

Abstract

Components of the chromatin remodelling switch/sucrose nonfermentable (SWI/SNF) complex are recurrently mutated in tumors, suggesting that altering the activity of the complex plays a role in oncogenesis. However, the role that the individual subunits play in this process is not clear. We set out to develop an inhibitor compound targeting the bromodomain of BRD9 in order to evaluate its function within the SWI/SNF complex. Here, we present the discovery and development of a potent and selective BRD9 bromodomain inhibitor series based on a new pyridinone-like scaffold. Crystallographic information on the inhibitors bound to BRD9 guided their development with respect to potency for BRD9 and selectivity against BRD4. These compounds modulate BRD9 bromodomain cellular function and display antitumor activity in an AML xenograft model. Two chemical probes, BI-7273 (1) and BI-9564 (2), were identified that should prove to be useful in further exploring BRD9 bromodomain biology in both in vitro and in vivo settings.
Introduction
Chromatin remodelling complexes regulate nucleosome positioning along DNA.1 These complexes are required for a variety of processes, including chromatin organization, transcriptional regulation, decatenation of chromatids during mitosis, and DNA repair.2 The mammalian switch/sucrose nonfermentable (SWI/SNF) complex is one of four mammalian chromatin remodelling complexes. Recurrent inactivating mutations in certain subunits of this complex have been identified in different cancers. Despite its known roles in tumor suppression, the mammalian SWI/SNF complex has recently received attention as a potential target for therapeutic inhibition.3 This stems from the recognition that residual SWI/SNF complexes are critical for the growth of genetically defined cancers, including SWI/SNF mutant and Max mutant tumors as well as acute leukemias.4,5 In acute leukemias, it was found that the SWI/SNF complex supports an oncogenic transcriptional program. In the absence of the SWI/SNF ATPase Brg1, leukemic cells arrest in G1 and differentiate. A recent study highlighted a role of another SWI/SNF subunit, BRD9, in leukemia growth. The BRD9 bromodomain (BD) was shown to be required for the proliferation of acute myeloid leukemia (AML) cells.6
Over the past decade, chemical probe compounds have been shown to be invaluable in the elucidation of protein function.7,8 We set out to develop a probe compound targeting the BD of BRD9 in order to evaluate the function of this domain within the SWI/SNF complex. BDs are protein-binding domains with an affinity to lysine-acetylated target proteins.9 The acetyl-lysine binding pockets of these domains have been shown to be amenable to inhibition by drug-like small molecules, and the activity of several inhibitors directed against bromodomain and extra-terminal motif (BET) containing proteins (BRD2, BRD3, BRD4, and BRD-T) is being clinically assessed in cancer, including hematopoietic malignancies,10,11 and atherosclerosis (http://www.resverlogix.com/blog/tag/atherosclerosis/). A key selectivity parameter in designing our tool compounds was to avoid activity against BET family proteins because of the pleiotropic effects that BET inhibitors exert on various cellular processes.12
Recently, three BRD9 inhibitors have been published in the literature: LP99,13I-BRD9,14 and ketone “compound 28”15 (Supporting Information Table 3). LP99 is the first published potent and selective inhibitor of BRD9 and BRD7 [KD (BRD9, ITC) = 99 nM vs KD (BRD7, ITC) = 909 nM]; its structure is based on a methylquinolinone scaffold.13 Cellular target engagement was demonstrated using a quantitative NanoBRET assay [IC50 (BRD9-H3.3) = 5.1 μM and IC50 (BRD9-H4) = 6.2 μM]. I-BRD9, which is derived from a thienopyridone scaffold, is a potent cell-active selective binder to BRD9 [KD (BRD9, DiscoveRx) = 1.9 nM vs KD (BRD7, DiscoveRx) = 380 nM; IC50 (BRD9-H3.3, NanoBRET) = 158 nM], but it presents some residual affinity toward other BET family members [KD (BRD4-BD1, DiscoveRx) = 1400 nM].14 The reason for its high BRD9/BRD7 selectivity is currently not well understood. Additionally, I-BRD9 was shown to bind to endogenous BRD9 in a chemoproteomic assay. Finally, ketone “compound 28” was developed starting from a keto-indolizine BAZ2A/B chemical probe; the compound is potent and selective toward BRD9 and BRD7 [KD (BRD9, ITC) = 68 nM vs KD (BRD7, ITC) = 368 nM] and shows cellular activity at 1 μM in a fluorescence recovery after photobleaching (FRAP) assay.15,16
In this article, we describe the discovery and development of a potent and selective BRD9 BD inhibitor series based on a new scaffold arising from two parallel screening approaches consisting of fragment-based screening and virtual screening of proprietary libraries. In particular, we report the structure-based design of BRD9 inhibitor 1 (BI-7273), which was previously demonstrated to mimic genetic perturbation of BRD9.61 also targets BRD7 BD, which is a BD protein that has been found in a subclass of SWI/SNF remodelling complexes (PBAF)17 and shares high sequence homology with BRD9.18 We further describe a second BRD9 inhibitor, 2 (BI-9564),19−23 which displays enhanced selectivity against the BRD7 BD as well as improved pharmacokinetic properties when compared to those of 1. These two chemical probes, 1 and 2, should prove to be useful in further probing BRD9 BD biology in both in vitro and in vivo settings.
Results
Binding the BRD9 BD with an Isoquinolinone or a Pyridinone Scaffold
Two parallel screening approaches were followed to identify BRD9 BD binders. Three parallel biophysical assays, a differential scanning fluorimetry (DSF) assay, a surface plasmon resonance (SPR) assay, and a microscale thermophoresis (MST) assay, were used to screen our proprietary fragment library of around 1700 compounds against the BRD9 BD (Supporting Information Figures 1 and 2 and Table 1). It is of note that this is one of the first fragment screening applications described for the MST technology.24 The primary screening hits identified by these three screening methods were validated in an orthogonal binding assay using heteronuclear single quantum coherence nuclear magnetic resonance (15N HSQC NMR) (Supporting Information Figures 1–3). Seventy-seven hits showed significant cross peak shifts in the 2D 1H/ 15N HSQC NMR spectra, and 55 of these compounds were successfully soaked into the crystals of the BRD9 BD. The observed hit rate in MST screening was significantly higher than that with the other two techniques (124 primary hits for MST vs 36 and 45 for DSF and SPR, respectively), with only a 10% overlap in the hits identified by SPR and DSF. Alternative assay formats or sensitivities of the MST instrument (Monolith NT.015 prototype) may underlie the observed differences in the hit rate. However, despite the higher primary hit rate, MST screening yielded 38 hits validated by HSQC NMR, from which 29 were identified solely by this technology. From these 29 hits, 14 co-crystal structures could be obtained, indicating the significant value of pursuing single-technology hits in addition to hits identified by several orthogonal primary screening techniques. After quantification of the binding affinity of the 77 compounds by SPR, 12 compounds displayed a dissociation constant (KD) below 100 μM. Additionally, a proprietary high-concentration screening (HiCoS) library of ∼74 500 compounds was screened by Glide docking,25 followed by BRD9 BD pharmacophore mapping and finally filtering based on molecular weight (MW < 280) and lipophilicity (clogP < 2) (Supporting Information Figure 4). This virtual screening led to the selection of 208 available compounds, which were measured in DSF and SPR (% ctrl) assays. The binding affinity (KD) of the hits was quantified using SPR (Supporting Information Figure 1), leading to the discovery of 23 additional candidates, of which 11 compounds had their binding mode elucidated by X-ray co-crystal structure determination. All 23 compounds were resynthesized and had their binding affinity confirmed by SPR. Eleven compounds had a KD below 100 μM by SPR (Supporting Information Figure 1). Structure-based medicinal chemistry was then initiated using the X-ray co-crystal structures of the fragments obtained in the BRD9 BD (Supporting Information Figures 5–8 and Table 2).
The methylpyridopyrimidinone or dimethylpyridinone scaffold compound series was selected as a promising starting point based on their potential exit vectors and binding affinities [KD (3, SPR) = 37.5 μM, KD (4, SPR) = 9.1 μM] (Figure 1a, with the scaffold highlighted). The binding mode of the two compounds showed similar binding in the BRD9 anchor region (Figures 1b,c and Supporting Information Figures 5–8). The carbonyl of the pyridinone functionality makes two hydrogen bonds with the protein: a direct H-bond with the N δ2H2 side chain of Asn100 and an H-bond to Tyr57 via a conserved water molecule. A methyl group (either from the N-methyl or from the methyl alpha to the carbonyl) occupies a small lipophilic pocket surrounded by four conserved water molecules. The binding mode of these small acetylated lysine mimic binders is typical of those reported for other BDs.9 Indeed, similar binding activity was observed toward BET family members [e.g., IC50 (3, BRD4-BD1) = 80.2 μM and IC50 (4, BRD4-BD1) = 3.7 μM] for both fragments, highlighting the challenge of achieving high selectivity against BRD4. Additionally, the aromatic pyridinone core in compounds 3 and 4 makes a π-stacking interaction with the Tyr106 in the BRD9 BD. The distance between the aromatic core and Tyr106 was measured at 3.4 and 3.7 Å for 3 and 4, respectively (optimal aromatic C–aromatic C parallel offset stacking is 3.4–3.6 Å).26 Finally, in the specific case of compound 4, we observe a C–H π-interaction between Ile53 and the phenyl moiety (distance CH3–phenyl = 3.6 Å), which could be beneficial for improving potency toward BRD9 (Figure 1c).
Figure 1.

Binding mode of the methylpyridopyrimidinone 3 or dimethylpyridinone 4 scaffold in BRD9 BD (compound 3: PDB code 5F2P; compound 4: PDB code 5F25). (a) Structures and binding affinities of compounds 3 and 4 identified by FBS screening and virtual screening of HiCos library, respectively. (b, c) Binding mode of (b) 3 and (c) 4 in the BRD9 BD: H-bond to Asn100, water-bridged interaction with Tyr57, π-stacking with Tyr106, and C–H π-interaction with Ile53. Important amino acids for binding to the BRD9 BD are indicated. ZA channel, anchor region, and N-side are indicated in blue.
In parallel to the biophysical SPR assay, we developed a biochemical assay measuring the inhibitory effect of our compounds on the binding between acetylated histone H3 and BRD9 BD. This assay showed good correlation with the data obtained by SPR [KD (3, SPR) = 37.5 μM vs IC50 (3) = 48.9 μM; KD (4, SPR) = 9.1 μM vs IC50 (4) = 9.4 μM]. As the aim of the project was to develop a potent and selective inhibitor, we developed in vitro peptide displacement assays to routinely monitor the inhibitory effect of our compounds on the binding between BRD9, BRD7, BRD4-BD1, BRD4-BD2, or BRD2-BD1 BD and acetylated histone H3 or H4.
Structure-Based Design of BRD9 BD Inhibitors
Acetylated histones bind to their BD partners via their N-acetylated lysine tails, in particular interacting with a conserved asparagine (for BRD9 BD it is Asn100) in the anchor region and along a region previously referred to as the “N-side”27 (Figure 1b). Considering that this binding mode is conserved among all BDs, we decided to concentrate on improving potency by growing the initial fragment toward the less conserved “ZA channel” area (Figure 1b), as we anticipated that this approach would give us a greater chance of successfully achieving selectivity against BET family members.
In order to optimize the ZA channel linker, we used the dimethylpyridinone scaffold as an anchor binder. Both methyl and carbonyl functions on this scaffold are essential for effective binding to the BRD9 BD; indeed, removal of one of the methyl groups [position 1 (N-Me) or 3] or removal of the carbonyl moiety (e.g., replacement of anchor binder by 2,6-dimethylpyridine) led to a loss of binding. In the ZA channel of the BRD9 BD, we identified Phe44 (Figures 1b,c) as an amino acid of interest and hypothesized that addressing this interaction could improve potency. Compound 4 showed a suboptimal edge-to-face interaction (also referred to as T-stacking) with Phe44 [distance Phe44–phenyl (4) = 4.67 Å vs optimal aromatic C–aromatic C edge-to-face distance = 3.6–3.9 Å26]. It is reported that introduction of electron-donating groups on the facially substituted phenyl improves T-stacking.28 Following this principle, we replaced the amide functional group by a methylene dimethylamine (compound 5, Table 1), which led to an 8-fold increase in potency. Introduction of additional electron-donating groups onto the phenyl ring further enhanced potency by improving T-stacking with Phe44 (compounds 6–10, Table 1). Using 4-(dimethylamino)methyl-3,5-dimethoxyphenyl as a ZA channel linker (compound 7) increased the potency ∼170-fold compared to that of initial starting hit 4 [IC50 (7, BRD9) = 54 nM vs IC50 (4, BRD9) = 9338 nM]. It was possible to further boost potency by addressing the backbone carbonyl of His42 with either a hydroxyl moiety or an amine moiety (compounds 11 and 12, Table 1). The azetidine substituent provided the best vector to the His42 carbonyl without interfering with the optimal T-stacking of the dimethoxyphenyl ZA channel linker with Phe44. 4-(3-Aminoazedinylmethyl)-3,5-dimethoxyphenyl dimethylpyridinone (12) exhibited low nanomolar activity [IC50 (12, BRD9) = 9 nM]. Binding of the structurally closely related compound 11 was confirmed by X-ray co-crystal structure determination (Figure 2a and Supporting Information Figures 9 and 10 and Table 2). The anchor region binding is similar to the one observed with fragment 4: the pyridinone core binds through its carbonyl to Asn100 and to the conserved water molecule bound to Tyr57, a methyl group occupies the small lipophilic pocket, and the pyridinone makes a π-stacking interaction with Tyr106. In the ZA channel, the dimethoxyphenyl linker adopts a conformation that permits optimal T-stacking with Phe44, with the distance between the aromatic core and Phe44 now being the ideal 3.9 Å, while keeping the C–H π-interaction with Ile53, with the distance between aromatic core and Ile53 being 3.6 Å. The azedin-3-ol forms an H-bond to His42; additionally, for this molecule, we observe an induced fit with Phe47 closing onto the molecule due to a C–H π-interaction with the methoxy group of compound 11.29
Table 1. SAR for BRD9 Activity in the ZA Channela.


*Alpha format, mean value; number of measurements: 2–5.
Figure 2.

(a–c) Binding mode of BRD9 BD inhibitors (a) 11, (b) 1, and (c) 2 [compound 11: PDB code 5F1L; compound 1: PDB code 5EU1; compound 2: PDB code 5F1H]. (d) X-ray co-crystal structure of 1 in BRD9 (not shown) aligned with BRD4-BD1 (compound 1: PDB code 5EU1). (a) Binding mode of compound 11 in BRD9 BD: H-bond to Asn100, water-bridged interaction with Tyr57, π-stacking with Tyr106, C–H π-interaction with Ile53, T-stacking with Phe44, H-bond to His42, and induced fit Phe47/C–H π-interaction. (b) Binding mode of compound 1 in BRD9 BD:6 two H-bonds to Asn100, water-mediated hydrogen bond with Tyr57, π-stacking with Tyr106, C–H π-interaction with Ile53, and T-stacking with Phe44. (c) Binding mode of compound 2 in BRD9 BD: two H-bonds to Asn100, water-bridged interaction with Tyr57, π-stacking with Tyr106, C–H π-interaction with Ile53, T-stacking with Phe44,and induced fit Phe47/C–H π-interaction. (d) Clash between 1 and BRD4-BD1 BD amino acids (Trp81, Gln85, and Leu92) is shown (brown circles).
While improving the potency toward BRD9, we also observed an enhancement of the selectivity against the first BD of BRD4 (BRD4-BD1) compared to that of starting hit 4, which showed higher potency for the off-target BRD4-BD1 than for BRD9. Encouraged by these results, we investigated modifications in the anchor region in order to further improve selectivity. As stated previously, we considered it to be essential that our final BRD9 chemical probes were inactive against BRD4-BD1 and other BET family members to avoid any misinterpretation of biological results [i.e., IC50 (BET family members) > 100 μM]. After comparing the surface of BRD9 and BRD4-BD1, we hypothesized that enhanced selectivity could be achieved by forcing a clash with key amino acids in the anchor region or the ZA channel of BRD4-BD1. This could be done by introducing substituents at the 4 or 6 position on the pyridine-2-one core of our inhibitors to force a change in the torsion angle between the anchor binder ring and the ZA channel linker ring (Table 2). A methyl group at position 4 (or 6) forced a twist between the 2 aryl moieties, “anchor binder” pyridine-2-one and “ZA channel linker” (measured torsion angle approximately −60° vs −27° to −44° when hydrogen is present), and indeed translated to an improvement of selectivity against the BET family [compounds 13 (BI-7189)6 and 14, Table 2]. On the basis of the same principle, an aromatic ring merged to the pyridinone scaffold gave an improved selectivity against the BET family and concomitantly improved the π-stacking interaction with Tyr106 in the BRD9 anchor region [compounds 15 (BI-7271)6 to 19, Table 2). The most efficient inhibitor was 2-methyl-2,7-naphthyridin-1-one compound 1 with a 3-fold increase in affinity for BRD9 and 50-fold increase in selectivity against BRD4-BD1 over compound 7 (selectivity BRD9 vs BRD4-BD1: ∼100-fold for compound 7 and >5200-fold for 1). 1 forms an additional positive interaction with the carbonyl of Asn100 in BRD9; indeed, the presence of a nitrogen atom at position 7 on the naphthyridinone ring acidifies the CH bond at position 8, permitting an interaction with the carbonyl side chain of Asn100 (Figure 2b and Supporting Information Figures 11 and 12 and Table 2). This translates into an improved potency for BRD9 BD. 1 displays no measurable activity toward BET family BDs up to a concentration of 100 μM in our biochemical Alpha assay, which can be explained by a potential clash of the anchor part of the molecule with Leu92 in BRD4-BD1 and a clash of the trisubstituted phenyl linker of the molecule with Gln85 and Trp81 in BRD4-BD1 (Figure 2d, superimposition of 1 with the BRD4-BD1 surface).
Table 2. SAR for BRD9, BRD4-BD1, BRD4-BD2, BRD2-BD1, and BRD7 Activitya.


*Alpha format, mean value; number of measurements: 2–5.
Finally, while investigating modifications on the ZA linker part, we discovered that para-substituted dimethoxy groups resulted in an enhanced selectivity for BRD9 over its closest homologue, BRD7 (Table 3). We believe that this selectivity might stem from differences in flexibility between the two proteins. On the basis of the protein melting properties shown in the thermal shift assay, we hypothesized that the BRD7 BD was more flexible and dynamic in solution than BRD9. 2 was slightly less potent toward BRD9 compared to that of 1 in the Alpha assay, but it showed improved selectivity against BRD7 (45-fold more potent for BRD9 vs BRD7) and, most importantly, remained inactive toward BET family members (Scheme 1). The binding mode of 2 in BRD9 BD was confirmed by X-ray co-crystal structure determination (Figure 2c and Supporting Information Figures 13 and 14 and Table 2). 2 bound with an induced fit of Phe47 in the ZA channel, similar to that for compound 11. Synthesis of para-substituted dimethoxy analogues of 2, which could directly address the carbonyl of His42 with either a benzyl amine linker or sulphonamide linker at the 4 position of the ZA linker, yielded compounds with an improved BRD9 potency and similar selectivity profile compared to those of 2. However, these compounds later presented less attractive pharmacokinetic properties (e.g., low permeability or high efflux ratio), which made them unsuitable for in vivo testing.
Table 3. SAR for BRD9, BRD4-BD1, BRD4-BD2, BRD2-BD1, and BRD7 Activitya.
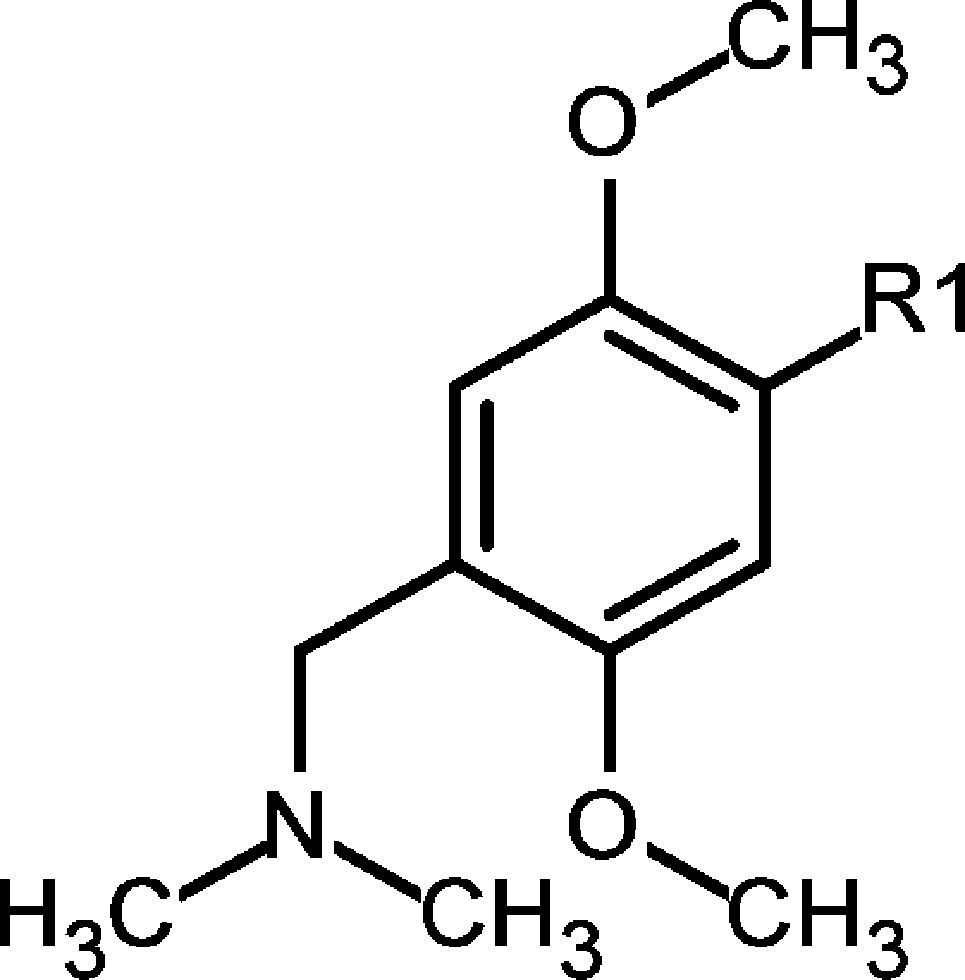

*Alpha format, mean value; number of measurements: 2–5.
Scheme 1. Synthesis of 2.

Reagents and conditions: (a) NaH, MeI, DMF, rt, 71%; (b) HNMe2·HCl, NaOAc, AcOH, NaBH(OAc)3, DCM, rt, 89%; (c) B2pin2, KOAc, Pd(dppf)Cl2, 1,4-dioxane, 90 °C, 63%; (d) Pd(dppf)Cl2·DCM, Na2CO3(aq), DMF, 100 °C, 17%
Finally, binding affinities of 1 and 2 toward BRD9 were measured by isothermal titration calorimetry (ITC) and bromoKdELECT (Figure 3, Table 4, and Supporting Information Figures 17 and 18). High-affinity binding to BRD9 BD was confirmed for both compounds [KD (1, ITC) = 15.4 nM and KD (2, ITC) = 14.1 nM]. 1 and 2 were selected as BRD9 BD chemical probes for further in vitro and in vivo profiling.
Figure 3.

ITC analysis of compounds 1 and 2 (T = 293.15 K). (a) Compound 1 binds with a KD value of 15.4 nM (ΔH = −12.1 kcal/mol) and (b) 2 binds with a KD value of 14.1 nM (ΔH = −11.2 kcal/mol).
Table 4. Summary of Properties of 1 and 2.

1 and 2 Are Potent, Selective, and Cell-Permeable BRD9 BD Inhibitors
Target engagement in the cell was demonstrated in a semiquantitative FRAP assay16 using a green fluorescent protein–BRD9 fusion protein expressed in U2OS cells. 2 showed inhibition of BRD9 in cells at 100 nM, whereas 1 was active in the cell at 1 μM (with 1 μM being the lowest concentration tested) (Figure 4a–d and Table 4). No compound-related toxicity was observed in U2OS cell lines after 24 h.
Figure 4.

FRAP assay using U2OS cells transfected with GFP–BRD9. (a) Recovery half times of wild-type (wt) cells treated with DMSO in the absence or presence of 2.5 μM SAHA or treated with 1 at 1 μM and SAHA as indicated. In addition, cells expressing GFP–BRD9 with a BD-inactivating mutation (N100F) were analyzed. Significant differences relative to cells treated with SAHA (p < 0.0001) are shown by ****. (b) Time dependence of fluorescence recovery in the bleached area of cells expressing wt or mutant GFP–BRD9 with the corresponding treatments shown in (a). (c) Recovery half times of cells expressing wt GFP–BRD9 treated with various concentrations of DMSO and 2 in the presence or absence of SAHA as indicated. Cells expressing the GFP–BRD9 mutant (N100F) were treated as indicated. Significant differences relative to cells treated with SAHA (p < 0.0001) are shown by ****. (d) Time dependence of fluorescence recovery in the bleached area of cells expressing wt or mutant GFP–BRD9 with the corresponding treatments shown in (c). Curves represent averaged data of at least 20 replicates. 1 shows potency (100% inhibition) at 1 μM in the BRD9 FRAP assay. 2 shows potency (∼90% inhibition) at 0.1 μM in the BRD9 FRAP assay. Both compounds showed no toxicity in U2OS cells after 24 h. The N100F construct is a negative control BRD9 mutant in which Asn100 is replaced by Phe100 and therefore acetylated histone cannot bind because of the lack of interaction to the anchor Asn and because of steric hindrance. SAHA is added to the mixture to increase the signal-to-noise ratio by inhibiting the deacetylation of histones.
To assess selectivity, the compounds were profiled against a variety of other BDs. BD selectivity was checked by differential scanning fluorimetry for 48 BDs followed by ITC KD determination (Table 4 and Supporting Information Figures 15, 16, 19, and 20). The same selectivity pattern was observed using the bromoMAX/bromoKdMAX technology (32 BDs screened; Supporting Information Figures 17 and 18). High selectivity against BET family members was confirmed, with KD values exceeding 10 μM. Aside from BRD9, highly homologous BRD7 and CECR2 were the only two BDs identified as additional targets (Table 4 and Supporting Information Figures 19 and 20). Although 2 showed potency at 1 μM in a BRD7 FRAP assay (Table 4 and Supporting Information Figure 22), pleasingly, no cellular inhibition of the CECR2 BD was observed at this concentration (Table 4 and Supporting Information Figure 23). CECR2 has been described as being part of the CECR2-containing-remodeling factor (CERF) complex. Aside from its BD, CECR2 contains an AT hook motif, a DNA-binding motif with a preference for A/T-rich regions,30 that might contribute to the reduced displacement from chromatin by 2 in the FRAP assay compared to BRD9/7, which do not possess any additional DNA or chromatin binding domains.
Further profiling was conducted to assess the selectivity over a range of targets, particularly kinases and G-protein coupled receptors (GPCRs). Concentrations of compound 2 of less than 5 μM showed no activity against 324 kinases, and at 10 μM, an inhibition >40% was observed for only 2 out of 55 GPCRs (Table 4 and Supporting Information Figure 24). 1 showed an inhibition below 40% against 31 kinases at 10 μM.
Inhibition of Cellular Proliferation by 1 and 2
The cellular response to BRD9 inhibition was assessed in a broad cancer cell line panel. Treatment of the panel with 2 resulted in selective growth inhibition of a significant proportion of AML cell lines tested (Supporting Information Figure 31). This result was in agreement with decreased proliferation in the murine AML RN2 cell line following treatment with 1.6
CECR2 mRNA expression was hardly detectable in any of the eight sensitive cell lines, which, in addition to the lack of cellular activity of 2 in the CECR2 FRAP assay (Supporting Information Figure 23), suggests that the antiproliferative effect was not due to the effects of the compound on this BD. The most sensitive cell line was human acute myeloid eosinophilic leukemia cell line EOL-1 [EC50 (2, EOL-1) = 800 nM; EC50 (1, EOL-1) = 1400 nM] (Figure 5, Supporting Information Figure 31).
Figure 5.
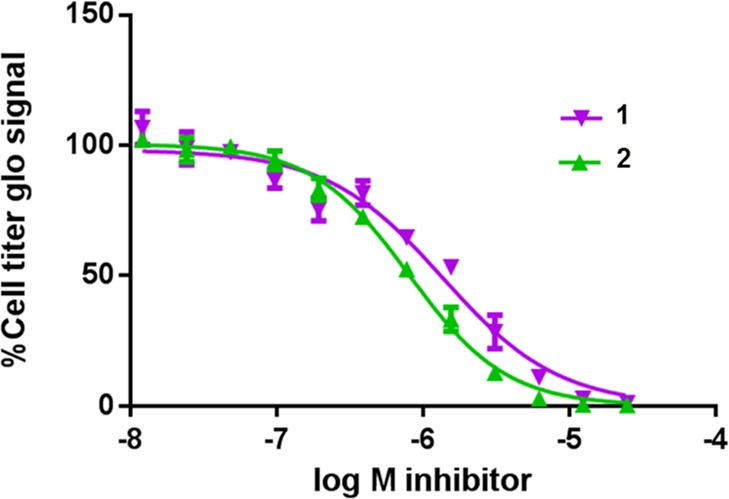
1 and 2 block EOL-1 cell proliferation with EC50’s of 1400 and 800 nM, respectively.
As observed from the phenotype of murine cells exposed to 1,6 BRD9 inhibition translated into a potent but only partial inhibition of MYC expression in AML cell lines (Supporting Information Figure 32a–d). The reason for the inhibition of MYC expression is currently not understood. Complete suppression of MYC expression, observed at higher concentrations with some of the less selective compounds, was likely due to the effects of the compounds on BET family members [e.g., IC50 (13, BRD4-BD1) = 31 μM, IC50 (15, BRD4-BD1) = 24 μM] (Supporting Information Figure 32a,b).
BRD9 can fulfill its cellular function in murine leukemia cells when its BD is exchanged with that of BRD4.6 These domain-swap experiments were used to demonstrate that the antiproliferative activities of 1 in this cell line are due to its effect on the BRD9 BD. Using 2 in such domain-swap experiments up to 5 μM concentration, we could further confirm that BRD9 BD is responsible for mediating the antiproliferative effects of 2. However, we also noted a degree of off-target effects of 2 in this murine leukemia cell line when it was used at higher concentrations (data not shown). Nonetheless, these findings suggested that 2 is a suitable chemical probe for probing the in vitro and in vivo functions of BRD9.
Pharmacokinetic Profiling of 1 and 2
Compounds 1 and 2 both showed attractive ADME/PK profiles for in vivo proof-of-concept studies, namely, high solubility at pH 6.8, moderate to high in vitro metabolic stability, low plasma protein binding, and no cytochrome P450 inhibition, together with moderate to high absorptive permeability and moderate in vivo plasma clearances upon i.v. dosing (Table 4 and Supporting Information Figures 25 and 28). Despite elevated efflux ratios in the Caco-2 transporter assay, both compounds displayed high oral bioavailability (Table 4 and Supporting Information Figures 25–30). In order to explore the potential of 2 and 1 as in vivo chemical probes, female BomTac:NMRI-Foxn1nu mice were given two doses orally (20 and 180 mg/kg p.o.) and the compound concentration in plasma over time was measured. Dose-dependent but nonlinear AUCs were obtained for both compounds, achieving exposures that were higher compared to the EC50 levels determined for both compounds in proliferation assays with EOL-1 cells (Figure 5). 2 presented an exposure over time that was twice that of 1 and a higher oral bioavailability (Table 4). This justified in vivo efficacy experiments in mice at the highest dose of 2 (180 mg/kg p.o.) in the disseminated EOL-1 AML mouse model (summary of properties: Table 4 and Supporting Information Figures 26, 27, 29, and 30).
Efficacy of 2 in a Disseminated Mouse Model of AML
Prior to performing efficacy studies, 1 week tolerability studies were carried out using 180 mg/kg of 2 in non-tumor-bearing female CIEA-NOG mice. Continuous daily dosing of the selected dose was well-tolerated, with a medium weight change on day 7 of −3.8% (n = 4).
The human acute myeloid eosinophilic leukemia cell line EOL-1 was chosen for in vivo experiments as it was the most sensitive cell line in vitro to BRD9 inhibition. A disseminated model, which more closely reflects the clinical situation compared to that of a subcutaneous xenograft, was chosen to assess the efficacy of 2. EOL-1 cells (107), stably transduced with a luciferase-expressing vector to allow continuous assessment of tumor load by bioluminescence, were injected in the tail vein of CIEA-NOG mice. Oral treatment with 180 mg/kg of 2 was initiated on day 5 and applied daily (q.d.) with an interruption at days 18 and 19. A significant (p = 0.0086) reduction in tumor growth (measured in average radiance [p/s/cm2/sr]) compared to that of controls was observed on day 18, resulting in a median tumor growth inhibition (TGI) value of 52% (Figure 6a). Imaging data on day 18 provided evidence of a significantly reduced disease burden (Figure 6b) in mice treated with compound 2. The animals were closely monitored for clinical signs and were sacrificed when the disease burden exceeded a prespecified grade as a surrogate end point for survival. An increase in tumor burden in the control mice resulted in body weight loss at the end of the study. On day 18, dosing with 2 was interrupted for 2 days, as continuous body weight loss was observed, and a body loss of 15% was observed in one mouse. The body weight loss might be explainable by an increased tumor burden because on this day the control group showed a median body weight loss of −11 compared to −8 in the treated group; however, tolerability issues with the compound cannot be ruled out (Figure 6c). In the disseminated EOL-1 mouse model, the median survival of the vehicle (Natrosol 0.5%) treated control animals was 20 days. Median survival of the animals treated with 180 mg/kg 2 resulted in a modest but significant additional survival benefit of 2 days compared to survival of the control group (Figure 6d). Pharmacokinetic analysis of plasma samples taken on the last day of treatment showed that high systemic exposure had been reached, with an AUC0–last of 268 000 nM·h, as expected from single-dose PK experiments in mice (Table 4). The mean total plasma concentrations of compound 2 exceeded the EC50 of 800 nM from the EOL-1 cellular proliferation assay for 20 h following dosing. While the antileukemia effects of 2 in this model are modest, these experiments demonstrate that 2 is a suitable probe to evaluate the effects of in vivo modulation of BRD9 BD activity.
Figure 6.

Efficacy and tolerability of BRD9 inhibitor 2 in a xenograft model of human AML. Data for panels a–d was collected in the same in vivo experiment. (a) CIEA-NOG mice were injected intravenously with 107 EOL-1 AML cells. Starting on day 5 after cell injection, mice were orally treated with vehicle (daily) or with 180 mg/kg 2 (qd5–17 and qd20–22). With the help of bioluminescence imaging, the tumor burden was assessed in each animal on days 5, 8, 11, 14, and 18 and calculated as median average radiance [p/s/cm2/sr]. One-sided decreasing Mann–Whitney tests were used to compare tumor volumes. (b) Bioluminescence imaging on day 18: (upper) vehicle control and (lower) 180 mg/kg 2. (c) Average body weight changes as a percentage of the initial weight. (d) Kaplan–Meier curve showing prolonged survival of animals treated with 180 mg/kg 2 (vehicle = black line; 180 mg/kg 2 = blue line). The p-value was calculated by Mann–Whitney test.
Discussion
Epigenetic modifications have been linked to many diseases, in particular, cancer and immune/inflammatory disorders. The molecular machinery required to read and modify chromatin has been shown to comprise large protein complexes where the activities of multiple domains are coordinated. The need to identify the activities that drive disease pathology has spurred the development of high-quality inhibitors to probe the function of individual domains in the context of the native cellular complexes.
In this article, we focused on the BD of BRD9, a subunit of the SWI/SNF chromatin remodeling complex. Our study describes how a combination of FBS and virtual screening of proprietary libraries allowed us to identify a new scaffold class of BRD9 BD inhibitors. Among the current BET-sparing BRD9 BD inhibitors, 2 and 1 displayed the greatest cellular potency. This was principally achieved following a structure-guided chemical optimization that led to the introduction of 2-methyl-2,7-naphthyridin-1-one as an anchor region binder. Optimization of the compounds focused on addressing all key interactions as efficiently as possible, keeping the overall size of the final molecule small and the ligand efficiency high [MW (1) = MW (2) = 353.4 Da; ligand efficiency (1) = 0.41 and LE (2) = 0.38; lipophilic ligand efficiency (1) = 5.7 and LLE (2) = 5.7]. It is of note that CECR231 was the only in vitro off-target identified outside the BRD9/BRD7 subfamily; however, at 1 μM, no cellular inhibition of this BD was observed in the CECR2 FRAP assay. The overall ADME properties of the two compounds permit them to be used in in vivo experiments. Early work in a disseminated mouse model of AML showed efficacy for 2 (at 180 mg/kg) with a median TGI value of 52% on day 18, which translated into an additional survival benefit compared to that of the control group.
Three structurally unrelated BRD9 inhibitors have been recently published in the literature: LP99,13I-BRD9,14 and ketone “compound 28” (Supporting Information Table 3).15,19 As selective, potent compounds with cellular activity, 1 and 2 will prove to be invaluable as tools to further explore BRD9 biology (Table 4). The ADME parameters of these two inhibitors will, in addition, allow the scientific community to elucidate the role of BRD9, either as a single agent or in combination with other inhibitors, in both in vitro and in vivo settings. Compound 2 is available to the scientific community via the SGC consortium23 as a BRD9/BRD7 potent, selective, cell-permeable, and noncytotoxic probe compound (http://www.thesgc.org/chemical-probes/BI-9564). We believe that this molecular probe will provide a useful tool to broaden the study of chromatin regulators not only in oncology but also potentially in additional therapeutic areas such as neurology, immunology,14,32 and inflammation.13
Experimental Section
Unless otherwise indicated, all reactions were carried out in standard commercially available glassware using standard synthetic chemistry methods. Air- and moisture-sensitive reactions were performed under an atmosphere of dry nitrogen or argon with dried glassware. Commercial starting materials were used without further purification. Solvents used for reactions were of commercial dry, extra-dry, or analytical grade. All other solvents used were reagent grade.
Preparative RP-HPLC was carried out on an Agilent or Gilson system using columns from Waters (Sunfire C18 OBD, 5 or 10 μm, 20 × 50 mm, 30 × 50 mm, or 50 × 150 mm; X-Bridge C18 OBD, 5 or 10 μm, 20 × 50, 30 × 50, or 50 × 150 mm) or YMC (Triart C18, 5, or 10 μm, 20 × 50 or 30 × 50 mm). Unless otherwise indicated, compounds were eluted with MeCN/water gradients using either acidic (0.2% HCOOH or TFA) or basic water (5 mL 2 M NH4HCO3 + 2 mL of NH3 (32%) brought up to 1 L with water).
NMR experiments were recorded on Bruker Avance 400 and 500 MHz spectrometers at 298 K. Samples were dissolved in 600 μL of DMSO-d6 or CDCl3, and TMS was added as an internal standard. One-dimensional 1H spectra were acquired with 30° excitation pulses and an interpulse delay of 4.2 s with 64k data points and 20 ppm sweep width. One-dimensional 13C spectra were acquired with broadband composite pulse decoupling (WALTZ16) and an interpulse delay of 3.3 s with 64k data points and a sweep width of 240 ppm. Processing and analysis of 1D spectra were performed with Bruker Topspin 2.0 software. No zero filling was performed, and spectra were manually integrated after automatic baseline correction. Chemical shifts are reported in ppm on the δ scale.
Analytical LC/MS data [LC/MS(BAS1)] were measured on an Agilent HPLC 1100 series with an Agilent LC/MSD SL detector using a Waters X-Bridge (C18, 2.5 μm, 2.1 × 20 mm) column (part no. 186003201) and solvents A [20 mM aqueous NH4HCO3/NH3 (pH 9)] and B [acetonitrile HPLC grade] as eluent (additional settings: flow, 1 mL/min; injection volume, 5 μL; column temp, 60 °C). Standard gradient was as follows: 0.00 min, 10% B; 0.00–1.50 min, 10% → 95% B; 1.50–2.00 min, 95% B; and 2.00–2.10 min, 95% → 10% B. For some intermediates, analytical LC/MS data was measured using different methods: LC/MS(INT1) was measured on a Shamadzu HPLC LC-20AB, SPD-M20A 190–370 nm system using a Luna C18(2) (5 μm, 50 × 2 mm) column and solvents A [H2O containing 0.0375% TFA] and B [acetonitrile HPLC grade containing 0.018% TFA] as eluent (additional settings: flow, 0.8 mL/min; column temp, 40 °C). Standard gradient was as follows: 0.00 min, 10% B; 0.00–4.00 min, 10% → 80% B; 4.00–4.90 min, 80% B; 4.90–4.92 min, 80% B → 10% B; and 4.92–5.50 min, 10% B. LC/MS(INT2) was measured on an Agilent HPLC 1200 Series (DAD 200–400 nm) with an Agilent 6120 MS system using a Luna C18(2) (3 μm, 30 × 2 mm) column and solvents A [H2O containing 0.0375% TFA] and B [acetonitrile HPLC grade containing 0.018% TFA] as eluent (additional settings: flow, 1.0 mL/min; column temp, 50 °C). Standard gradient was as follows: 0.00 min, 10% B; 0.00–1.15 min, 10% → 80% B; 1.15–1.55 min, 80% B; 1.55–1.56 min, 80% B → 10% B; and 1.56–2.99 min, 10% B.
HRMS data were recorded using a Thermo Scientific Orbitrap Elite hybrid ion trap/orbitrap spectrometer system with an Ultimate 3000 series LPG-3400XRS pump system. Mass calibration was performed using the Pierce LTQ Velos ESI positive ion calibration solution from Thermo Scientific (lot PF200011, product no. 88323)
The purity of the biologically evaluated compounds was determined by LC/MS (for all compounds) and Q-NMR (for compounds 1 and 2) to be above >95%.
Compound 3 is commercially available from, e.g., ChemDiv.
Selected Experimental Procedures
4-Bromo-2-methyl-1,2-dihydro-2,7-naphthyridin-1-one (24)
Sodium hydride (3.41 g, 142 mmol) was added slowly to a cooled solution (0 °C) of 4-bromo-1,2-dihydro-2,7-naphthyridin-1-one (23) (16.0 g, 71.1 mmol; commercial from Activate) in DMF (300 mL), and the resulting mixture was stirred for 0.5 h. Methyl iodide (40.4 g, 285 mmol) was added slowly, and stirring was continued for 2 h. The reaction mixture was quenched with ice water, whereupon the product precipitates. The solid was collected by filtration, washed, and dried in vacuo to give pure 4-bromo-2-methyl-1,2-dihydro-2,7-naphthyridin-1-one (24) (12.0 g, 50.2 mmol, 71%). 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.87 (d, J = 5.6 Hz, 1H), 8.26 (s, 1H), 7.62 (d, J = 5.6 Hz, 1H), 3.53 (s, 3H); LC/MS (BAS1): [M + H]+ = 239/241; tR = 0.92.
[(4-Bromo-2,5-dimethoxyphenyl)methyl]dimethylamine (26)
A solution of NaOAc (6.10 g, 44.9 mmol), AcOH (2.45 g, 40.8 mmol), and dimethylamine hydrochloride (6.98 g, 85.7 mmol) in DCM (160 mL) was stirred for 10 min at rt. 4-Bromo-2,5-dimethoxybenzaldehyde (25) (10.0 g, 40.8 mmol) was added, and stirring was continued. After 30 min, sodium triacetoxyborohydride (17.2 g, 81.6 mmol) was added in one portion, and the reaction mixture was stirred at rt for 16 h. A saturated NaHCO3 solution was added, and the layers were separated. The aqueous layer was extracted three times with DCM. The combined organic layer was dried over MgSO4, filtered, and evaporated to give [(4-bromo-2,5-dimethoxyphenyl)-methyl]dimethylamine (26) (10.0 g, 36.5 mmol, 89%). 1H NMR (500 MHz, DMSO-d6) δ 7.17 (d, J = 1.4 Hz, 1H), 7.06 (s, 1H), 3.78 (d, J = 1.5 Hz, 3H), 3.74 (d, J = 1.4 Hz, 3H), 3.35 (s, 2H), 2.16 (s, 6H); LC/MS (BAS1): [M + H]+ = 274/276; tR = 1.11 min.
{[2,5-Dimethoxy-4-(tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl}dimethylamine (27)
[(4-Bromo-2,6-dimethoxyphenyl)methyl]dimethylamine (26) (7.80 g, 28.5 mmol) and bis(pinacolato)diboron (21.7 g, 85.5 mol) were dissolved/suspended in 1,4-dioxane (150 mL) under N2. Potassium acetate (8.43 g, 49.6 mmol) and Pd(dppf)Cl2 (1.00 g, 1.37 mmol) were added, and the mixture was stirred at 90 °C for 8 h. After cooling to rt, the mixture was concentrated and the residue was taken up in DCM. Water was added, the layers were separated, and the aqueous phase was extracted three times with DCM. The combined organic layer was dried over Na2SO4, filtered, and evaporated. The crude product was purified by preparative RP-HPLC using a MeCN/water (0.2% TFA added to the water) gradient as eluent to give the TFA salt of {[2,6-dimethoxy-4-(tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl}-dimethylamine (27), which was transferred into the corresponding hydrochloride by dissolving and stirring in HCl/MeOH for 30 min (5.80 g, 18.1 mmol, 63%). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H, prot. amine), 7.24 (s, 1H), 7.16 (s, 1H), 4.24 (d, J = 5.4 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 2.70 (d, J = 4.9 Hz, 6H), 1.29 (s, 12H); LC/MS (BAS1): [M + H]+ = 240 (ester cleaved); tR = 0.70 min.
4-{4-[(Dimethylamino)methyl]-2,5-dimethoxyphenyl}-2-methyl-1,2-dihydro-2,7-naphthyridin-1-one (2)
4-Bromo-2-methyl-1,2-dihydro-2,7-naphthyridin-1-one (24) (7.44 g, 31.1 mmol), 27 (10.0 g, 31.1 mmol), and Pd(dppf)Cl2·DCM (2.54 g, 3.11 mmol) were suspended in DMF (100 mL) under argon. A degassed Na2CO3 solution (2 N, 38.9 mL, 77.8 mmol) was subsequently added, and the resulting mixture was heated at 100 °C for 2 h. After cooling to rt, DMF was evaporated and a mixture of MeOH/DCM was added. All solids were filtered off, and the filtrate was evaporated again to give the crude material, which was purified by flash chromatography on SiO2 using a MeOH/DCM gradient as eluent to give 4-{4-[(dimethylamino)methyl]-2,5-dimethoxy-phenyl}-2-methyl-1,2-dihydro-2,7-naphthyridin-1-one (2) (3.40 g, 9.62 mmol, 31%). Further purification was achieved by preparative RP-HPLC (X-Bridge C18 50 × 100 mm, 10 μm) using a MeCN/water gradient as eluent to give highly pure material (1.90 g, 5.38 mmol, 17%). 1H NMR (500 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.64 (d, J = 5.6 Hz, 1H), 7.74 (s, 1H), 7.12 (s, 1H), 7.03 (d, J = 5.6 Hz, 1H), 6.93 (s, 1H), 3.75 (s, 3H), 3.63 (s, 3H), 3.58 (s, 3H), 3.48–3.44 (m, 2H), 2.22 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 161.1, 151.7, 151.4, 150.8, 150.6, 142.0, 138.5, 128.4, 122.2, 120.0, 118.6, 115.1, 113.6, 113.5, 57.1, 56.5, 56.2, 45.8 (2C), 36.8. HRMS (CI+): calcd for C20H24N3O3 (MH+), 354.18122; found, 354.18091; Δ −0.88 ppm; LC/MS (BAS1): [M + H]+ = 354; tR = 0.91 min.
Acknowledgments
The authors are grateful for financial support by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. Boehringer Ingelheim is grateful for financial support by the Austrian Research Promotion Agency FFG (grants 836644, 842876, and 847818). A.F.H. is supported by a Boehringer Ingelheim Fonds Ph.D. Fellowship. C.R.V. is supported by a Burroughs-Wellcome Fund Career Award, a National Institutes of Health grant (NCI RO1 CA174793), and the Pershing Square Sohn Cancer Research Alliance. The authors thank Thomas Arnhold, Alexandra Beran, Helmut Berger, Adrian Carter, Sonia-Lucia Cesana, Yunhai Cui, Wolfgang Egermann, Norbert Eidkum, Andreas Fischer, Gerlinde Flotzinger, Michael Galant, Gerhard Gmaschitz, Eric Haaksma, Eva-Marie Haupt, Wolfgang Hela, Hans Hoffmann, Gudrun Illibauer, Matthias Klemencic, Martina Kohla, Roland Kousek, Norbert Kraut, Rebecca Langlois, Moriz Mayer, Jürgen Moll, Oliver Petermann, Franziska Popp*, Nikolai Pototschnig, Carlos Roberto Ramirez-Santa Cruz, Heidi Roth*, Samira Selman, Bernadette Sharps, Gabriella Siszler, Christian Smethurst, Michaela Streicher, Diane Thompson, Anika Weiss, Alexander Weiss-Puxbaum (*BI Global Skill Center Alternative Lead Identification).
Glossary
Abbreviations Used
- AML
acute myeloid leukemia
- BAF
BRG1- or HRBM-associated factors, also called SWI/SNF-A
- BD
bromodomain
- BET
bromodomain and extra-terminal
- DSF
differential scanning fluorimetry
- FBS
fragment-based screening
- FRAP
fluorescence recovery after photobleaching
- HiCoS library
high-concentration screening library
- HSQC NMR
heteronuclear single quantum coherence nuclear magnetic resonance
- ITC
isothermal titration calorimetry
- LE
ligand efficiency
- LLE
lipophilic ligand efficiency
- MST
microscale thermophoresis
- MW
molecular weight
- PBAF
polybromo-associated BAF, also called SWI/SNF-B
- SPR
surface plasmon resonance
- SWI/SNF
switch/sucrose non-fermentable
- TGI
tumor growth inhibition
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01865.
Description of all biophysical and biological methods and results, FBS screening and virtual screening, X-ray co-crystal structure, selectivity data (Bromoscan, TM shift, ITC, GPCR, kinase data for 1 and 2), FRAP assay data, PK profiles i.v. and p.o. for 1 and 2, cell results, and chemistry experimental procedures for compounds 1 and 4–22 (PDF)
SMILES strings and IC50 data for 1–22 (CSV)
Accession Codes
Atomic coordinates of BRD9 BD bound to compounds 1 (PDB code 5EU1),62 (PDB code 5F1H), 3 (PDB code 5F2P), 4 (PDB code 5F25), and 11 (PDB code 5F1L) have been deposited with the Protein Data Bank
Author Contributions
L.J.M. supervised the medicinal chemistry team. L.J.M, M.K., and M.H.H. wrote the manuscript. L.J.M and S.S. designed synthetic strategies. M.K. designed biological experiments and supervised the biology team. G.Ba., D.F., D.K., and H.N. determined and analyzed crystal structures. X.-L.C. performed the virtual screening of the HiCoS library. O.F. supervised ITC and FRAP assays. D.F. designed and supervised the crystal soaking experiments. T.G. designed and supervised the biochemical Alpha assays. A.F.H. carried out the BD-swap allele experiments. M.H.H. designed and supervised the animal experiments. D.K. and H.N. supervised the FBS screening. S.Kn., S.M., and G.Bo. supervised the collaboration with SGC. P.K. and S.Ko. synthesized the compounds and developed chemistry routes. C.R. performed all of the FRAP assays. K.R. supervised the SPR assays. O.S. supervised PK studies. S.S. wrote the synthesis in the Supporting Information. C.T. supervised protein production and performed ITC experiments and KD determination. C.R.V. designed the BD-swap allele experiments. M.Z. performed the NMR experiments and supervised the DSF, MST, and NMR measurements of the FBS screening. A.Z. supervised the production of protein and DSF assays. M.P. was responsible for the biology strategy. D.M. was responsible for the medicinal chemistry strategy
The authors declare the following competing financial interest: all authors except O.F., S.Kn., S.M., C.R., C.T., A.F.H., and C.R.V. were full-time employees of Boehringer Ingelheim at the time this study was performed.
This manuscript posted ASAP on 3/10/2016. The amino acid Glu85 was changed to Gln85 and the revised version was reposted on 3/22/2016.
Supplementary Material
References
- Whitehouse I.; Flaus A.; Cairns B. R.; White M. F.; Workman J. L.; Owen-Hughes T. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature 1999, 400, 784–787. 10.1038/23506. [DOI] [PubMed] [Google Scholar]
- Liu B.; Yip R. K. H.; Zhou Z. Chromatine remodeling, DNA damage repair and aging. Curr. Genomics 2012, 13, 533–547. 10.2174/138920212803251373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Castex J.; Tallant C.; Owen D. R.; Martin S.; Aldeghi M.; Monteiro O.; Filippakopoulos P.; Picaud S.; Trzupek J. D.; Gerstenberger B. S.; Bountra C.; Willmann D.; Wells C.; Philpott M.; Rogers C.; Biggin P. C.; Brennan P. E.; Bunnage M. E.; Schüle R.; Günther T.; Knapp S.; Müller S. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 10, e1500723. 10.1126/sciadv.1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann A. F.; Vakoc C. R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–361. 10.1016/j.tig.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C.; Hargreaves D. C.; Hodges C.; Elias L.; Ho L.; Ranish J.; Crabtree G. R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann A. F.; Martin L. J.; Minder J.; Roe J.-S.; Shi J.; Steurer S.; Bader G., McConnell D.; Pearson M.; Gerstberger T.; Gottschamel T.; Thompson D.; Suzuki Y.; Koegl M.; Vakoc C. R.. A bromodomain-swap allele demonstrates that on-target chemical inhibition of BRD9 limits the proliferation of acute myeloid leukemia cells, submitted for publication.
- Bunnage M. E.; Piatnitski Chekler E. L.; Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frederiksen M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V. M; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Müller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Ross R.; Roth B.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundström M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P. C.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Herait P.; Dombret H.; Thieblemont C.; Facon T.; Stathis A.; Cunningham D.; Palumbo A.; Vey N.; Michallet M.; Recher C.; Rezai K.; Preudhomme C. O7.3 BET-bromodomain (BRD) inhibitor OTX015: Final results of the dose-finding part of a phase I study in hematologic malignancies. Ann. Oncol. 2015, 26, ii10. 10.1093/annonc/mdv085.3. [DOI] [Google Scholar]
- Moros A.; Rodríguez V.; Saborit-Villarroya I.; Montraveta A.; Balsas P.; Sandy P.; Martínez A.; Wiestner A.; Normant E.; Campo E.; Pérez-Galán P.; Colomer D.; Roué G. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. 10.1038/leu.2014.106. [DOI] [PubMed] [Google Scholar]
- Garnier J.-M.; Sharp P. P.; Burns C. J. BET bromodomain inhibitors: a patent review. Expert Opin. Ther. Pat. 2014, 24, 185–199. 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- Clark P. G. K.; Vieira L. C. C.; Tallant C.; Fedorov O.; Singleton D. C.; Rogers C. M.; Monteiro O. P.; Bennett J. M.; Baronio R.; Müller S.; Daniels D. L.; Mendez J.; Knapp S.; Brennan P.; Dixon D. J. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Bamborough P.; Bannister A. J.; Becher I.; Bit R. A.; Che K. H.; Chung C.-W.; Dittmann A.; Drewes G.; Drewry D. H.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Michon A.-M.; Molnar J.; Robson S. C.; Tomkinson N. C. O.; Kouzarides T.; Prinjha R. K.; Humphreys P. G. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. A.; Rogers C. M.; Fedorov O.; Tallant C.; Martin S.; Monteiro O. P.; Muller S.; Knapp S.; Schofield C. J.; Brennan P. E. Design and synthesis of potent and selective inhibitors of BRD7 and BRD9 bromodomains. MedChemComm 2015, 6, 1381–1386. 10.1039/C5MD00152H. [DOI] [Google Scholar]
- Philpott M.; Rogers C. M.; Yapp C.; Wells C.; Lambert J. P.; Strain-Damerell C.; Burgess-Brown N. A.; Gingras A. C.; Knapp S.; Müller S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenet. Chromatin 2014, 7, 14. 10.1186/1756-8935-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows A. E.; Smogorzewska A.; Elledge S. J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14280–14285. 10.1073/pnas.1009559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Cote A.; Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Brown P. J.; Müller S. Open access chemical probes for epigenetic targets. Future Med. Chem. 2015, 7, 1901–1917. 10.4155/fmc.15.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri E.; Petosa C.; McKenna C. E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2015, 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Theodoulou N. H.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Progress in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 2016, 11, 477–487. 10.1002/cmdc.201500540. [DOI] [PubMed] [Google Scholar]
- BI-9564 A chemical probe for BRD9 and BRD7. http://www.thesgc.org/chemical-probes/BI-9564 (accessed May 18, 2015).
- Linke P.; Amaning K.; Maschberger M.; Vallee F.; Steier V.; Baaske P.; Duhr S.; Breitsprecher D.; Rak A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screening 2015, 10.1177/1087057115618347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Meyer E. A.; Castellano R. K.; Diederich F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- Lawton G.; Witty D. R.. Progress in Medicinal Chemistry; Elsevier B.V.: Amsterdam, 2012, Vol. 51. [Google Scholar]
- Lee E. C.; Hong B. H.; Lee J. Y.; Kim J. C.; Kim D.; Kim Y.; Tarakeshwar P.; Kim K. S. Substituent Effects on the Edge-to-Face Aromatic Interactions. J. Am. Chem. Soc. 2005, 127, 4530–4537. 10.1021/ja037454r. [DOI] [PubMed] [Google Scholar]
- Picaud S.; Strocchia M.; Terracciano S.; Lauro G.; Mendez J.; Daniels D. L.; Riccio R.; Bifulco G.; Bruno I.; Filippakopoulos P. 9H-purine scaffold reveals induced-fit pocket plasticity of the BRD9 bromodomain. J. Med. Chem. 2015, 58, 2718–2736. 10.1021/jm501893k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banting G. S.; Barak O.; Ames T. M.; Burnham A. C.; Kardel M. D.; Cooch N. S.; Davidson C. E.; Godbout R.; McDermid H. E.; Shiekhattar R. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 2004, 14, 513–524. 10.1093/hmg/ddi048. [DOI] [PubMed] [Google Scholar]
- Kooistra M. K.; Leduc R. Y.; Dawe C. E.; Fairbridge N. A.; Rasmussen J.; Man J. H.; Bujold M.; Juriloff D.; King-Jones K.; McDermid H. E. Strain-specific modifier genes of Cecr2-associated exencephaly in mice: genetic analysis and identification of differentially expressed candidate genes. Physiol. Genomics 2012, 44, 35–46. 10.1152/physiolgenomics.00124.2011. [DOI] [PubMed] [Google Scholar]
- Cain C. Chromatińs rising tide. SciBX 2014, 7, 1–7. 10.1038/scibx.2014.545. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.