

Abstract
The interaction of innate immune cells with pathogens leads to changes in gene expression that elicit our body's first line of defense against infection. Although signaling pathways and transcription factors have a central role, it is becoming increasingly clear that epigenetic factors, in the form of DNA or histone modifications, as well as noncoding RNAs, are critical for generating the necessary cell lineage as well as context‐specific gene expression in diverse innate immune cell types. Much of the epigenetic landscape is set during cellular differentiation; however, pathogens and other environmental triggers also induce changes in histone modifications that can either promote tolerance or ‘train’ innate immune cells for a more robust antigen‐independent secondary response. Here we review the important contribution of epigenetic factors to the initiation, maintenance and training of innate immune responses. In addition, we explore how pathogens have hijacked these mechanisms for their benefit and the potential of small molecules targeting chromatin machinery as a way to boost or subdue the innate immune response in disease.
Short abstract
The March 2015 issue contains a Special Feature on the epigenetic mechanisms underlying health and disease. Epigenetic modifications to chromatin influence the transcriptional status of our genes. Thus, understanding the epigenetic mechanisms that regulate immune cell fate are of great importance as they will provide insight into not only how to boost immune responses but also alter harmful ones such as autoimmunity and cancer. Immunology and Cell Biology thanks the coordinators of this Special Feature ‐ Rhys Allan ‐ for his planning and input.
Introduction
The innate immune system is the body's first barrier against pathogen infection. At the core of every innate immune response is a signal‐, cell lineage‐specific and kinetically precise gene expression program. The products of such synchronized gene expression following microbial pattern or danger signal recognition by pattern recognition receptors on neutrophils, monocytes, macrophages, natural killer cells, basophils, dendritic cells or epithelial cells enable pathogen clearance, aid adaptive immunity, clear cellular debris and restore damaged tissues. Multiple regulatory mechanisms are in place to ensure context‐specific and appropriately pitched responses from these cells. Unrestrained innate immune responses and prolonged production of inflammatory mediators can lead to a wide range of diseases including inflammatory bowel disease, arthritis, sepsis and cancer.
A proportion of the specificity and correct timing of the innate immune response is dictated by the ability of pattern recognition receptors to activate defined signaling pathways and employ a specific set of transcription factors. However, a growing body of evidence demonstrates that epigenetic factors, in the form of covalent modifications on DNA or histones, are the critical link that enables or prevents access of these transcription factors to identical DNA sequences in the different immune cell types. Further, these epigenetic factors are essential for the recruitment of transcription machinery—either rapidly after pathogen sensing or in a delayed manner. 1 Importantly, histone modifications also prevent unwanted expression of potent mediators 2 and are implicated in the repression or enhancement of secondary gene programs triggered by re‐stimulation of innate immune cells. 3 , 4 , 5 , 6 , 7 Recent work has also demonstrated the essential role of noncoding RNAs in innate immune gene expression. 8 Collectively, the combination of DNA or histone modifications, controlled recruitment of transcription factors following signal transduction and noncoding RNAs all lead to gene expression that is kinetically defined and cell‐type specific. This allows an assault on pathogens, usually without deleterious inflammation.
Epigenetics is defined as heritable traits that are not linked to changes in the DNA sequence; however, in broader terms, epigenetics is used to describe the mechanisms by which chromatin‐associated proteins and posttranslational modifications of histones regulate transcription. Thirteen years ago, Allis and colleagues 9 put forward the ‘histone code’ hypothesis, which provided a model to explain how single and/or combinatorial posttranslational modifications on histones regulate gene transcription. They hypothesized that this code is as important as the DNA sequence itself. Since the conception of this hypothesis, largely due to the development of techniques such as Chromatin Immunoprecipitation sequencing, the field has witnessed unprecedented advance in our understanding of the numerous enzymes that contribute to the establishment of histone modifications, as well as the assorted effector proteins that bind them. Whether or not histone modifications constitute a strict ‘code’, it is clear that the elaborate combinations of posttranslational modifications on histone function tightly regulate cell‐specific gene transcription. Also, it has been argued that histone modifications are not truly ‘epigenetic’, as the nature of their heritability (a requirement in the classical definition of epigenetic) remains questionable. This is particularly relevant in cells of the immune system where direct heritability of induced epigenetic modifications has yet to be formally demonstrated. However, as we will discuss, pathogen‐induced epigenetic modifications, particularly in cells of the innate immune system, can influence secondary responses to the same or different pathogens at least in the short term (1 week to 3 months).
The necessity of epigenetic regulation in pathogen defense is substantiated by evidence that microorganisms have evolved to target epigenetic regulatory factors for evasion of immune attack, which we will discuss here. Further, dysregulation of many chromatin‐modifying enzymes is a recurrent and sentinel event in multiple diseases, signifying their crucial role in accurate gene expression. As a result, enzymes that ‘write’, ‘erase’ and ‘read’ histone tail and DNA modification are the most promising and intently pursued targets in drug discovery today. We will discuss rapidly accumulating evidence of their potential as targets in immunologic disease, for enhanced responses to pathogens and prevention of unwanted inflammation.
Epigenetics, the basics
Epigenetic (‘Epi’=outside of or above) regulation of gene expression is a dynamic process that establishes precise cellular development and function in genetically identical cells. Such regulation is brought about by embellishments on the DNA itself and on DNA‐associated histone proteins. DNA modifications primarily are CpG cytosine‐5 methylation 10 and 5‐hydroxymethylcytosine, 11 but hydroxylation, formylation and carboxylation have also been observed. 12 In eukaryotic cells, DNA is packaged into chromatin, the basic repeating unit of which is a nucleosome. A nucleosome consists of 147 bp nucleotides wrapped around a histone octamer, which is composed of two copies each of histone H2A, H2B, H3 and H4 (Figure 1). With the addition of another histone protein called the ‘linker’ H1, nucleosomes are packaged into progressively higher‐order structures to ultimately form chromosomes. Unstructured NH2‐terminal histone tails that protrude from the nucleosome are subject to covalent chemical modifications, which impact chromatin organization and function. Recent mass spectrometry analysis identified more than a dozen different types of posttranslational modifications on histone tails, 13 including acetylation, methylation, phosphorylation, sumoylation, citrullination and ubiquitination. 14 Histone modifications with the exception of methylation result in a change in the net charge of nucleosomes, loosening interactions between histones and DNA. This directly affects the levels of chromatin compaction, creating condensed ‘heterochromatic’ or more open ‘euchromatic’ regions and thus restricting or allowing access of transcription factors to promoters or enhancers on DNA. The effects of histone methylation on gene expression are dependent on the position of the amino acid residues that are methylated and whether the residues are mono‐, di‐ or tri‐methylated. Methylation of Histone 3, Lysine 4 (H3K4), H3K36 and H3K79 is often associated with active transcription, whereas methylation of H3K9, H3K27 or H4K20 is associated with transcriptional repression. 10 , 15
Figure 1.
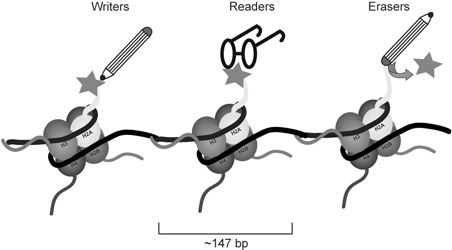
Writers, readers and erasers of histone covalent modifications. Schematic representation of DNA (black ribbon) wound around histone octamers. Each octamer is made up of two copies each of four histone proteins around which ~147 bp of DNA is wound. N‐terminal ‘tails’ of the histone proteins protrude from the core of the octamer and are the sites of reversible covalent modifications such as acetylation, methylation, phosphorylation and ubiquitination (all represented by a generic pink star). The gain of covalent modifications is catalyzed by histone‐modifying enzymes—that is, ‘writers’. ‘Readers’ recognize specific modifications and in doing so assist assembly of chromatin‐remodeling complexes at the sites of recognition, and ‘erasers’ catalyze removal of covalent modifications. A full color version of this figure is available at the Immunology and Cell Biology journal online.
A key facet of epigenetics is that these modifications can be stably maintained, yet adapt to changing developmental or environmental needs. This delicate task is accomplished by three main classes of enzymes: ‘writers’, which establish the epigenetic modifications (DNA and histone methyltransferases, histone acetyltransferases, kinases, and so on), ‘erasers’, which remove them (demethylases, histone deacetylases (HDACs), phosphatases) and ‘readers’, which interpret them by docking to modified histones through defined protein domains (Figure 1, see Table 1 for examples of epigenetic enzymes). The reader enzymes aid assembly of the appropriate transcriptional machinery at sites of recognition. Thus, not only do histone modifications determine the accessibility of DNA but are also directly responsible for recruiting transcriptional machinery to specific loci. Other contributors to epigenetic regulation of genes expression include nucleosome occupancy and positioning, 16 histone variants 17 and noncoding RNAs such as long noncoding RNAs (lncRNAs). 8
Table 1.
Writers, readers and erasers of the major histone covalent modifications in mammals
| Epigenetic modification | Writers | Readers | Erasers |
|---|---|---|---|
| DNA methylation | DNA methyltransferases (for example, DNMT1, DNMT3a, DNMT3b, DNMT3L) | Methyl‐CpG binding domains (for example, MBT 1‐6, MECP2); Kaiso and Kaiso‐like proteins with C2H2 type zinc finger (for example, ZBTB33, ZBTB4 and ZBTB38) | Passive DNA demethylase TET1–3; active DNA demethylases not known |
| Histone lysine (K) methylation Sites of mono/di/tri methylation: H3: K4, 9, 20, 27, 36, 79 H4: K20, 59 | Protein lysine methyltransferases (PKMTs) SET domain containing proteins (for example, PRDM2, SETD1A, SETD1B, MLL, KMT5B, DOT1L) non‐SET domain containing DOT1 (H3K79me methyltransferase) | Chromodomains; tudor domains; PHD fingers; MBT domains; ZF‐CW proteins; PWWP containing proteins; BAH domains; WD‐40; ankyrin repeat proteins | Histone demethylases: lysine‐specific demethylases (LSD1‐2); jumonji domain containing (for example, JMJD1‐8, JARID1‐2) |
| Histone arginine (R) methylation Sites of methylation: H3: R2, 17, 26 H4: R3 | Protein arginine methyltransferases (for example, PRMT2, 5, 6, 7) | Tudor domains; ADD; PHD fingers | Histone demethylases (eg: JMJD6) |
| Histone acetylation Sites of acetylation: H3: K4, 5, 9, 12, 14, 18, 23, 56 H4: K5, 8, 12, 14, 16 H2A: K5 H2B: K5, 12, 15, 20 | Histone acetyltransferases: Gcn5‐related N‐acetyltransferases (for example, PCAF, GCN5); MYST (for example, Tip60, MSL); P300/CBP; nuclear receptor co‐activators (SRC‐1) | Tandem PHD domains; tandem bromodomains; bromodomains; tandem PHD fingers | Histone deacetylases (HDAC class I and II); NAD+‐dependent sirtuins |
| Histone phosphorylation Sites of phosphorylation: H3: T (threonine) 3, 6, 11, 45; S10, 28; Y (tyrosine) 41 H4: S1, S47 H1 H2A: S1, 16, 139; T120 H2B: S14, 32, 36; Y37 | Ser/Thr kinases (for example, Janus kinases, PKCα/β, Haspin, Aurora B kinase) | Chromoshadow domain (for example, of HP1α); 14‐3‐3 proteins; BRCT proteins; BIR domains | Protein phosphatases (for example, protein serine/threonine phosphatases, tyrosine‐specific phosphatases; protein phosphatase 1D) |
| Histone ubiquitination Sites of ubiquitination: H2A: K119 H2B: K120 | Ubiquitin E2 conjugases, Ubiquitin E3 ligases | Unknown | Ubiquitin‐specific proteases; Ubiquitin carboxy‐terminal hydrolases (UCHs) |
Innate immune transcription regulation by epigenetic factors
Histone methylation
Histone lysine methylation (for example, H3K4, H3K9, H3K27, H3K36 and H4K20) promotes or represses transcription. 15 , 18 Histone methyltransferases (‘writers’) and demethylases (‘erasers’) collectively regulate the dynamic histone methyl landscape. A prominent role for H3K27 methylation has been described in transcriptional responses from innate immune cells. One of the first reports identifying the Jumonji (JMJ) catalytic domain containing JMJD3 as a histone H3K27me3 demethylase demonstrated a rapid induction of JMJD3 by proinflammatory stimuli. 19 It was subsequently shown that JMJD3 is recruited to the transcription start sites of >70% of lipopolysaccharide (LPS)‐induced genes. 20 Further, JMJD3 is essential for M2 macrophage polarization in response to helminth infection and chitin, although it is dispensable for M1 responses. 21 The histone methyltransferase G9a directs methylation of histone H3 on Lysine 9 (H3K9me). Di‐ or tri‐methylation of H3K9 is repressive not only by influencing DNA methylation and heterochromatin formation but also by prohibiting the ‘activating’ modification acetylation and actively recruiting transcriptional repressors of the Heterochromatin protein 1 family. 22 H3K9 methylation is found at a subset of promoters of inducible genes such as IL12b and CCL22 but this repressive mark is removed rapidly following LPS stimulation. 23 Similarly, H3K9 di‐methylation levels at type I interferon (IFN) and IFN responsive genes inversely correlate with the scope and amplitude of IFN and ISG expression. Professional IFN‐producing cells such as dendritic cells have significantly lower levels of H3K9 di‐methylation at these gene promoters compared with weak producers of IFN, such as fibroblasts, cardiac myocytes, or neuroblastoma cells. Further, genetic ablation of the H3K9 methyltransferase G9a enhanced IFN production by fibroblasts and their ability to suppress virus. 2 Toll‐like receptors (TLRs) and other sensors modify histones in a manner associated with the activation of transcription. Two of the main permissive modifications are H3 Lysine 4 trimethylation (H3K4me3) denoting an active promoter and H3 Lysine 36 tri‐methylation (H3K36me3) associated with active transcription. Dendritic cells stimulated with LPS upregulate H3K4me3, which is very stable for 2 h following stimulation, with the exception of about 30 loci that are lowly expressed before stimulation and become strongly induced after stimulation. 24 Similarly, macrophages upregulate H3K4me3 and H3K4me1 at the promoters and enhancers, respectively, of multiple genes following exposure to a range of stimuli, with the majority of acquired H3K4me3 returning to basal levels within a few hours. 4 , 25
Histone acetylation
Histone acetylation is a reversible posttranslational modification catalyzed by histone acetyltransferases (HATs) that transfer the acetyl moiety of acetyl‐CoA to lysine (K) residues. HDACs reverse this process. LPS‐stimulated macrophages show increased histone H4 acetylation (H4Ac), an indicator of open chromatin at numerous sites across the genome. The induced histone acetylation at lysines 5, 8 and 12 (H4K5/8/12Ac) usually occurs at promoters, within 1 h of stimulation, decreasing after 2 h. 25 In dendritic cells H4K27 acetylation was very dynamic over an LPS time course correlating with RNA polymerase II (Pol II) binding. 24 Histone acetylation is exclusively ‘read’ by bromodomains of which there are 46 bromodomain‐containing proteins in the human genome. Bromodomains of individual proteins have defined affinities to specific locations of lysine acetylation and recruit distinct proteins to the chromatin. 26 Hence, each subclass of bromodomain proteins likely has unique functions in regulating gene expression. Most work has focused on the role of the bromodomain and extra‐terminal (BET) subfamily of bromodomain‐containing proteins that consists of BRD2, BRD3, BRD4 and a testis‐specific BRDT. BET proteins connect histone acetylation state to transcriptional elongation machinery. 1 , 27 Only BRD4 can recruit positive transcription elongation factor b (P‐TEFb) through its C terminal domain and removes the pausing complex negative elongation factor (NELF) and 5,6‐dichloro‐1‐β‐d‐ribofuranosylbenzimidazole (DRB) sensitivity‐inducing factor (DSIF) allowing transcriptional elongation. 27 , 28 BRD4 can also co‐activate transcriptional activation of NF‐κB via specific binding to acetylated RelA. 29 Knockdown of BRD2, 3 or 4 proteins, however, results in reduced expression of multiple inflammatory cytokines in macrophages, suggesting that all three BET proteins control the transcription of inflammatory genes in LPS‐stimulated macrophages. 25 This is likely through the ability of BRD2, 3 and 4 to interact with the polymerase‐associated factor 1 complex, or other chromatin‐modifying proteins such as NSD3 or JMJD6. 30
Histone variants
Histone variants are non‐allelic forms of conventional histones. The exact role of histone variants in gene transcription is unclear, but the emerging picture is that the presence of histone variants confers novel structural and functional properties on the nucleosome. IFN treatment triggered robust H3.3 incorporation into activated genes, which persisted even after cessation of transcription. 17 Interestingly, this deposition was dependent on the histone methyltransferase Wolf‐Hirschhorn syndrome candidate 1 that interacts with histone cell cycle regulator HIRA, the H3.3‐specific histone chaperone. Indeed, Wolf‐Hirschhorn syndrome candidate 1 also interacted with BRD4 and P‐TEFb, demonstrating that deposition of histone variants can facilitate transcriptional elongation. 17
Noncoding RNAs
A number of recent papers have described essential roles for lncRNAs in innate immune gene expression. 8 , 31 The Fitzgerald laboratory identified lncRNA‐Cox2 as a highly inducible lncRNA in both macrophages and dendritic cells following microbial stimulation. LncRNA‐Cox2 is essential for controlling basal levels of IFN stimulatory genes (ISGs) and is also required for proinflammatory cytokine production following microbial challenge. LncRNA‐Cox2 mediates its repressive functions on ISGs through interactions with hnRNP‐A/B and A2/B1. Knockdown of lncRNA‐Cox2 or hnRNP‐A/B or A2/B1 resulted in decreased levels of RNA Pol II recruitment to the promoter of Ccl5 in macrophages. 8 LncRNAs have also been shown to be upregulated in the context of viral infection and downstream IFN production. Approximately 500 lncRNAs were differentially expressed following infection with severe acute respiratory syndrome coronavirus or influenza 32 and more than 200 lncRNAs were upregulated following treatment of human hepatocytes with type I IFN. 33 One lncRNA, lnRNA‐CMPK2, was a potent negative regulator of ISGs and its knockdown resulted in reduction of Hepatitis C virus replication. 33
Kinetics of gene induction, the epigenetic landscape and chromatin binding proteins
The precise kinetics of innate immune cell gene transcription following pathogen assault is intimately regulated by histone modifications and chromatin‐remodeling complexes. ATP‐dependent chromatin‐remodeling complexes are responsible for sliding of the nucleosomes, as well as for insertion and ejection of histone octamers, processes that are, like histone modification, important for transcriptional repression and activation. The remodeling complexes can be divided into four families: SWI/SNF, CHD (chromodomain and helicase‐like domain), ISWI and INO80 (including SWR1, or SRCAP in mammals). Promoters of primary response genes (PRGs) such as Tnf, Fos and Nfkbia contain CpG islands which are refractory to DNA methylation, and histone tail modifications, both of which are commonly found at the promoters of actively transcribed genes (for example, H3K4me3 and H4Ac) as well as high levels of RNA Pol II association in naive macrophages, indicating that these chromatin specifications likely occur during lineage commitment. 1 , 25 Thus, macrophages are preprogramed to enable expression of a defined set of PRGs within minutes of cell activation. 34 , 35 These genes do not require SWI/SNF‐mediated chromatin remodeling or de novo protein synthesis for their activation, as they are transcriptionally primed and their chromatin state is permissive of rapid access by transcription factors. PRGs produce low levels of unspliced and unstable transcripts, and upon stimulation recruit the elongation factor P‐TEFb and switch to production of mature, processed mRNAs. 1 In contrast, late PRGS such as Ifnb1 and secondary response genes such as Il12b and Il6, which are transcribed hours after cell stimulation, possess low‐density CpG promoters, display low H3K4me3, H4Ac and RNA Pol II occupancy in naive macrophages and require the SWI/SNF complex for chromatin remodeling for transcription to take place. 34 , 35
Innate immune ‘memory’ driven by epigenetic and metabolic reprogramming
Traditionally, the innate immune system was considered to be perpetually naive, with immunological memory being the major feature of the adaptive immune system. However, the ability to remember and respond more vigorously to a second pathogen encounter has been described in organisms lacking T and B cells. Plants and invertebrates do not possess an adaptive immune system, which first appeared during evolution in jawless or early‐jawed vertebrates. 36 However, multiple studies have demonstrated that the immune system of plants and invertebrates can be primed by previous infections and mount stronger recall responses upon pathogen re‐challenge. 37 , 38 Now, examples of innate immune antigen‐ and non‐antigen‐specific ‘memory’ are rapidly emerging in mammals. 4 , 5 , 6 , 7 , 39 Innate monocytes and neutrophils from mice infected with attenuated Listeria monocytogenes were capable of ‘bystander’ killing of an unrelated pathogen (Leishmania major) upon secondary infection. 40 , 41 However, such priming was orchestrated primarily by IFNγ and other inflammatory mediators produced by memory T cells. 40 , 42 Certainly, other studies have now shown that memory T cells can trigger activation of innate immune cells following re‐infection. 43 , 44 Examples of innate immune boosting by means that are independent of adaptive immunity have also emerged. A combination of aerosolized TLR agonists could protect mice against bacterial pneumonia and influenza infection 45 , 46 and infection with H. polygyrus significantly inhibited type 1 diabetes in non‐obese diabetic mice through CD25‐ and interleukin (IL)‐10‐independent mechanisms. 47 The Netea laboratory demonstrated that monocytes recovered from healthy volunteers who were vaccinated with Bacillus Calmette‐Guérin, a widely used live attenuated vaccine against tuberculosis, produced significantly higher levels of inflammatory cytokines following exposure to non‐mycobacterial bacteria and fungi. 7 Importantly, this non‐antigen‐specific innate immune ‘training’ was maintained up to 3 months post the initial vaccination. Further, they showed that monocyte immune ‘training’ was completely independent of T and B cells, as severe combined immunodeficiency mice vaccinated 2 weeks prior with Bacillus Calmette‐Guérin survived significantly longer after a lethal inoculum of Candida albicans. 7 Moreover, Rag1‐deficient mice were protected from a secondary challenge of C. albicans or LPS following priming with C. albicans. These findings suggest that ‘training’ of innate immune cells in mammals could be cell intrinsic, or at the very least independent of the adaptive immune system.
One mechanism that appears to drive the ‘training’ of innate immune cells to respond differently to secondary stimulation is pathogen‐induced epigenetic reprogramming (Figure 2). Until recently, it was thought that cell lineage‐ and signal‐specific gene expression programs are fundamentally predetermined during cellular differentiation. However, recent evidence demonstrated that terminally differentiated cells such as monocytes and macrophages can acquire additional histone modifications upon pathogen exposure that affect gene expression upon subsequent stimulation. TLR4 activation by LPS induces histone modifications that lead to altered and repressed gene expression upon secondary LPS stimulation. 3 , 48 Many of these pathogen‐induced epigenetic changes in macrophages, particularly mono‐methylation of lysine(K)‐4 on Histone 3 (H3K4me1) at enhancers, persist despite washout of the stimulus and removal of the transcription factors responsible for the initial deposition. Moreover, H3K4me1 was associated with a faster and stronger induction of multiple genes upon nonspecific restimulation. 4
Figure 2.
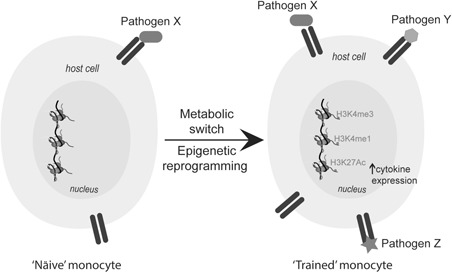
Epigenetic reprogramming in training of innate immune cells. Upon pathogen X recognition by a receptor, naive monocytes undergo epigenetic reprogramming and a metabolic shift, and become primed to respond more robustly to nonspecific (Pathogens X, Y and Z) secondary stimulation. A full color version of this figure is available at the I mmunology and Cell Biology journal online.
C. abicans‐induced innate immune training was associated with changes in the activating H3K4me3 at certain gene promoters in peritoneal macrophages 7 days post initial infection. In addition, a methyltransferase inhibitor prevented this induced training. 6 A follow‐up study by the same group showed that β‐glucan, the cell wall component of C. albicans, could induce changes in H3K4me3 as well as H3K27ac in human monocytes 7 days after washout. Without a direct comparison to induced H3K4me3 or H3K27Ac after acute stimulation it remains difficult to interpret whether these are maintained epigenetic modifications or are just demonstrative of active transcription in these cells triggered by other mechanisms. Nonetheless, many genes with altered H3K4me3 or H3K27ac profiles 1 week after C. albicans exposure were involved in innate immune signaling, and a large proportion with enhanced H3K4me3 and H3K27Ac were associated with glycolysis, 5 raising an interesting potential of a metabolic switch in innate immune training. Multiple epigenetic modifications have a well‐established link to central metabolism, as histone‐modifying enzymes require metabolites as substrates or cofactors: demethylases and TET proteins are Fe(II) and α‐ketoglutarate dioxygenases; HDACs are (NAD)‐dependent enzymes; and S‐adenosyl methionine is required for function of DNA/histone methyltransferases. Therefore, these enzymes are likely sensitive to fluctuations in these metabolites. 49 Interplay between metabolism and epigenetics would allow the relative metabolic activity of the cell to feed back into transcriptional regulation in an effort to maintain homeostasis. In fact, it has been proposed that epigenetic processes may initially have been a means to transduce metabolic events into phenotypic results. 50 This is well documented in cancer cells that switch to anaerobic metabolism (the ‘Warburg effect’) and exhibit multiple epigenetic imbalances. 49 Interestingly, macrophages and other innate immune cells are frequently found in inflamed sites, which are characterized by low oxygen levels and therefore may also rely heavily on the relationship between metabolism and epigenetics for gene expression. Certainly, activation of TLRs, notably TLR4, leads to a switch from oxidative phosphorylation to glycolysis in immune cells. 51 Succinate, which is known to inhibit α‐ketoglutarate and Fe(II)‐dependent dioxygenases such as histone and DNA demethylases, as well as prolyl hydroxylases, 50 is elevated in inflammation and sustains IL‐1β production through hypoxia‐inducible factor (HIF)‐1α stabilization. 52 Also, differentiation of monocytes to macrophages results in a change in abundance of enzymes responsible for peroxisomal β‐oxidation pathway, glycine, serine and threonine metabolism and the tricarboxylic acid cycle. 48 β‐Glucan‐trained monocytes exhibited reduced oxygen consumption, enhanced glucose consumption, increased production of lactate and an increased NAD+/NADH ratio, 5 which may be responsible for the observed alterations in H3K4me3, H3K27Ac and H3K4me1 in these cells. 5 , 48
It is unknown whether these pathogen‐induced ‘epigenetic’ changes are maintained for the life of the infected cell (or in daughter cells or hematopoietic stem cells, to be truly ‘epigenetic’) for an innate immune memory of pathogen infection in vivo. Cells of the innate immune system have been generally thought to be short lived. However, tissue macrophages have been shown to live for months if not years, particularly at sites of inflammation or tumors, 53 but whether they proliferate remains unclear. If demonstrated to be long lived, it is to be expected that trained immunity through epigenetic and metabolic mechanisms will have important consequences for the design of vaccination strategies. Moreover, an individual's history of infection may influence the function of their innate immune system, at least in the short term, through altered epigenetics.
Pathogen subversion of the host innate immune response
Coevolution of the host and pathogen, driven by conflicting interests, is akin to an evolutionary arms race. It is in the host's interest to detect and stop the progression of an infection early by mounting a timely inflammatory response; conversely, it is important for the pathogen to subvert the innate immune system—that is, the first defense response of the host in order to establish an infection. The pathogen must also prevent an excessive inflammatory reaction not only to avoid elimination but also to ensure its own survival by keeping the host alive. To this end, pathogens have evolved strategies to disrupt host immune signaling cascades that culminate in drastic transcriptional upregulation of several proinflammatory and other immune response genes. Inhibition of NF‐κB, MAPK and JAK/STAT signaling and modulation of protein ubiquitylation are well‐documented strategies of host immune evasion.
As we saw earlier, the proinflammatory transcriptional response is formulated by underlying complex and multistep processes of histone modification and chromatin remodeling. The elaborate nature of such epigenetic control provides the pathogen with substantial opportunity to manipulate host gene expression to its own advantage. An example of bacteria epigenetically manipulating the host innate immune response for nonpathogenic survival can be seen in the mammalian gut. Commensal bacteria are essential for the induction/maintenance of DNA methylation at the TLR4 gene in large intestinal epithelial cells, resulting in reduced TLR4 expression and thus avoiding an excessive inflammatory reaction. 54 , 55 Pathogens, which face a different selection pressure to commensals, have evolved proteins that interfere, interact with or mimic components of the host's epigenetic machinery, often resulting in subversion of the host innate immune response. Broadly, this is achieved by changing the chromatin architecture in three ways: host histone modification using the host's own epigenetic writer enzymes, interfering directly with enzymes of the host's chromatin‐remodeling machinery, or by manufacturing proteins that specifically recognize host histone targets. We will review these strategies with the help of relevant examples below (Figure 3,Table 2).
Figure 3.
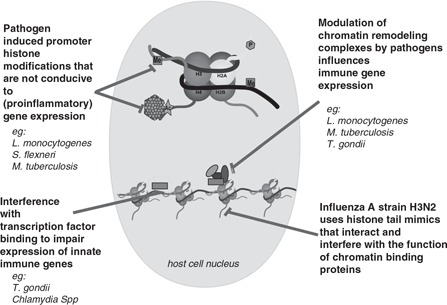
Schematic representation of various strategies employed by pathogens to modulate the host innate immune gene expression response to their advantage. Top: a histone octamer around which DNA (black ribbon) is wrapped. ‘Tails’ of histone proteins are the sites of reversible covalent modifications like methylation (shown as Me), phosphorylation (P) and acetylation (Ac) catalyzed by histone‐modifying enzymes, that is, writers (textured hexagon). Bottom: DNA wound around four histone octamers. Transcription factor binding is shown as a pink rectangle; and chromatin‐remodeling complexes are represented by a group of green, blue, purple and pink shapes. A full color version of this figure is available at the Immunology and Cell Biology journal online.
Table 2.
Various strategies employed by pathogens for modulation of the host epigenome to thwart inflammatory responses
| Pathogen | Effector molecule | Host molecule interacted with/involved | Mode of action | Epigenetic modification induced | Studied in: |
|---|---|---|---|---|---|
| Listeria monocytogenes | LntA (listeria nuclear targeted protein A) | BAHD1 (chromatin repressor) | LntA interacts with chromatin repressor BAHD1 in host nucleus 64 , 65 | ↑H3Ac at ISGs | Mouse fibroblasts, in vivo studies |
| Listeriolysin O (LLO), pore‐forming toxin | Host cell membrane | unknown | Global H3deP and H4deAc 56 , 57 | HeLa cells | |
| Bacterial protein lnlB dependent | SIRT2 (HDAC) | Met‐dependent enrichment of SIRT2 to gene promoters, including ISGs 58 | H3K18 deacetylation | HeLa cells, in vivo mouse studies | |
| Shigella flexneri | OspF (dually specific phosphatase) | MAPKs | OspF dephosphorylates MAPK, thus preventing promoter H3S10P 59 | ↓ H3S10P at NF‐κB responsive genes (for example IL8, CCL20) | HeLa cells, in vivo rabbit studies |
| Mycobacterium tuberculosis/ avium | LpqH (19‐kDa lipoprotein) | TLR2 | LpqH activates MAPK pathway via TLR2 → TF C/EBP induction, recruitment and possible exclusion of SWI/SNF chromatin remodelers at gene promoters 62 , 63 , 94 | ↓ Histone acetylation at promoters of CIITA, HLA‐DR | Human THP‐1 monocytic cells, mouse macrophage‐like RAW264.7 cells and in vivo mouse studies |
| Clymedia trachomatis | NUE (nuclear effector) | Chromatin | NUE localization to chromatin 95 | In vitro methylation of H2B, H3, H4 | HeLa cells, 3T3 cell line |
| Anaplasma phagocytophilum | AnkA (ankyrin‐repeat‐containing A) | HDAC1 | AnkA binding to DNA at AT rich regions 69 | ↑ HDAC1 expression and binding to host defense gene promoters and 69 , 96 ↓ H3Ac at host defense genes 69 | Acute monocytic leukemia THP‐1 cell line |
| Bacillus anthracis | BaSET (SET domain containing) | H1 lysine | Localization to and methylation of histone H1 | H1K trimethylation 67 | HeLa cells, human embryonic kidney (HEK293T), mouse macrophage RAW264.7 cell lines |
| Toxoplasma gondii | Unknown | BRG‐1 (brahma‐related gene‐1, a catalytic subunit of chromatin‐remodeling complexes) | Unknown (phenotype rescued by treatment with HDAC inhibitors) | ↓ H3Ac and H4Ac 97 at CIITA promoter region | Mouse bone‐marrow‐derived macrophages |
| unknown | Global ↓ H3S10 phorphorylation and prevention of H3K9 and H3K14 acetylation at the IL‐10 promoter 60 , 61 | Mouse bone‐marrow‐derived macrophages | |||
| Streptococcus pyogenes | Ser/Thr phosphatase Sp‐STP | Host chromatin 98 | Unknown | Unknown | Human carcinoma cell lines |
| Ligionella pneumophila | RomA methyltransferase | H3 | RomA catalyzes H3K14 methylation, preventing H3K14 acetylation 66 | Infection causes a switch from acetylated to methylated H3K14 | Human monocyte (THP‐1), human alveolar epithelial (A549) cell lines |
| Influenza A strain H3N2 | NS1 carboxy‐terminal (nonstructural protein) | PAFI transcriptional elongation complex CHD1 chromatin‐remodeling complex | Interaction with PAFI 70 | Reduction of PAF1 and RNA Pol II enrichment at gene bodies | A549 cell lines |
Pathogens that induce histone covalent modifications in the host
L. monocytogenes infection causes a drastic and global dephosphorylation of H3 and deacetylation of H4, accompanied by repression of a subset of proinflammatory and immunity genes. 56 , 57 L. monocytogenes also causes histone deacetylase‐SIRT2‐dependent histone deacetylation at promoters of ISGs, and in fact is heavily reliant on host SIRT2 for infection, as SIRT2‐null mutants are resistant to L. monocytogenes infection. 58 The human intestinal pathogen S. flexnei phosphatase, OspF dephosphorylates host MAPKs, thereby preventing MAPK‐dependent phosphorylation of histone H3S10 at select gene promoters, lending them inaccessible to NF‐κB mediated upregulation. 59 By an unknown mechanism, infection with T. gondii also causes loss of phosphorylation and acetylation at H3 at the Tnf promoter, resulting in impaired recruitment of transcription factors and Pol II binding, and subsequent inability of the cell to upregulate Tnf upon LPS stimulation or secondary infection. 60 , 61 , 62
Pathogens that interfere with the host's chromatin‐remodeling machinery
M. tuberculosis counters the host IFN‐γ‐induced inflammatory response by repressing IFN stimulatory MHC class II (HLA‐DR) and its transactivator protein CIITA. TLR2‐mediated downstream MAPK signaling leads to binding of a transcriptional repressor C/EBP to the CIITA promoter region, keeping out the chromatin remodeler complex SWI/SNF. The CIITA‐regulated HLA‐DR is also repressed, with promoter enrichment of HDAC containing chromatin complexes. 62 , 63 Interestingly, L. Monocytogenes secretory protein LntA directly interacts with the chromatin repressor BAHD1, probably dislodging BAHD1 from ISG promoters and subsequently upregulating some ISG. 64 , 65 This seemingly counterintuitive strategy is proposed to be a mechanism used by the pathogen to fine‐tune host IFN I and II response to infection, given that, although required for bacterial virulence, constitutive LntA expression leads to faster bacterial clearance, and BAHD1 deletion heterozygous mice are more resistant to infection compared with wild‐type siblings. 65
Pathogen enzymes that use host histone proteins as substrates
Several bacterial pathogens, despite lacking histones or higher‐order chromatin structures, produce histone‐modifying proteins. For instance, the SET‐domain‐containing protein RomA, required for pathogenesis of Legionella pneumonia, localizes to several gene promoters including innate immune gene promoters and catalyzes a previously unreported H3K14 methylation genome‐wide, leading to global gene repression. 66 BaSET, of Bacillus anthracis, is required for virulence and methylates Lysine residues on H1, leading to repression of various NF‐κB target gene promoters. 62 , 67 The ankyrin‐repeat‐containing proteins are yet another class of eukaryotic protein mimics found in intracellular pathogens of the Anaplasma, Ehrlichia, Ricketssia, Orientia, Coxiella and Legionella species. A. phagocytophilum, a tick‐transmitted pathogen causing human granulocytic analpasmosis, propagates within the primary antimicrobial defense cells, neutrophils. A. phagocytophilum infection leads to a decrease in H3 acetylation at a subset of defense gene promoters and to an overall increase in expression of HDAC1 and HDAC2. Consistent with this, treatment with HDAC1 inhibitors severely restricts the bacterium's ability to survive in the host, suggesting that the pathogen may survive the harsh environment of the neutrophil by HDAC1‐mediated deacetylation and suppression of host defense genes. 68 It has been suggested that an ankyrin‐repeat‐containing bacterial secretory protein that binds AT‐rich chromatin regions is responsible for HDAC recruitment to relevant gene promoters in the host 68 , 69
Pathogen‐derived proteins that ‘mimic’ host histone tails
Pathogens employ various types of molecular mimicry to evade the host immune response. In recent years, evidence of pathogens using molecular mimicry of host histone proteins to modify transcriptional response to infection has emerged. The carboxy terminal of the Influenza A strain H3N2 protein NS1 (nonstructural protein) shares resemblance with Histone H3 tails. These NS1‐histone‐like tails associate with the polymerase‐associated factor 1 transcriptional elongation complex as well as with the CHD1 chromatin‐remodeling complexes of the host. Like histone H3 tails, the NS1 tails bind to the polymerase‐associated factor 1 complex unmodified or after lysine methylation, but not upon lysine acetylation. Such binding inhibits elongation of virally induced genes, presumably by occluding polymerase‐associated factor 1 and RNA Pol II from gene bodies. 70
How, or if indeed, these mechanisms specifically achieve silencing of immune and proinflammatory gene sets in the host is not well understood. In many examples cited here, expression of a wide array of genes apart from immune‐related genes is affected. 56 , 57 , 59 Presumably, such broad‐ranging effects of epigenetic interference by the pathogen may not face elimination so far as not fatal to the host.
Potential of targeting chromatin‐modifying enzymes as anti‐inflammatory therapeutics
Increasingly it is being recognized that disrupted epigenetic processes have an instrumental role in pathogenesis of several major diseases. Although the role of epigenetic modifications in cancer etiology and progression is well established, direct evidence of a dysregulated epigenetic landscape in chronic, immune‐based diseases is rapidly emerging. 71 , 72 Studies in monozygotic twins minimize the confounding effects of genetic heterogeneity in disease etiology and have implicated epigenetic discordance between disease‐affected and ‐nonaffected twins in inflammatory in diseases like type 1 diabetes, 73 Systemic Lupus Erythematosus (SLE) 74 and asthma. 75 Genome‐wide association studies have identified single‐nucleotide polymorphisms in chromatin‐interacting proteins as significant susceptibility loci for inflammatory diseases: variants in histone reader proteins are associated with incidence of Crohn's disease and multiple sclerosis, 76 , 77 variants within the DNA methylation writer DNMT3A with Crohn's disease 76 and the histone demethylase JARID1A (KDM5A) with ankylosing spondylitis. 78
Our fast evolving understanding of the role of chromatin‐modifying enzymes in dictating the precise gene transcription program in homeostatic as well as detrimental innate immunity and inflammation raises the exciting possibility of targeting chromatin‐modifying enzymes to combat human immune‐based diseases (Table 3). Targeting of epigenetic enzymes makes it possible to regulate subsets of genes with similar function and kinetics, giving an advantage over targeting of single inflammatory cytokines.
Table 3.
Inhibitors of epigenetic eraser enzymes and reader domains with applications in treating inflammatory diseases
| Target protein type | Target molecules | Inhibitory molecule | Subjects/models | Effects |
|---|---|---|---|---|
| Histone acetylation erasers | HDACs | Phenylbutyrate | Human subjects | Anti‐inflammatory in Crohn's disease 81 and Ulcerative Colitis 82 |
| SAHA and Valproic Acid | Sulfate sodium‐ and trinitrobenzene sulfonic acid‐induced colitis mouse models | Reduction in colonic pre‐inflammatory cytokine production and decreased severity of Colitis 99 | ||
| Givinostat (ITF‐2357) | Human subjects | Anti‐inflammatory in juvenile idiopathic arthritis 83 | ||
| SAHA | Rodent model of arthritis | Anti‐inflammatory in rheumatoid arthritis 100 | ||
| Phenylbutryate and TSA | Adjuvant‐induced rat arthritis model 101 | Reduction in TNFα production in RA affected tissues and in arthritic scores | ||
| MPT0G009 (3‐[1‐(4‐methoxybenzenesulfonyl)‐2,3‐dihydro‐1 H‐indol‐5‐yl]‐N‐hydroxyacrylamide) | Human RA fibroblast‐like synoviocytes adjuvant‐induced arthritic mouse model 102 | Inhibits cytokine release and causes a global increase in H3 acetylation in human Anti‐arthritic | ||
| ITF‐2357 | LEW1.WR1 rat which develops inflammation and type 1 Diabetes post infection with Kilham rat virus 103 | Reduction of virus‐induced inflammation and prevention of type 1 diabetes | ||
| HDAC1 | MS‐275 | Rat model of autoimmune prostatitis 104 mouse model of periodontitis 105 mouse models of arthritis 100 | Anti‐inflammatory effects | |
| HDAC3 | MI192 | Human PBMCs from RA patients 106 | Reduction in TNFα production and dose dependent suppression of IL‐6 | |
| HDACs | ITF‐2357 suppresses | LEW1.WR1 rat which develops inflammation and Type 1 Diabetes post infection with Kilham rat virus 103 | Reduction of virus‐induced inflammation and prevention of type 1 diabetes. | |
| Histone methylation erasers | JMJD3 and UTX (H3K27me3‐specific demethylase) | GSK‐J1 and J4 | Primary human macrophages 107 | Reduction in LPS‐induced proinflammatory cytokine production |
| Histone acetylation readers | BET | I‐BET762 | Mouse BMDM 25 Mouse model of Bacteria induced sepsis | Downregulation of proinflammatory genes upon LPS stimulation; Protection from endotoxic shock and sepsis |
| I‐BET151 | Mouse model of Bacteria induced sepsis 87 | Reduction in IL6; protection from sepsis | ||
| JQ1 | In vivo mouse 88 | Anti‐inflammatory; protection from LPS‐induced death | ||
| MS417 | Mouse model of HIV‐associated nephropathy, 108 rat model of autoimmune Neuritis 109 and prostitis 104 | Anti‐inflammatory |
Abbreviations: BET, bromodomain and extra‐terminal subfamily; HDAC, histone deacetylase; SAHA, suberanilohydroxamic acid; TSA, trichostatin A.
Inhibitors of histone demethylases as anti‐inflammatory agents
As outlined above, H3K27me3 suppresses the expression of multiple proinflammatory genes in macrophages. These studies suggest that modulating the ‘eraser’ of these suppressive modifications, JMJD3 demethylase, by small molecules may be one way to curtail inflammation. However, this possibility is complicated by the fact that regulation of proinflammatory genes by JMJD3 may be independent of its demethylase activity. 20 Also, the degree of sequence similarity among the JMJC domains of histone demethylases made it unclear whether small molecule inhibitors could exhibit adequate substrate specificity. GlaxoSmithKline answered this challenge by identifying a highly selective inhibitor (GSK‐J1 and a cell‐permeable GSK‐J4) of a lysine‐specific demethylase UTX and JMJD3 that acted as an α‐ketoglutarate mimic. 79 GSK‐J4 prevented demethylation of the repressive H3K27me3, and reduced RNA Pol II recruitment, which prevented transcription of TNF and other inflammatory cytokines in LPS‐treated human monocytes. 79 Interestingly, a single knockdown of either UTX or JMJD3 did not reduce TNF, suggesting that these demethylases act together in the control of cytokine transcription.
HDAC inhibitors as anti‐inflammatory agents
Until recently it remained unclear whether histone acetylation was an active regulator of transcription or just a passive by‐product. Some recent and elegant single‐cell analyses revealed that histone H3 lysine‐27 acetylation at a gene locus alters downstream transcription kinetics by as much as 50%, affecting two temporally separate events. First, acetylation enhances the search kinetics of transcriptional activators, and later the acetylation accelerates the transition of Pol II from initiation to elongation. 80 In mammals, HDACs are divided into three classes on the basis of their cellular localization and tissue distribution. Class I HDACs are ubiquitously expressed and are predominately nuclear. Class II HDACs are both nuclear and cytoplasmic and only expressed in certain tissues. Class III HDACs, also called sirtuins (SIRT1–7), are NAD+‐dependent enzymes.
Although successfully used for the treatment of cancer, research now suggests that targeting certain HDAC (‘erasers’) could be utilized for treatment of inflammatory diseases such as asthma, rheumatoid arthritis, IBDs and some virus infections. Indeed, various Class I as well as Class II HDAC targeting inhibitors like trichostatin A, Vorinostat (suberanilohydroxamic acid), phenylbutyrate and givinostat have shown anti‐inflammatory effects both in vitro and in vivo (reviewed in Table 3). 81 , 82 , 83 , 84 Recently it was shown that butyrate exposure of mouse colonic lamina propria macrophages leads to an increase in expression of proinflammatory mediators NO, IL‐6 and IL‐12 but not of TNF, and to an increase in H3K9Ac levels at the promoter regions of these genes in mouse bone‐marrow‐derived macrophages. 85 In contrast to the above‐described pan‐inhibitors of HDACs, specific class and isoform HDAC inhibitors have been identified for HDAC1 and HDAC3, and show anti‐inflammatory effects in animal models of inflammatory diseases and in peripheral blood mononuclear cells (PBMCs) 81 from rheumatoid arthritis (RA) patients, respectively (Table 3). Despite the reasonable success of HDACs as anti‐inflammatory agents, their exact mode of epigenetic regulation as anti‐inflammatory agents in vivo is unclear, the elucidation of which is further confounded by the fact that most HDACs act on both histone or nonhistone substrates and that HDAC inhibition leads to both gene expression and suppression in a cell‐context‐dependent manner (reviewed in Adcock 86 ).
BET inhibitors as anti‐inflammatory agents
Through their function as epigenetic ‘readers’ and their central role in the recruitment of transcriptional machinery, the BET family of bromodomain‐containing proteins is critical for the expression of multiple genes, including those involved in tumor cell growth and inflammation, making them very attractive therapeutic targets. Moreover, targeting epigenetic ‘readers’ seemed an appealing way to specifically interrupt the interpretation of epigenetic modifications without altering the overall epigenetic landscape of the cell, which could conceivably occur by targeting ‘writers’ or ‘erasers’. Bromodomain modules share a conserved fold that comprises a left‐handed bundle of four α‐helices that surround a central acetylated lysine‐binding site. I‐BET762 (also known as GSK525762A) and GSK525768A (which is the (R)‐enantiomer of I‐BET762) were identified initially through a screen for upregulation of APOA1. Upon subsequent chemoproteomics involving immobilization of the compounds on a matrix, followed by affinity purification of interacting proteins from cell extracts and liquid chromatography‐tandem mass spectrometry, the interacting proteins were identified as BRD2, 3 and 4. I‐BET762 acts as a histone mimic and competitively inhibits the binding of BET proteins to acetylated histone peptides, and has a low affinity toward other bromodomain family members, making it a specific inhibitor of the BET subfamily. 25 Treatment of mouse bone‐marrow‐derived macrophages with I‐BET762 selectively inhibited activation of a subset of LPS‐inducible cytokines, chemokines and several transcription factors required for an inflammatory response. 25 LPS‐inducible and I‐BET susceptible genes showed significantly reduced enrichment of the BET proteins BRD2, 3 and 4, as well as P‐TEFb and Pol II, demonstrating that I‐BET762 successfully prevented assembly of chromatin‐activating and elongation‐promoting complexes at these promoters. The vast majority of genes that were suppressed by I‐BET were late PRGs and secondary response genes with low CpG, low H4Ac, low H3K4me3 and low Pol II at their promoters in naive macrophages. PRGs or housekeeping genes with high CpG, high H3K4me3 and high H4Ac could not be inhibited with I‐BET treatment, possibly because of the inability of I‐BET, acting as a histone mimic, to outcompete the preexisting levels of acetylation at those loci. Importantly, I‐BET administration (30 mg kg−1, intravenous) also prevented LPS‐induced endotoxic shock and bacteria‐induced sepsis in mice, 25 highlighting its potential as an anti‐inflammatory agent. A second class of BET family bromodomain inhibitor, I‐BET151, with improved pharmacokinetics, 30 was also shown to reduce levels of circulating IL‐6 and protected mice from LPS‐induced death. 87 Independent studies using an alternative pan‐BET inhibitor, JQ1, also observed suppression of proinflammatory cytokine induction and rescued mice from LPS‐induced death. 88 Finally, in murine macrophages, MS436, a compound that preferentially targets the first bromodomain of BRD4, blocked the transcriptional activity of BRD4 in the NF‐κB‐directed production of nitric oxide and IL‐6. 89
BET family members have also been implicated in the replication of the viral genome and in the transcriptional regulation of multiple viral proteins. For example, BRD4 competes with the HIV transactivator protein Tat for P‐TEFb binding, 90 which results in repression of Tat‐mediated transactivation of the HIV promoter. Further, BRD2 modulates HIV transcription by associating with the E2F1 transcription factor, which binds together with NF‐κB to the HIV enhancer to repress HIV transcription. 91 This suggested that BET inhibitors could reverse HIV latency. 92 Awakening of latent HIV means that the virus can be completely eradicated using antiviral agents, which suggests that BET bromodomains could be potential new targets for HIV induction strategies. 93
Although these pan‐BET inhibitor studies show great preclinical promise and also aid in investigating the biology of bromodomain‐containing proteins, specific BET isoform inhibitors that solely target BRD4, 3, 2 or BRDT may eventually be required for specific indications with limited side effects.
Concluding remarks
The field of epigenetics within immunology is rapidly emerging. This is illustrated by recent discoveries of new classes of chromatin‐modifying enzymes, greater insight into the function of some of these chromatin‐associated proteins in immune cells, findings of somatic mutations in genes coding for epigenetic machinery in immune‐based disorders and the development of highly potent and specific small molecule inhibitors to epigenetic enzymes that demonstrate potency in immune cells. Along with this, our idea of innate immunity is swiftly changing with the developing concept that the innate immune system may bear some ‘memory’ of previous pathogen encounters. These new data, along with strong interest and hasty progress from drug companies and academic consortiums (http://www.thesgc.org/epigenetics) to target epigenetic factors, could be effectively utilized for improved treatment of diseases associated with innate immunity. Pathogens that have been difficult to target through conventional vaccination strategies could benefit from ‘training’ of the innate immune system via epigenetic manipulation. Further, we believe that adjusting whole subsets of inflammatory genes rather than individual inflammatory mediators through druggable epigenetic enzymes would better serve the multitude of inflammatory disorders that currently lack effective therapies. Future understanding of the plethora of epigenetic modifiers, newly developed chemical probes, as well as the ongoing documentation of epigenetic landscapes in innate immune cells through such initiatives as the BLUEPRINT consortium (http://www.blueprint-epigenome.eu) and NIH Roadmap Epigenomics Mapping Consortium (http://www.roadmapepigenomics.org) will help achieve this goal.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1. Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal‐dependent transcriptional elongation. Cell 2009; 138: 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fang TC, Schaefer U, Mecklenbrauker I, Stienen A, Dewell S, Chen MS et al Histone H3 lysine 9 di‐methylation as an epigenetic signature of the interferon response. J Exp Med 2012; 209: 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foster SL, Hargreaves DC, Medzhitov R. Gene‐specific control of inflammation by TLR‐induced chromatin modifications. Nature 2007; 447: 972–978. [DOI] [PubMed] [Google Scholar]
- 4. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S et al Latent enhancers activated by stimulation in differentiated cells. Cell 2013; 152: 157–171. [DOI] [PubMed] [Google Scholar]
- 5. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V et al mTOR‐ and HIF‐1alpha‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014; 345: 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quintin J, Saeed S, Martens JH, Giamarellos‐Bourboulis EJ, Ifrim DC, Logie C et al Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012; 12: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S et al Bacille Calmette‐Guerin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA 2012; 109: 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL et al A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013; 341: 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 10. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13: 484–492. [DOI] [PubMed] [Google Scholar]
- 11. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al Tet proteins can convert 5‐methylcytosine to 5‐formylcytosine and 5‐carboxylcytosine. Science 2011; 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E et al Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011; 146: 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J et al Combinatorial patterning of chromatin regulators uncovered by genome‐wide location analysis in human cells. Cell 2011; 147: 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13: 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaffney DJ, McVicker G, Pai AA, Fondufe‐Mittendorf YN, Lewellen N, Michelini K et al Controls of nucleosome positioning in the human genome. PLoS Genet 2012; 8: e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarai N, Nimura K, Tamura T, Kanno T, Patel MC, Heightman TD et al WHSC1 links transcription elongation to HIRA‐mediated histone H3.3 deposition. EMBO J 2013; 32: 2392–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell 2006; 125: 213–217. [DOI] [PubMed] [Google Scholar]
- 19. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine‐27 demethylase Jmjd3 links inflammation to inhibition of polycomb‐mediated gene silencing. Cell 2007; 130: 1083–1094. [DOI] [PubMed] [Google Scholar]
- 20. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L et al Jmjd3 contributes to the control of gene expression in LPS‐activated macrophages. EMBO J 2009; 28: 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y et al The Jmjd3‐Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 2010; 11: 936–944. [DOI] [PubMed] [Google Scholar]
- 22. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001; 292: 110–113. [DOI] [PubMed] [Google Scholar]
- 23. Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 2002; 16: 2219–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M et al A high‐throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell 2012; 47: 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW et al Suppression of inflammation by a synthetic histone mimic. Nature 2010; 468: 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte‐Lovejoy D et al Histone recognition and large‐scale structural analysis of the human bromodomain family. Cell 2012; 149: 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P‐TEFb and stimulates RNA polymerase II‐dependent transcription. Mol Cell 2005; 19: 523–534. [DOI] [PubMed] [Google Scholar]
- 28. Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N et al BRD4 coordinates recruitment of pause release factor P‐TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon‐stimulated genes. Mol Cell Biol 2013; 33: 2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF‐kappaB via specific binding to acetylated RelA. Mol Cell Biol 2009; 29: 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI et al Inhibition of BET recruitment to chromatin as an effective treatment for MLL‐fusion leukaemia. Nature 2011; 478: 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carpenter S, Ricci EP, Mercier BC, Moore MJ, Fitzgerald KA. Post‐transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol 2014; 14: 361–376. [DOI] [PubMed] [Google Scholar]
- 32. Peng X, Gralinski L, Armour CD, Ferris MT, Thomas MJ, Proll S et al Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio 2010; 1: pii: e00206‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kambara H, Niazi F, Kostadinova L, Moonka DK, Siegel CT, Post AB et al Negative regulation of the interferon response by an interferon‐induced long non‐coding RNA. Nucleic acids Res 2014; 42: 10668–10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramirez‐Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR et al A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 2009; 138: 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramirez‐Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN et al Selective and antagonistic functions of SWI/SNF and Mi‐2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev 2006; 20: 282–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boehm T, McCurley N, Sutoh Y, Schorpp M, Kasahara M, Cooper MD. VLR‐based adaptive immunity. Annu Rev Immunol 2012; 30: 203–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurtz J. Specific memory within innate immune systems. Trends Immunol 2005; 26: 186–192. [DOI] [PubMed] [Google Scholar]
- 38. Sun JC, Ugolini S, Vivier E. Immunological memory within the innate immune system. EMBO J 2014; 33: 1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun JC, Beilke JN, Lanier LL. Immune memory redefined: characterizing the longevity of natural killer cells. Immunol Rev 2010; 236: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Narni‐Mancinelli E, Campisi L, Bassand D, Cazareth J, Gounon P, Glaichenhaus N et al Memory CD8+ T cells mediate antibacterial immunity via CCL3 activation of TNF/ROI+ phagocytes. J Exp Med 2007; 204: 2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeffrey K. Rechallenging immunological memory. Nat Med 2007; 13: 1142. [DOI] [PubMed] [Google Scholar]
- 42. Soudja SM, Chandrabos C, Yakob E, Veenstra M, Palliser D, Lauvau G. Memory‐T‐cell‐derived interferon‐gamma instructs potent innate cell activation for protective immunity. Immunity 2014; 40: 974–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strutt TM, McKinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD et al Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med 2010; 16: 558–564 1p following 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014; 346: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duggan JM, You D, Cleaver JO, Larson DT, Garza RJ, Guzman Pruneda FA et al Synergistic interactions of TLR2/6 and TLR9 induce a high level of resistance to lung infection in mice. J Immunol 2011; 186: 5916–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tuvim MJ, Gilbert BE, Dickey BF, Evans SE. Synergistic TLR2/6 and TLR9 activation protects mice against lethal influenza pneumonia. PLoS ONE 2012; 7: e30596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mishra PK, Patel N, Wu W, Bleich D, Gause WC. Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL‐10 in the absence of a Th2‐type response. Mucosal Immunol 2013; 6: 297–308. [DOI] [PubMed] [Google Scholar]
- 48. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F et al Epigenetic programming of monocyte‐to‐macrophage differentiation and trained innate immunity. Science 2014; 345: 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013; 153: 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal 2011; 15: 551–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY et al TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 2014; 15: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G et al Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 2013; 496: 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue‐resident macrophages. Nat Immunol 2013; 14: 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, Arditi M. TLR signaling at the intestinal epithelial interface. J Endotoxin Res 2003; 9: 322–330. [DOI] [PubMed] [Google Scholar]
- 55. Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of toll‐like receptor‐4 and MD‐2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol 2001; 167: 1609–1616. [DOI] [PubMed] [Google Scholar]
- 56. Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C et al Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA 2007; 104: 13467–13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hamon MA, Cossart P. K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore‐forming toxins. Infect Immun 2011; 79: 2839–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eskandarian HA, Impens F, Nahori M‐A, Soubigou G, Coppée J‐Y, Cossart P et al A role for SIRT2‐dependent histone H3K18 deacetylation in bacterial infection. Science 2013; 341: 1238858. [DOI] [PubMed] [Google Scholar]
- 59. Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C et al An injected bacterial effector targets chromatin access for transcription factor NF‐kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 2007; 8: 47–56. [DOI] [PubMed] [Google Scholar]
- 60. Leng J, Butcher BA, Egan CE, Abi Abdallah DS, Denkers EY. Toxoplasma gondii prevents chromatin remodeling initiated by TLR‐triggered macrophage activation. J Immunol 2009; 182: 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leng J, Denkers EY. Toxoplasma gondii inhibits covalent modification of histone H3 at the IL‐10 promoter in infected macrophages. PLoS ONE 2009; 4: e7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium tuberculosis 19‐kDa lipoprotein inhibits IFN‐gamma‐induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol. 2006; 176: 4323–4330. [DOI] [PubMed] [Google Scholar]
- 63. Wang Y, Curry HM, Zwilling BS, Lafuse WP. Mycobacteria inhibition of IFN‐gamma induced HLA‐DR gene expression by up‐regulating histone deacetylation at the promoter region in human THP‐1 monocytic cells. J Immunol 2005; 174: 5687–5694. [DOI] [PubMed] [Google Scholar]
- 64. Lebreton A, Job V, Ragon M, Le Monnier A, Dessen A, Cossart P et al Structural basis for the inhibition of the chromatin repressor BAHD1 by the bacterial nucleomodulin LntA. MBio 2014; 5: e00775–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A et al A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 2011; 331: 1319–1321. [DOI] [PubMed] [Google Scholar]
- 66. Rolando M, Sanulli S, Rusniok C, Gomez‐Valero L, Bertholet C, Sahr T et al Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 2013; 13: 395–405. [DOI] [PubMed] [Google Scholar]
- 67. Mujtaba S, Winer BY, Jaganathan A, Patel J, Sgobba M, Schuch R et al Anthrax SET protein: a potential virulence determinant that epigenetically represses NF‐kappaB activation in infected macrophages. J Biol Chem 2013; 288: 23458–23472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rennoll‐Bankert KE, Dumler JS. Lessons from Anaplasma phagocytophilum: chromatin remodeling by bacterial effectors. Infect Disord Drug Targets 2012; 12: 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Garcia‐Garcia JC, Rennoll‐Bankert KE, Pelly S, Milstone AM, Dumler JS. Silencing of host cell CYBB gene expression by the nuclear effector anka of the intracellular pathogen Anaplasma phagocytophilum . Infect Immun 2009; 77: 2385–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marazzi I, Ho JSY, Kim J, Manicassamy B, Dewell S, Albrecht RA et al Suppression of the antiviral response by an influenza histone mimic. Nature 2012; 483: 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shanmugam MK, Sethi G. Role of epigenetics in inflammation‐associated diseases. Subcell Biochem 2013; 61: 627–657. [DOI] [PubMed] [Google Scholar]
- 72. Liu A, La Cava A. Epigenetic dysregulation in systemic lupus erythematosus. Autoimmunity 2014; 47: 215–219. [DOI] [PubMed] [Google Scholar]
- 73. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D et al Identification of type 1 diabetes–associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011; 7: e1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Javierre BM, Fernandez AF, Richter J, Al‐Shahrour F, Martin‐Subero JI, Rodriguez‐Ubreva J et al Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010; 20: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Runyon RS, Cachola LM, Rajeshuni N, Hunter T, Garcia M, Ahn R et al Asthma discordance in twins is linked to epigenetic modifications of T cells. PLoS ONE 2012; 7: e48796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Franke A, McGovern DP, Barrett JC, Wang K, Radford‐Smith GL, Ahmad T et al Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42: 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L et al Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Wordsworth BP. The histone demethylase JARID1A is associated with susceptibility to ankylosing spondylitis. Genes Immun 2011; 12: 395–398. [DOI] [PubMed] [Google Scholar]
- 79. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G et al A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488: 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stasevich TJ, Hayashi‐Takanaka Y, Sato Y, Maehara K, Ohkawa Y, Sakata‐Sogawa K et al Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 2014. advance online publication. [DOI] [PubMed] [Google Scholar]
- 81. Segain JP, de la Blétière DR, Bourreille A, Leray V, Gervois N, Rosales C et al Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn's disease. Gut 2000; 47: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Luhrs H, Gerke T, Muller JG, Melcher R, Schauber J, Boxberge F et al Butyrate inhibits NF‐kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol 2002; 37: 458–466. [DOI] [PubMed] [Google Scholar]
- 83. Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med. 2011; 17: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chung YL, Lee MY, Wang AJ, Yao LF. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther 2003; 8: 707–717. [DOI] [PubMed] [Google Scholar]
- 85. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA 2014; 111: 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Adcock IM. HDAC inhibitors as anti‐inflammatory agents. Br J Pharmacol 2007; 150: 829–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F et al Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I‐BET151 (GSK1210151A). Bioorg Med Chem Lett 2012; 22: 2968–2972. [DOI] [PubMed] [Google Scholar]
- 88. Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 2013; 190: 3670–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J et al Structure‐guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem 2013; 56: 9251–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P‐TEFb‐interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci USA 2007; 104: 13690–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kundu M, Guermah M, Roeder RG, Amini S, Khalili K. Interaction between cell cycle regulator, E2F‐1, and NF‐kappaB mediates repression of HIV‐1 gene transcription. J Biol Chem 1997; 272: 29468–29474. [DOI] [PubMed] [Google Scholar]
- 92. Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G et al Reactivation of latent HIV‐1 by inhibition of BRD4. Cell reports 2012; 2: 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Halper‐Stromberg A, Lu C‐L, Klein F, Horwitz Joshua A, Bournazos S, Nogueira L et al Broadly neutralizing antibodies and viral inducers decrease rebound from HIV‐1 latent reservoirs in humanized mice. Cell 2014; 158: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pennini ME, Liu Y, Yang J, Croniger CM, Boom WH, Harding CV. CCAAT/enhancer‐binding protein beta and delta binding to CIITA promoters is associated with the inhibition of CIITA expression in response to Mycobacterium tuberculosis 19‐kDa lipoprotein. J Immunol 2007; 179: 6910–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pennini ME, Perrinet S, Dautry‐Varsat A, Subtil A. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis . PLoS Pathog 2010; 6: e1000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Garcia‐Garcia JC, Barat NC, Trembley SJ, Dumler JS. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum . PLoS Pathog 2009; 5: e1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lang C, Hildebrandt A, Brand F, Opitz L, Dihazi H, Luder CG. Impaired chromatin remodelling at STAT1‐regulated promoters leads to global unresponsiveness of Toxoplasma gondii‐infected macrophages to IFN‐gamma. PLoS Pathog 2012; 8: e1002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Agarwal S, Agarwal S, Jin H, Pancholi P, Pancholi V. Serine/threonine phosphatase (SP‐STP), secreted from Streptococcus pyogenes, is a pro‐apoptotic protein. J Biol Chem 2012; 287: 9147–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F et al Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol 2006; 176: 5015–5022. [DOI] [PubMed] [Google Scholar]
- 100. Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L et al Anti‐rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen‐induced arthritis in rodents. Br J Pharmacol 2007; 150: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chung Y‐L, Lee M‐Y, Wang A‐J, Yao L‐F. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther 2003; 8: 707–717. [DOI] [PubMed] [Google Scholar]
- 102. Hsieh IN, Liou JP, Lee HY, Lai MJ, Li YH, Yang CR. Preclinical anti‐arthritic study and pharmacokinetic properties of a potent histone deacetylase inhibitor MPT0G009. Cell Death Dis 2014; 5: e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hara N, Alkanani AK, Dinarello CA, Zipris D. Histone deacetylase inhibitor suppresses virus‐induced proinflammatory responses and type 1 diabetes. J Mol Med (Berl) 2014; 92: 93–102. [DOI] [PubMed] [Google Scholar]
- 104. Zhang Z‐Y, Schluesener HJ. HDAC inhibitor MS‐275 attenuates the inflammatory reaction in rat experimental autoimmune prostatitis. Prostate 2012; 72: 90–99. [DOI] [PubMed] [Google Scholar]
- 105. Cantley MD, Bartold PM, Marino V, Fairlie DP, Le GT, Lucke AJ et al Histone deacetylase inhibitors and periodontal bone loss. J Periodontal Res 2011; 46: 697–703. [DOI] [PubMed] [Google Scholar]
- 106. Gillespie J, Savic S, Wong C, Hempshall A, Inman M, Emery P et al Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3‐selective inhibitor reduces interleukin‐6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum 2012; 64: 418–422. [DOI] [PubMed] [Google Scholar]
- 107. Kruidenier L, Chung C‐w, Cheng Z, Liddle J, Che K, Joberty G et al A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488: 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L et al Down‐regulation of NF‐κB transcriptional activity in HIV‐associated kidney disease by BRD4 inhibition. J Biol Chem 2012; 287: 28840–28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhang ZY, Zhang Z, Schluesener HJ. MS‐275, an histone deacetylase inhibitor, reduces the inflammatory reaction in rat experimental autoimmune neuritis. Neuroscience 2010; 169: 370–377. [DOI] [PubMed] [Google Scholar]