesis of 1-[2-[4-(Dimethylaminomethyl)-1-piperidyl]ethyl]-3-(1H-pyrrolo[2,3-b]pyridin-5-yl)pyrazolo[3,4-d]pyrimidin-4-amine (9d)
60 mg of 8d was added to a 20 mL microwave vial. 5 mL of water was added, followed by 5 mL of TFA, and the mixture heated at 100 °C for 1 h. The mixture was concentrated in vacuo to give a light brown oil which was used without further purification. The crude compound was dissolved in 2 mL of DCM. N,N-dimethyl-1-(4-piperidyl)methanamine (1.5 equiv, 0.245 mmol, 34.8 mg) was added followed by a drop of acetic acid. The mixture was allowed to stir for 10 min, then sodium triacetoxyborohydride (1.5 equiv, 0.245 mmol, 51.9 mg) was added, and the mixture allowed to stir for 2 h. The mixture was concentrated in vacuo, and the product purified by column chromatography, MeOH/DCM (5–10% then 10% with 5–20 drops of NH3 aq./100 mL) to give a light orange solid (15.3 mg, 0.0365 mmol, 15%). 1H NMR (500 MHz, MeOD) δ 8.52 (s, 1H), 8.29 (d, J = 2.0, 1H), 8.28 (s, 1H), 7.52 (d, J = 3.5, 1H), 6.62 (d, J = 3.5, 1H), 4.64 (t, J = 6.4, 2H), 3.25 (d, J = 11.7, 2H), 3.13 (t, J = 6.3, 2H), 2.96 (d, J = 7.2, 2H), 2.84 (s, 6H), 2.37 (t, J = 11.4, 2H), 1.88 (m, 1H), 1.80 (d, J = 13.1, 2H), 1.36–1.31 (m, 2H); 13C NMR (126 MHz, MeOD) δ 161.8, 158.6, 155.5 (CH), 154.3, 148.2, 143.7, 141.8 (CH), 128.7 (CH), 127.1 (CH), 120.7, 100.6 (CH), 98.2, 62.7 (CH2), 56.3 (CH2), 52.2 (CH2), 43.4 (CH2), 42.7 (CH3), 30.9 (CH), 28.4 (CH2); MS (ES +ve) [M + H]+: 420.2; HRMS (ES +ve), C22H29N9 [M + H]+: calculated 420.25404, found 420.254249.
1-[2-[4-(Dimethylamino)-1-piperidyl]ethyl]-3-iodo-pyrazolo[3,4-d]pyrimidin-4-amine (10a)
150 mg (0.398 mmol) of 4 was added to a 10 mL microwave tube. 2.5 mL of water and 2.5 mL of TFA were added, and the mixture heated to 100 °C for 1 h. The mixture was concentrated in vacuo to give a white solid which was used without further purification. The aldehyde intermediate was suspended in 3 mL of DCM. 4-(N,N-dimethylamino)piperidine (1.5 equiv, 0.598 mmol, 76.6 mg) was added followed by a drop of acetic acid and the mixture allowed to stir for 10 min. Sodium triacetoxyborohydride (1.5 equiv, 0.598 mmol, 126.8 mg) was added, and the mixture allowed to stir for 17 h overnight. The mixture was concentrated in vacuo, and the product purified by column chromatography, MeOH/DCM (0–10% then 5–20 drops of NH3 aq. per 100 mL) to give a light orange/brown solid (163.8 mg, 0.405 mmol, 99%). 1H NMR (500 MHz, MeOD) δ 8.22 (s, 1H), 4.49 (t, J = 6.4, 2H), 3.32 (s, 3H), 3.15 (d, J = 12.1, 2H), 2.96 (ddd, J = 16.0, 8.0, 4.0, 1H), 2.91 (t, J = 6.4, 2H), 2.72 (s, 6H), 2.15 (td, J = 12.0, 2.0, 2H), 2.04–1.96 (m, 2H), 1.54 (qd, J = 12.2, 3.9, 2H); 13C NMR (126 MHz, MeOD) δ 158.07 (C), 155.67 (CH), 153.70 (C), 103.59 (C), 86.97 (C), 63.18 (CH), 55.86 (CH2), 51.38 (CH2), 44.37 (CH2), 39.31 (CH3), 26.42 (CH2); MS (ES +ve) [M + H]+: 416.2.
tert-Butyl N-[4-[4-amino-1-[2-[4-(dimethylamino)-1-piperidyl]ethyl]pyrazolo[3,4-d]pyrimidin-3-yl]-2-methoxy-phenyl]carbamate (11a, aka eCF509)
To a solution of 10a (50 mg, 0.1205 mmol) in dioxane/water (4.5 mL/0.5 mL) were added [4-(tert-butoxycarbonylamino)-3-methoxy-phenyl]boronic acid (1.5 equiv, 48.3 mg, 0.181 mmol), potassium carbonate (1.5 equiv, 25.0 mg, 0.181 mmol), and triphenylphosphine (20 mol %, 9.5 mg), followed by palladium acetate (5 mol %), and the mixture heated in the microwave at 120 °C for 1 h. EtOAc (50 mL) and water (50 mL) were added to the mixture, and the organic layer separated. The aqueous layer was washed with EtOAc (20 mL, ×2), and the organics combined, dried over anhydrous MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography, MeOH/DCM (0–10% then 5–20 drops of NH3 aq. per 100 mL) to give a light brown solid (23.1 mg, 0.0453 mmol, 38%). 1H NMR (500 MHz, MeOD) δ 8.27 (s, 1H), 8.08 (d, J = 8.2, 1H), 7.30 (d, J = 1.8, 1H), 7.26 (dd, J = 8.2, 1.9, 1H), 4.56 (t, J = 6.7, 2H), 3.98 (s, 3H), 3.14 (d, J = 11.9, 2H), 2.94 (t, J = 6.7, 2H), 2.39 (m, 7H), 2.14 (dd, J = 12.0, 10.0, 2H), 1.90 (d, J = 12.5, 2H), 1.57 (s, 9H), 1.49 (qd, J = 12.1, 3.6, 2H); 13C NMR (126 MHz, MeOD) δ 158.53 (C), 155.40 (CH), 154.12 (C), 153.46 (C), 149.24 (C), 145.02 (C), 128.78 (C), 127.38 (C), 120.43 (CH), 119.56 (CH), 110.28 (CH), 97.73 (C), 80.18 (C), 62.29 (CH), 56.22 (CH2), 55.07 (CH3), 52.20 (CH2), 44.00 (CH2), 40.06 (CH3), 27.24 (CH2), 27.21 (CH3); MS (ES +ve) [M + H]+: 511.3; HRMS (ES +ve), C26H38N8O3 [M + H]+: calculated 511.31396, found 511.3151.
Biological Studies
General Methods
MCF7, MDA-MB-231, and SYF cells were grown in Dulbecco’s modified eagle medium (DMEM), supplemented with serum (10% fetal bovine serum) and l-glutamine (2 mM), and incubated in a Heracell 240i tissue culture incubator at 37 °C and 5% CO2.
Time-Lapse Cell Growth Assay
Cells were plated in 96-well Nunc black optical-bottom plates (Thermo Scientific) at 1000 cells/well in 100 μL of DMEM medium containing 10% FBS and 2 mM l-glutamine and incubated for 48 h in an incubator at 37 °C and 5% CO2. The media was replaced with fresh media containing 100 μM concentration of 7a–l, PP1, or DMSO (0.1% v/v), and the plates imaged in the IncuCyte ZOOM system. Cell growth was monitored at sequential time points over 5 d using the bright-field microscopic images acquired by the IncuCyte ZOOM system. Cell confluence determination was performed by the IncuCyte software.
Dose–Response Viability Assay
Cells were plated in 96-well plates at 2000 cells/well in 100 μL of DMEM medium containing 10% FBS and 2 mM l-glutamine and incubated for 48 h in an incubator at 37 °C and 5% CO2. After 48 h, the media was aspirated from each well and replaced with 95 μL of fresh medium. Compounds, including DMSO, were prepared at 20× in DMEM medium in a separate 96-well intermediate plate. 5 μL from the intermediate plate was then added to each well containing cells. Untreated cells were incubated with DMSO (0.1% v/v). After 5 d, PrestoBlue cell viability reagent (10 μL) was added to each well, and the plates incubated for 60–90 min. Fluorescence emission was detected using an Envision fluorescence plate reader (excitation 540 nm, emission 590 nm). All conditions were normalized to the untreated cells (100%), and curves were fitted using a four parameter logistic fit with minimum value constrained to zero using GraphPad Prism software, to calculate EC50 values.
Apoptosis Assay
Cells were plated in 96-well Nunc black optical-bottom plates (Thermo Scientific) at 3000 cells/well in 100 μL of DMEM medium containing 10% FBS and 2 mM l-glutamine and incubated for 48h in an incubator at 37 °C and 5% CO2. The media was replaced with 95 μL of fresh media containing NucView 488 from Biotium at 1 μM concentration, and drugs or DMSO added along a concentration gradient, as described in the cell viability assay, and the plates imaged in the IncuCyte. Cell growth was monitored over 5 days using bright-field and NucView 488 fluorescence (excitation 460 nm, emission 524 nm) microscopy. Cell confluence and apoptotic count (positive NucView 488 emission signal threshold above background signal) were performed by the IncuCyte software. The numbers generated from the applied confluence and NucView 488 labeled cells masks were divided so as to create a ratio of number of apoptotic cells to cell confluence. The data were then normalized to DMSO to account for the decreased number of cells found in higher concentrations of drug treatment.
Western Blotting
Cells were plated at 1 × 106 cells/well in 2 mL of DMEM medium containing 10% FBS and 2 mM l-glutamine in 6-well plates and incubated at 37 °C with 5% CO2. After 24 h, the media was aspirated and replaced with 2 mL of DMEM medium containing 0.1% FBS and 2 mM l-glutamine and the cells incubated for a further 24 h. Two μL of compounds dissolved in DMSO at appropriate concentration was then added to each well and plates incubated for 1.5 h. 222 μL of FBS was then added to each well (giving a final concentration of 10%), and cells incubated for 1 h. Cell lysates were then prepared using 100 μL of cell lysis buffer (1% Triton X-100, 50 mM HEPES, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM Na3VO4, 10% glycerol and protease and phosphatase inhibitors) per well. The total cell protein concentration in each lysate was determined using precision red advanced protein reagent #2 from Cytoskeleton. Lysates were normalized to 2 mg/mL and boiled at 100 °C for 3 min in SDS-PAGE sample buffer (40% glycerol, 8% SDS, 0.1 M DTT, 0.25 M Tris-HCl, pH 6.8). Samples were resolved by SDS-PAGE using BioRad 4–15% precast gels over 60 min at 140 V and transferred to PVDF membranes over 150 min at 210 mA. Nonspecific antibody binding was blocked by incubation of membranes for 1 h at room temperature using western blocking reagent solution (Roche) prior to adding primary antibodies in 0.5% blocking buffer at 4 °C overnight. Membranes were washed with TBS/T (×3, 5 min) and then secondary antibody linked to horseradish peroxidase (HRP) added for 1 h at room temperature. Following further washing with TBS/T (×3, 5 min) and TBS (×2, 5 min), HRP was detected by peroxidase enhanced chemiluminescence (POD ECL from Roche) and bands visualized using X-ray film or the ChemiDoc MP Imaging System from BioRad.
Cell Migration Assay
Cells were plated at 50,000 cells/well in 100 μL of DMEM medium containing 10% FBS and 2 mM l-glutamine in a 96-well ImageLock plate from Essen BioScience and left overnight to adhere in an incubator at 37 °C and 5% CO2. Scratch wounds were created in each well using the WoundMaker supplied by Essen BioScience, and each well washed with media (100 μL, ×2) to remove floating cells. 95 μL of fresh media was added in each well. Compounds, including DMSO, were prepared at 20× in DMEM medium in a separate plate, and 5 μL was then added to each well containing cells. Untreated cells were incubated with DMSO (0.1% v/v). Images were recorded every 30 min using the IncuCyte-ZOOM for 24 h. Analysis of cell migration into the wound was performed using the IncuCyte software.
Kinase Screening Assays - IC50 Determination
Radioisotope-based assay ([γ-33P] ATP) consisting of measuring 33P incorporation on the substrate (poly [Glu, Tyr] 4:1) relative to DMSO. Compound IC50 values were determined from 10-point, 1:3 dilution curves starting at 10 μM, by Reaction Biology Corp, with 10 μM ATP.
Evaluation of ATP Competition and Ki Value for 11a against SRC
Materials: Inhibitor: 10 mM stock solution of 11a in DMSO. Kinase reaction buffer: 20 mM HEPES-HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.1 mM Na3VO4, 0.02 mg/mL BSA, 2 mM DTT, and 1% DMSO. Kinase: SRC; recombinant human full length cytoplasmic domain (accession no. NP_005408), C-terminal His-tagged, expressed in insect cells, MW = 62.3 kDa. Substrate for kinase: poly [Glu, Tyr] 4:1. Reaction conditions: 0.6 nM SRC, 0.2 mg/mL poly [Glu, Tyr] 4:1, and varied ATP. Experimental procedure: The kinase assays were performed at room temperature. 11a was added as 10-point dose IC50 mode into the enzyme/substrate mixture using acoustic technology with 20 min preincubation. Then the five concentrations of ATP were added to initiate the reaction. The activity was monitored every 5–15 min for a time course study. The ATP, substrate, and 11a concentrations tested were as follows: (i) ATP concentrations tested: 1, 5, 15, 75, and 150 μM ATP; (ii) substrate concentration: constant at 0.2 mg/mL; (iii) 11a concentrations tested: 10-point dose IC50 with 3-fold serial dilution started at 100 nM; (iv) time points measured: 0, 5, 10, 15, 20, 30, 45, 60, 75, 90, 105, and 120 min.
Docking Studies
Crystal structures of SRC and ABL were examined for their suitability for docking. PDB 3GEQ was selected to represent SRC as it contained pyrazolopyrimidine ligand PP2, which most closely resembles compounds PP20 and 11a, and its DFG motif is in the active conformation. Similarly, PDB 4TWP was selected as the ABL structure as its DFG motif is also flipped in the ATP-interacting position. The side chain of Asp381 in the ABL DFG motif was manually rotated in PyMol to match the ATP-binding mode of the equivalent Asp in SRC. ABL in this structure also contains a T316I mutation which was reverted to wild type. Models of PP20 and 11a were built using the structure and binding mode of PP2 as a template. Water molecules and other heteroatoms were removed from the optimized structures, and the program PDB 2PQR 1.865 used to assigned position-optimized hydrogen atoms, utilizing the additional PropKa66 algorithm with a pH of 7.4 to predict protonation states. The MGLTools 1.5.4 utility prepare_receptor4.py was used to assign Gasteiger charges to atoms. Hydrogen atoms were assigned to compound structures using OpenBabel 2.3.2,67 utilizing the -p option to predict the protonation states of functional groups at pH 7.4. The MGLTools utility prepare_ligand4.py68 was used to assign Gasteiger charges and rotatable bonds. Autodock 4.2.369 was used to automatically dock the compounds into the ATP binding pocket of the crystal structures. A grid box that encompassed the maximum dimensions of the cognate ATP ligand plus 12 Å in each direction was used. The starting translation and orientation of the ligand and the torsion angles of all rotatable bonds were set to those of the modeled compounds. The Autogrid grid point spacing was set at 0.2 Å. The Autodock parameter file specified 10 Lamarckian genetic algorithm runs, 15,000,000 energy evaluations and a population size of 300.
Zebrafish PD/Toxicology Assay
Transgenic Tg(brn3c:mGFP) zebrafish embryos were collected from breeding pairs and reared at 28 °C in E3 embryo media. One dpf embryos were treated with 11a at different doses (10–750 μM) at 20 hpf, 36 hpf and 48 hpf, or DMSO (0.1% v/v). Zebrafish embryos were imaged by fluorescent microscopy at 72 hpf. Safety assays: Wild-type zebrafish embryos were collected from AB-TPL breeding pairs and reared at 28 °C in E3 embryo media. One dpf embryos were treated with 11a or dasatinib at 100 μM, and DMSO (0.1% v/v) as negative control, for 4 h before being washed off and replaced with fresh E3 media. For PP20 treatment, the fish were incubated for 2 h post-amputation then replaced with fresh E3 media. The embryos were left to develop in E3 media at 28 °C for 2 d, after which, they were imaged by light microscopy. Zebrafish husbandry was performed under Home Office License in compliance with the Animals (Scientific Procedures) Act 1986 and approved by the University of Edinburgh Ethics Committee.
In Vivo PK Analysis
Three female cluster of differentiation 1 (CD1) mice, 25–30 g, were dosed per administration route per time point, per compound. 11a was administered either orally or intravenously (typical dose level of 10 mg compound per kg body weight). Animals were given free access to food throughout the study. At the following time points, the animals were anaesthetised, blood collected in heparinised tubes, and animals sacrificed: (i) oral dosing: 0.08, 0.25, 0.5, 1, 2, 4, and 8 h post-dose; (ii) IV dosing: 0.08, 0.25, 0.5, 1, 2, 4, and 8 h post-dose.
In Vivo PD Study
Tumor xenografts were generated in mice by injection of 2 million HCT116 cells subcutaneously. Tumors were allowed to grow until 3–4 mm in diameter. A daily dose of 50 mg/kg of 11a in pure water was administered by oral gavage. Mice were sacrificed 3 h after the last dose and tumors excised, fixed in 4% formaldehyde in 0.1 M phosphate buffer (pH 7.2), and embedded in paraffin. Sections were cut using a Reichert-Jung 1150/Autocut microtome to perform phospho-SRC immunochemistry. Antigen retrieval was performed using heat treatment under pressure in a microwave oven for 10 min in 10 mM citrate buffer pH = 6. Sections were blocked for endogenous peroxidase followed by incubation with antiphospho-SRC antibody (Cell Signaling Technology) (1:200 dilution) at 4 °C overnight. Staining was developed using EnVision (Dako) and diaminobenzidene (Dako) before slides were counterstained in hematoxylin, dehydrated, and mounted in dibutyl phthalate xylene. Slices were imaged on a NanoZoomer digital slide scanner, Hammamatsu. Staining was scored by a single experienced observer, blinded to treatment, using a weighted histoscore method.
Acknowledgments
We are grateful to the MRC, Wellcome Trust (ISSF 2012/13), and Edinburgh BioQuarter (now Sunergos Innovations) for financial support. C.F. is grateful to MRC for a DTP scholarship. J.C.D., V.G.B., and M.C.F. were supported by the ERC Advanced Investigator grant (no. 294440 Cancer Innovation). R.D. and L.H. were funded by the Wellcome Trust (WT84637). E.E.P. was funded by the MRC and the ERC (no. 643092 ZF-MEL-CHEMBIO). N.O.C. and A.U.B. thanks RCUK and IGMM, respectively, for an Academic Fellowship. We thank Dr John Dixon (JD International Consulting) for his expert advice.
Glossary
Abbreviations Used
- ATP
adenosine triphosphate
- CD3OD
deuterated methanol
- CD1
cluster of differentiation 1
- CML
chronic myeloid leukemia
- CYP
cytochrome P450
- DFG
aspartate-phenylalanine-glycine
- DMF
dimethylformamide
- DMEM
Dulbecco’s modified eagle’s media
- DCM
dichloromethane
- DMPK
drug metabolism and pharmacokinetics
- DMSO
dimethyl sulfoxide
- dpf
days post-fertilization
- EC50
half-maximal effective concentration
- FAK
focal adhesion kinase
- GFP
green fluorescent protein
- HLM
human liver microsomes
- IC50
half-maximal inhibitory concentration
- PD
pharmacodynamics
- rt
room temperature
- SAR
structure–activity relationship
- SFK
SRC family of kinases
- SYF
murine embryonic fibroblasts SRC–/–, YES–/–, FYN–/–
- TLC
thin-layer chromatography
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00065.
Synthesis and characterization of compounds 2 to 12a–y, in vitro ADME-Tox methods and supporting Figures S1–S12 and Table S1 (PDF)
seven-day time-lapse motion picture of MCF7 cell proliferation under treatment (addition at 48 h) with 100 μM of 8d with Nucview488 (apoptosis fluorescent marker) (AVI)
seven-day time-lapse motion picture of MCF7 cell proliferation under treatment (addition at 48 h) with 100 μM of 9d with Nucview488 (apoptosis fluorescent marker) (AVI)
Additional compound data (XLSX)
Author Present Address
# J.T.W.: Centre for Innovative Cancer Therapeutics, Ottawa Hospital Research Institute, Ottawa, Ontario, Canada.
The authors declare the following competing financial interest(s): The authors declare that a patent application (No. GB1508747.1) is pending on compounds 11-12.
Supplementary Material
References
- Hughes J. P.; Rees S.; Kalindjian S. B.; Philpott K. L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Screening for drug discovery. Nature 2002, 418, 453–459. 10.1038/418453a. [DOI] [PubMed] [Google Scholar]
- Holdgate G.; Holdgate G.; Geschwindner S.; Breeze A.; Davies G.; Colclough N.; Temesi D.; Ward L. Biophysical methods in drug discovery from small molecule to pharmaceutical. Methods Mol. Biol. 2013, 1008, 327–355. 10.1007/978-1-62703-398-5_12. [DOI] [PubMed] [Google Scholar]
- McInnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Vistoli G.; Pedretti A.; Testa B. Assessing drug-likeness--what are we missing?. Drug Discovery Today 2008, 13, 285–294. 10.1016/j.drudis.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Hoelder S.; Clarke P. A.; Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A.; Wee S.; Lengauer C. Why is cancer drug discovery so difficult?. Nat. Rev. Drug Discovery 2007, 6, 115–120. 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- Paul S. M. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discovery 2010, 9, 203–214. 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Carragher N.; Unciti-Broceta A.; Cameron D. Advancing cancer drug discovery towards more agile development of targeted combination therapies. Future Med. Chem. 2012, 4, 87–105. 10.4155/fmc.11.169. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I.; Landis J. Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discovery 2004, 3, 711–716. 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Creedon H.; Gómez-Cuadrado L.; Tarnauskaitė Ž.; Balla J.; Canel M.; MacLeod K. G.; Serrels B.; Fraser C.; Unciti-Broceta A.; Tracey N.; Le Bihan T.; Klinowska T.; Sims A. H.; Byron A.; Brunton V. Identification of novel pathways linking epithelial-to-mesenchymal transition with resistance to HER2-targeted therapy. Oncotarget 2016, 7, 11539–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoon I.; Blanc V. Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol. Biol. 2013, 1045, 1–27. 10.1007/978-1-62703-541-5_1. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: beyond proof of principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Clavel C. M.; Zava O.; Schmitt F.; Kenzaoui B. H.; Nazarov A. A.; Juillerat-Jeanneret L.; Dyson P. J. Thermoresponsive chlorambucil derivatives for tumour targeting. Angew. Chem., Int. Ed. 2011, 50, 7124–7127. 10.1002/anie.201101133. [DOI] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Macleod K. G.; Rybski W.; Fraser C.; Torres-Sánchez C.; Patton E. E.; Bradley M.; Carragher N. O.; Unciti-Broceta A. Extracellular palladium-catalyzed dealkylation of 5-fluoro-1-propargyl-uracil as a bioorthogonally-activated prodrug approach. Nat. Commun. 2014, 5, 3277. 10.1038/ncomms4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson; Fraser C.; J C.; Rybski W.; Torres-Sánchez C.; Bradley M.; Patton E. E.; Carragher N. O.; Unciti-Broceta A. Development and bioorthogonal activation of palladium-labile prodrugs of gemcitabine. J. Med. Chem. 2014, 57, 5395–5404. 10.1021/jm500531z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. A.; Uhlik M. T.; Moxham C. M.; Tomandl D.; Sall D. J. Modern phenotypic drug discovery is a viable, neoclassic pharma strategy. J. Med. Chem. 2012, 55, 4527–4538. 10.1021/jm201649s. [DOI] [PubMed] [Google Scholar]
- Eder J.; Sedrani R.; Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discovery 2014, 13, 577–587. 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- Carragher N. O.; Brunton V. G.; Frame M. C. Combining imaging and pathway profiling: an alternative approach to cancer drug discovery. Drug Discovery Today 2012, 17, 203–214. 10.1016/j.drudis.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases-the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Li Y.; Kim S. M.; Bossuyt W.; Liu P.; Qiu Q.; Wang Y.; Halder G.; Finegold M. J.; Lee J. S.; Johnson R. L. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1437–1442. 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.-B. S.; Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- Fujimoto H.; Onishi N.; Kato N.; Takekawa M.; Xu X. Z.; Kosugi A.; Kondo T.; Imamura M.; Oishi I.; Yoda A.; Minami Y. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006, 13, 1170–1180. 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- Wilhelm S.; Carter C.; Lynch M.; Lowinger T.; Dumas J.; Smith R. A.; Schwartz B.; Simantov R.; Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery 2006, 5, 835–844. 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Fliri A. F.; Loging W. T.; Volkmann R. A. Drug effects viewed from a signal transduction network perspective. J. Med. Chem. 2009, 52, 8038–8046. 10.1021/jm901001p. [DOI] [PubMed] [Google Scholar]
- Peters J. U. Polypharmacology - foe or friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Greuber E. K.; Smith-Pearson P.; Wang J.; Pendergast A. M. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren N. K.; Foos G.; Hauser C. A.; Pasquale E. B. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat. Cell Biol. 2006, 8, 815–825. 10.1038/ncb1438. [DOI] [PubMed] [Google Scholar]
- Allington T. M.; Galliher-Beckley A. J.; Schiemann W. P. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009, 23, 4231–4243. 10.1096/fj.09-138412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Henn H.; Patsialou A.; Wang Y.; Warren M. S.; Condeelis J. S.; Koleske A. J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. H.; Brunton V. G.; Unciti-Broceta A. AXL inhibitors in cancer: A medicinal chemistry perspective. J. Med. Chem. 2016, 59, 3593. 10.1021/acs.jmedchem.5b01273. [DOI] [PubMed] [Google Scholar]
- Hanke J. H.; Gardner J. P.; Dow R. L.; Changelian P. S.; Brissette W. H.; Weringer E. J.; Pollok B. A.; Connelly P. A. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and Fyn-dependent T cell activation. J. Biol. Chem. 1996, 271, 695–701. 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Tatton L.; Morley G. M.; Chopra R.; Khwaja A. The Src-selective kinase inhibitor PP1 also inhibits Kit and Bcr-Abl tyrosine kinases. J. Biol. Chem. 2003, 278, 4847–4853. 10.1074/jbc.M209321200. [DOI] [PubMed] [Google Scholar]
- Jester B. W.; Gaj A.; Shomin C. D.; Cox K. J.; Ghosh I. Testing the promiscuity of commercial kinase inhibitors against the AGC kinase group using a split-luciferase screen. J. Med. Chem. 2012, 55, 1526–1537. 10.1021/jm201265f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinér P.; Alao J. P.; Söderlund J.; Sunnerhagen P.; Grøtli M. Preparation of 3-substituted-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amines as RET kinase inhibitors. J. Med. Chem. 2012, 55, 4872–4876. 10.1021/jm3003944. [DOI] [PubMed] [Google Scholar]
- Apsel B.; Blair J. A.; Gonzalez B.; Nazif T. M.; Feldman M. E.; Aizenstein B.; Hoffman R.; Williams R. L.; Shokat K. M.; Knight Z. A. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A.; Bocci G.; La Motta C.; Ferrari S. M.; Fallahi P.; Corrado A.; Fioravanti A.; Sartini S.; Orlandi P.; Piaggi S.; Corti A.; Materazzi G.; Galleri D.; Ulisse S.; Fontanini G.; Danesi R.; Da Settimo F.; Miccoli P. CLM29, a multi-target pyrazolopyrimidine derivative, has anti-neoplastic activity in medullary thyroid cancer in vitro and in vivo. Mol. Cell. Endocrinol. 2014, 393, 56–64. 10.1016/j.mce.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Schenone S.; Radi M.; Musumeci F.; Brullo C.; Botta M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: an old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. 10.1021/cr400270z. [DOI] [PubMed] [Google Scholar]
- Schindler T.; Sicheri F.; Pico A.; Gazit A.; Levitzki A.; Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol. Cell 1999, 3, 639–648. 10.1016/S1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- Knowles P. P.; Murray-Rust J.; Kjaer S.; Scott R. P.; Hanrahan S.; Santoro M.; Ibáñez C. F.; McDonald N. Q. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 2006, 281, 33577–33587. 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- Bowler F. R.; Diaz-Mochon J. J.; Swift M. D.; Bradley M. DNA analysis by dynamic chemistry. Angew. Chem., Int. Ed. 2010, 49, 1809–1812. 10.1002/anie.200905699. [DOI] [PubMed] [Google Scholar]
- Hsieh A. C.; Liu Y.; Edlind M. P.; Ingolia N. T.; Janes M. R.; Sher A.; Shi E. Y.; Stumpf C. R.; Christensen C.; Bonham M. J.; Wang S.; Ren P.; Martin M.; Jessen K.; Feldman M. E.; Weissman J. S.; Shokat K. M.; Rommel C.; Ruggero D. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C.; Carragher N. O.; Unciti-Broceta A. eCF309: a potent, selective and cell-permeable mTOR inhibitor. MedChemComm 2016, 7, 471–477. 10.1039/C5MD00493D. [DOI] [Google Scholar]
- Zhang C. H.; Zheng M. W.; Li Y. P.; Lin X. D.; Huang M.; Zhong L.; Li G. B.; Zhang R. J.; Lin W. T.; Jiao Y.; Wu X. A.; Yang J.; Xiang R.; Chen L. J.; Zhao Y. L.; Cheng W.; Wei Y. Q.; Yang S. Y. Design, synthesis, and structure-activity relationship studies of 3-(phenylethynyl)-1h-pyrazolo[3,4-d]pyrimidin-4-amine derivatives as a new class of src inhibitors with potent activities in models of triple negative breast cancer. J. Med. Chem. 2015, 58, 3957–3974. 10.1021/acs.jmedchem.5b00270. [DOI] [PubMed] [Google Scholar]
- Frame M. C. Src in cancer: deregulation and consequences for cell behaviour. Biochim. Biophys. Acta, Rev. Cancer 2002, 1602, 114–130. 10.1016/S0304-419X(02)00040-9. [DOI] [PubMed] [Google Scholar]
- Carragher N. O.; Westhoff M. A.; Fincham V. J.; Schaller M. D.; Frame M. C. A novel role for FAK as a protease-targeting adaptor protein: regulation by p42 ERK and Src. Curr. Biol. 2003, 13, 1442–1450. 10.1016/S0960-9822(03)00544-X. [DOI] [PubMed] [Google Scholar]
- Dar A. C.; Lopez M. S.; Shokat K. M. Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem. Biol. 2008, 15, 1015–1022. 10.1016/j.chembiol.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvold K. R.; Santos S. M.; Breen M. E.; Lachacz E. J.; Steffey M. E.; Soellner M. B. Exquisitely specific bisubstrate inhibitors of c-Src kinase. ACS Chem. Biol. 2015, 10, 1387–1391. 10.1021/cb501048b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton E. E.; Dhillon P.; Amatruda J. F.; Ramakrishnan L. Spotlight on zebrafish: translational impact. Dis. Models & Mech. 2014, 7, 731–733. 10.1242/dmm.017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo V. E.; Varshney G. K.; Lee M.; Bupp S.; Xu L.; Shinn P.; Crawford N. P.; Inglese J.; Burgess S. M. Phenotype-driven chemical screening in zebrafish for compounds that inhibit collective cell migration identifies multiple pathways potentially involved in metastatic invasion. Dis. Models & Mech. 2015, 8, 565–576. 10.1242/dmm.018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T.; Roeser T.; Staub W.; Baier H. A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development 2005, 132, 2955–2967. 10.1242/dev.01861. [DOI] [PubMed] [Google Scholar]
- Qiu Z.; Cang Y.; Goff S. P. c-Abl tyrosine kinase regulates cardiac growth and development. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1136–1141. 10.1073/pnas.0913131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chislock E. M.; Ring C.; Pendergast A. M. Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1-mediated survival. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12432–12437. 10.1073/pnas.1304188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasatinib (Sprycel), Summary Basis of Approval; U.S. Food and Drug Administration: Silver Spring, MD, 2006.
- Sander T.; Freyss J.; von Korff M.; Reich J. R.; Rufener C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. 10.1021/ci800305f. [DOI] [PubMed] [Google Scholar]
- Kamath A. V.; Wang J.; Lee F. Y.; Marathe P. H. Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS-354825): a potent oral multi-targeted kinase inhibitor against SRC and BCR-ABL. Cancer Chemother. Pharmacol. 2008, 61, 365–376. 10.1007/s00280-007-0478-8. [DOI] [PubMed] [Google Scholar]
- Animal experiments were performed under Home Office License in compliance with the Animals (Scientific Procedures) Act 1986 and approved by the University of Edinburgh Ethics Committee.
- Steegmann J. L.; Cervantes F.; le Coutre P.; Porkka K.; Saglio G. Off-target effects of BCR-ABL1 inhibitors and their potential long-term implications in patients with chronic myeloid leukemia. Leuk. Lymphoma 2012, 53, 2351–2361. 10.3109/10428194.2012.695779. [DOI] [PubMed] [Google Scholar]
- Nygaard H. B.; Wagner A. F.; Bowen G. S.; Good S. P.; MacAvoy M. G.; Strittmatter K. A.; Kaufman A. C.; Rosenberg B. J.; Sekine-Konno T.; Varma P.; Chen K.; Koleske A. J.; Reiman E. M.; Strittmatter S. M.; van Dyck C. H. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimer's Res. Ther. 2015, 7, 35. 10.1186/s13195-015-0119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvold K. R.; Steffey M. E.; Fox C. C.; Soellner M. B. Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 2012, 7, 1393–1398. 10.1021/cb300172e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green T. P.; Fennell M.; Whittaker R.; Curwen J.; Jacobs V.; Allen J.; Logie A.; Hargreaves J.; Hickinson D. M.; Wilkinson R. W.; Elvin P.; Boyer B.; Carragher N.; Plé P. A.; Bermingham A.; Holdgate G. A.; Ward W. H.; Hennequin L. F.; Davies B. R.; Costello G. F. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol. Oncol. 2009, 3, 248–261. 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creedon H.; Brunton V. G. Src kinase inhibitors: promising cancer therapeutics?. Crit. Rev. Oncog. 2012, 17, 145–159. 10.1615/CritRevOncog.v17.i2.20. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, 522–525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Robertson A. D.; Jensen J. H. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct., Funct., Genet. 2005, 61, 704–721. 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosconati S.; Forli S.; Perryman A. L.; Harris R.; Goodsell D. S.; Olson A. J. Virtual screening with autodock: theory and practice. Expert Opin. Drug Discovery 2010, 5, 597–607. 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.