

Abstract
Limited orbital granulomatosis with polyangiitis (GPA) is uncommon and its diagnosis may be delayed, especially when isolated lacrimal involvement is the initial presentation, because clinical manifestations are non-specific and systemic diagnostic criteria are not applicable. Making an early diagnosis despite the absence of systemic progression is extremely important because in some cases the disease is locally destructive, with irreversible visual and functional loss, and it can be refractory to corticosteroids and conventional immunosuppressive drugs to induce remission. The authors report an unusual limited form of orbital GPA in a 35-year-old woman presenting with bilateral dacryoadenitis, evolving later to locally aggressive bilateral orbital pseudotumour leading to proptosis, extraocular myositis, diplopia and medial deviation of the nasal septum. She had never had systemic manifestations but her disease was persistently active and unresponsive to corticosteroids and immunosuppressors. The aim of this paper is to provide further evidence of aggressive and refractory limited forms of GPA.
Background
Granulomatosis with polyangiitis (GPA) is an autoimmune small vessel vasculitis first described in 1931 by Heintz Klinger, and was formerly known by the eponymous name, Wegener's Granulomatosis, after Friedrich Wegener more clearly delineated this entity in 1936, distinguishing it from polyarteritis nodosa. The 1990 American College of Rheumatology (ACR) criteria for diagnosis of GPA distinguished GPA from other vasculitides with 88% sensitivity and 92% specificity if two or more of these criteria were present: (1) nasal or oral inflammation (painful or painless oral ulcers or purulent or bloody nasal discharge), (2) respiratory radiographic abnormalities consistent with respiratory tissue destruction (eg, nodules, infiltrates and cavities), (3) abnormal urinary sediment (microscopic haematuria with or without red cell casts) and (4) histological granulomas within an artery or in the perivascular area of an artery or arteriole. ACR criteria have limitations because they may not distinguish GPA from mimics of microscopic polyangiitis and vasculitis. With the identification of serological testing for antineutrophil cytoplasmatic antibody (ANCA) levels in the 1980s, the diagnosis of GPA is based on a combination of clinicopathological criteria and serological test results. A cytoplasmic ANCA (c-ANCA) with staining pattern directed against proteinase 3 is present in more than 90% of patients with GPA and may play a role in pathogenesis, but is not essential to cause the disease. Positive ANCA serology is not necessary for the diagnosis of GPA if the clinical and histological findings are suggestive of the disease. Common histopathological features of GPA are necrosis, granuloma formation and vasculitis of small-to-medium-sized vessels.1–3
GPA is a rare disease with an incidence of from 7 to 12 new cases per million inhabitants per year (about 10 cases per million in Northern Europe), distinctly higher among Caucasians, particularly those of northern European ancestry. There is equal frequency in males and females. The peak incidence is between 45 and 60 years. GPA is very rare in childhood and young adults.4
The aetiology of GPA may be related with infectious, environmental, chemical, toxic and pharmacological factors. Infectious triggers include infections of ears, nose and respiratory tract, and Staphylococcus aureus nasal carriage. Environmental triggers described are dust inhalation, exposure to silica, pollution, smoking, metals (mercury and lead) and inhaled toxins and chemicals. Medicines triggering drug-induced ANCA-vasculitis are cefotaxime, minocycline, antithyroid medication, antitumour necrosis factor α agents, clozapine, thioridazine, allopurinol, hydralazine, phenytoin, sulfasalazine and others.3 5 6 GPA was also reported among siblings and associated with the HLA-DP, SERPINA1 (gene encoding for α1-antitrypsin) and PRTN3 (gene encoding for proteinase 3) genes.4 7
GPA classically involves the upper respiratory tract, lungs and kidneys. However, it can involve any organ system, with variable clinical presentation and course of disease. Respiratory tract involvement occurs in up to 85% and renal in 70–80% of patients. Ocular involvement has been reported in 50–60% of patients and may be the presenting feature in 8–16% of cases.8 Ocular GPA may occur alone or, more frequently, as a component of a multisystemic presentation. Clinical manifestations of ocular GPA result from inflammation of ocular structures including extraocular muscles, lacrimal glands, orbital fat, globe, optic nerve and orbital nerves.9
The authors report an unusual limited form of orbital GPA in a 35-year-old woman presenting with bilateral inflammatory dacryoadenitis, evolving later to advanced locally aggressive bilateral orbital pseudotumour, without systemic progression but persistently refractory to the conventional therapies that induce remission.
Case presentation
A 35-year-old Caucasian woman presented with a 2-month history of progressive malaise, recurrent subfebrile episodes, chronic rhinosinusitis, chronic nasal congestion, serous nasal discharge, intermittent epistaxis, bilateral epiphora and conjunctival hyperaemia. She also developed right orbital swelling, pain and limitation of right extraocular movements with diplopia, 1 week before admission. There were no other symptoms.
Medical history was significant for dyslipidaemia, essential hypertension and cervical intraepithelial neoplasia 2 treated with cervical conisation.
She was previously medicated with alendronic acid 70 mg/colecalciferol 2800 UI once weekly, calcium carbonate 1250 mg twice daily and omeprazole 20 mg once daily.
There was no relevant social or family history.
Ophthalmological examination (OE) revealed bilateral conjunctival hyperaemia; right orbital and eyelid pain, swelling and erythaema; unilateral right proptosis and limitation of extraocular movements (elevation and abduction) with diplopia. Visual acuity was normal. The OE was otherwise unremarkable.
Anterior rhinoscopy showed bilateral inferior turbinate hypertrophy, significant congestion, redness of mucosa and slight nasal septum deviation.
General examination was normal.
Investigations
Initial laboratory work up was significant for elevated inflammatory markers (leucocyte count 12 800/µL with 86.3% neutrophils, C reactive protein 29.7 mg/L, erythrocyte sedimentation rate 51 mm/1st hour) and positive c-ANCA of 1/160 (normal <1/20). Immunological study showed normal serum immunoglobulins (IgA, IgM and IgG) and complement; negative rheumatoid factor, antinuclear, antidouble-stranded DNA, anti-Sjögren's syndrome A/B and antithyroid antibodies. Liver and renal function tests, ionogram, serum ACE inhibitors and thyroid function were normal.
A first orbital MRI was performed revealing localised bilateral lacrimal gland enlargement (more severe on the right side) with homogeneous contrast enhancement (figure 1). Mucosal thickening of maxillary, ethmoidal and frontal sinuses, and adenoid hypertrophy, were also evident. There was no bone erosion.
Figure 1.
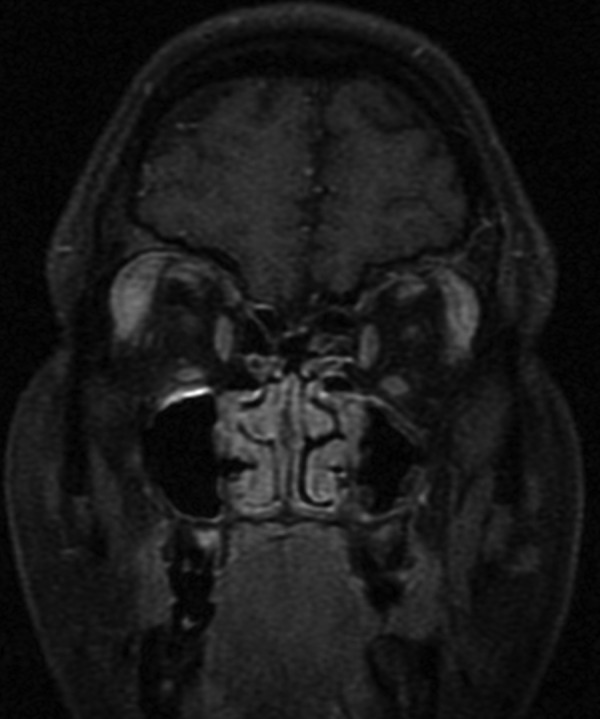
First orbital MRI showing localised bilateral lacrimal gland enlargement (more severe on the right side) with homogeneous contrast enhancement (coronal section, T1-weighted image fast spin-echo (FSE) fat-saturated contrast-enhanced).
A biopsy of the adenoid tissue was performed but the result was inconclusive, showing subepithelial non-granulomatous chronic inflammation.
An incisional biopsy from the right lacrimal gland through a small sub-brow incision was performed. It depicted acinar atrophy, tissue fibrosis, polymorphic inflammatory infiltration of lymphocytes, plasmocytes and neutrophils, and the classic triad tissue necrosis, vasculitis of small vessels and epithelioid granulomas, consistent with a diagnosis of GPA (figure 2A, B). The biopsy tissue was negative for acid-fast bacilli (Ziehl-Neelsen stain) and fungal hyphae (Periodic acid-Schiff stain); bacterial, mycobacterial and fungal cultures were sterile; Congo red staining was negative for amyloid; immunohistochemical stain was negative for lymphoma of the orbit.
Figure 2.
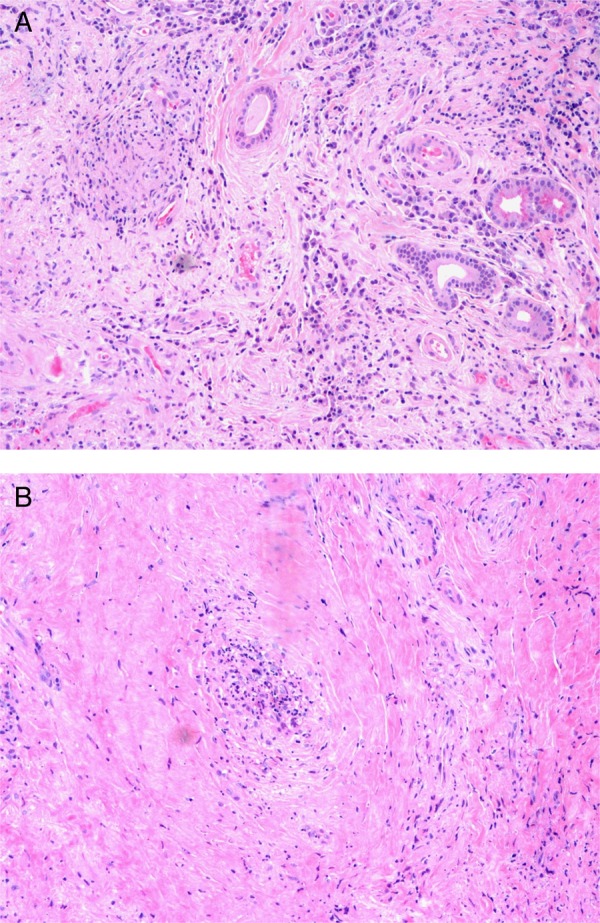
Biopsy of right lacrimal gland. (A) Acinar atrophy, tissue fibrosis, polymorphic inflammatory infiltration of lymphocytes, plasmocytes and neutrophils, and the classic triad tissue necrosis, vasculitis of small vessels and epithelioid granulomas, consistent with a diagnosis of GPA. (B) Vasculitis lesion. GPA, granulomatosis with polyangiitis.
A systemic work up including brain and chest contrast-enhanced CT, pulmonary function tests and urinalysis, was normal.
Differential diagnosis
The differential diagnosis included infectious, inflammatory and neoplastic disorders. Infectious orbital cellulitis was initially suspected but antibiotic treatment did not improve local inflammatory signs and subsequently histochemical stains and cultures ruled out bacterial, mycobacterial and fungal infection.
Several systemic inflammatory conditions associated with orbital inflammatory disease were considered, including Graves’ orbitopathy, GPA, sarcoidosis, eosinophilic GPA (EGPA), idiopathic orbital inflammation (IOI) and IgG4-related disease. Graves’ orbitopathy is a very common cause of unilateral and bilateral proptosis but endocrine manifestations were absent (normal serum thyroid hormone levels and negative antithyroid antibodies). Sarcoidosis was ruled out because pulmonary parenchymal lesions, bilateral hilar and mediastinal lymphadenopathy or erythaema nodosum were absent; ACE was normal; lacrimal gland histology revealed granulomas but also necrosis and vasculitis, and c-ANCAs were positive, all suggestive of GPA. EGPA was also initially suspected because of chronic rhinosinusitis, but it rarely involves the orbit, and there was neither peripheral nor lacrimal gland histological specimen eosinophilia. IOI is a common cause of orbital inflammation but it was ruled out because it is a diagnosis of exclusion when laboratory, radiographic and histological evaluation are negative for other causes. IgG4-related disease of the orbit was excluded because there was no increased number of IgG4-positive plasma cells on histology, and this, combined with the presence of necrosis and acute inflammation, was more suggestive of GPA.
Neoplastic disorders such as lymphoma (the most common orbital malignancy in adults) and rhabdomyosarcoma, were excluded by histology and immunohistochemical stain of lacrimal gland biopsy.
Outcome and follow-up
The patient was first seen in the Emergency Room and later in consultation by Internal Medicine and Ophthalmology.
She was initially treated with a course of antibiotics for a possible infectious orbital cellulitis, with no improvement of symptoms. Antihistamine for sinonasal symptoms and topical corticosteroids because of the presence of signs suggestive of orbital inflammation (ocular pain, epiphora, injection, proptosis and diplopia) were also administered, with only mild clinical improvement.
Clinical and imagiological findings and the detection of positive c-ANCA supported the diagnosis of bilateral dacryoadenitis suggestive of limited GPA, so oral prednisolone (1 mg/kg/day) and immunosuppressive therapy with azathioprine 2 mg/kg/day were started. Azathioprine was chosen for its safety profile since the patient was a young woman with a desire to become pregnant soon and because she had limited disease, with neither renal nor pulmonary manifestations. The prednisolone dose was tapered slowly, reaching 20 mg/day by the end of 2 months. This treatment was carried out for ∼2 years, maintaining regular follow-up, with clinical improvement, reasonable disease activity control and c-ANCA titre decrease and without systemic progression of the disease.
Subsequently, the patient experienced recurrent ophthalmological relapses, with right proptosis, bilateral epiphora, conjunctival hyperaemia and chemosis. She also developed bilateral ocular myositis with orbital pain that worsened with eye movement, leading to decreased ocular motility and diplopia. There was no significant elevation of inflammatory markers nor further increase in c-ANCA titre, and she remained without systemic involvement. Immediate oral and topical corticosteroid dose adjustments were made according to these clinical findings, with improvement of symptoms. A follow-up orbital MRI was performed showing an expanding bilateral orbital mass extending from the lacrimal gland to the optic canal, with involvement of the neighbouring extraocular muscles, namely contact along the superior rectus muscles and medial deviation of lateral rectus muscles. This orbital mass was larger on the right orbit, occupying the intraconal compartment, with medial deviation of the optic nerve and ocular proptosis (figure 3A–C). Given the clinical and imagiological worsening, azathioprine was stopped and new treatment was initiated with oral methotrexate (MTX) 7.5 mg/week with folic acid supplementation, keeping prednisolone 20 mg/day. Liver function test abnormalities arose after increasing the MTX dose, leading to discontinuation of this drug. Meanwhile, another follow-up orbital MRI was performed depicting progression of the orbital mass size, inducing bilateral proptosis (figure 4A–C) and medial deviation of the right lamina papyracea of the ethmoid bone (figure 4C). A combination of intravenous cyclophosphamide (CYP), dexamethasone 10 mg and mercaptoethane sulfonate (Mesna) 1.2 g to prevent cystitis was at this point started for remission induction. This therapy was given twice monthly over the following 3 months (7 administrations for a body surface area of 2 m2—total of 8 g of CYP). The patient was also taking trimetoprim-sulphamethoxazole, orally, three times per week, for Pneumocystis jirovecii prophylaxis. She remained with neither clinical nor imagiological improvement (a new orbital MRI was similar to that performed 3 months earlier). Clinical and laboratory tests to adjust the treatment and to check for the possible appearance of CYP-related side effects, such as severe leukopaenia or haemorrhagic cystitis, were performed regularly and remained negative.
Figure 3.
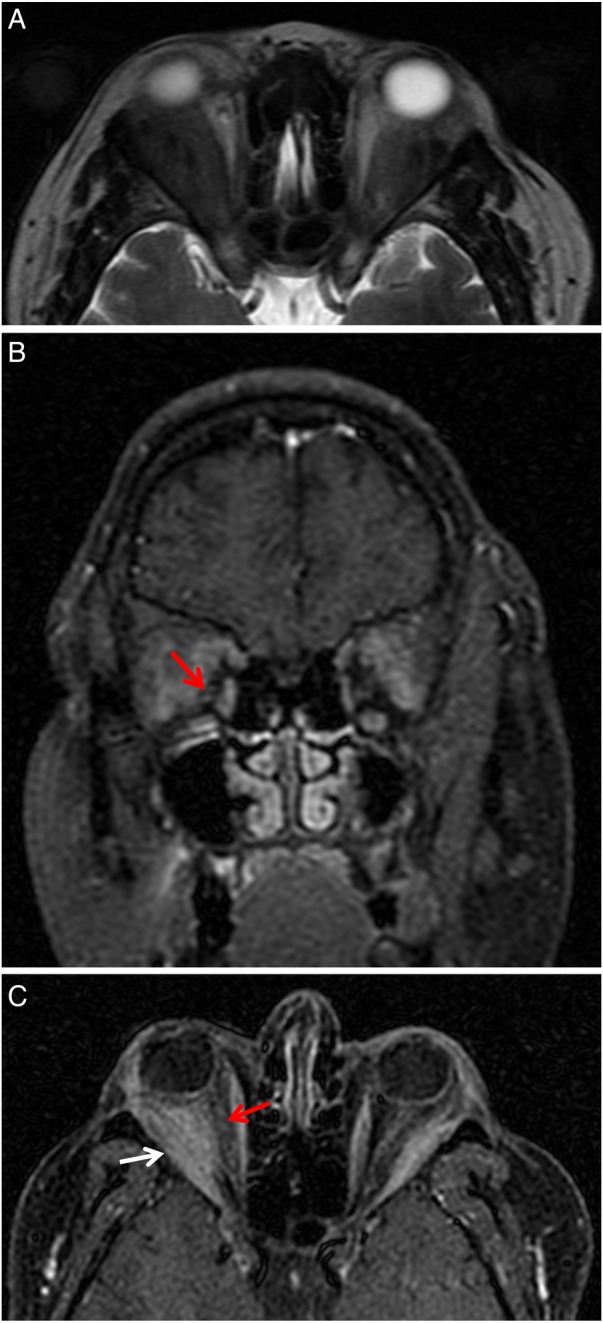
Orbital MRI showing expanding hypointense bilateral orbital mass suggestive of GPA extending from the lacrimal gland to the optic canal, with involvement of the neighbouring extraocular muscles, namely, the contact along the superior rectus muscles and medial deviation of lateral (white arrow) rectus muscles. This orbital mass was larger on the right orbit, occupying the intraconal compartment, with medial deviation of the optic nerve (red arrow) and ocular proptosis. (A) Axial section, T2-weighted image FSE. (B) Coronal section, T1-weighted image FSE fat-saturated contrast-enhanced. (C) Axial section, T1-weighted image FSE fat-saturated contrast-enhanced. FSE, fast spin-echo; GPA, granulomatosis with polyangiitis.
Figure 4.
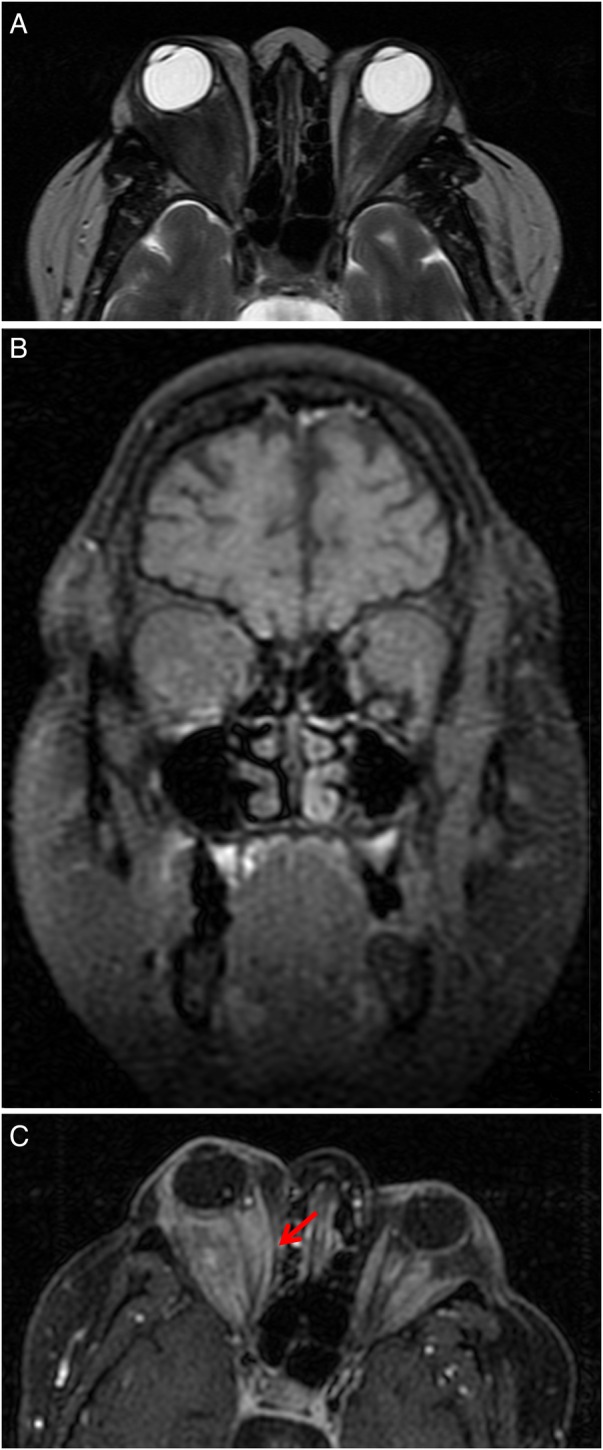
Orbital MRI revealing progression of the orbital mass size, inducing bilateral proptosis and medial deviation of the right lamina papyracea of the ethmoid bone (red arrow). (A) Axial section, T2-weighted image FSE. (B) Coronal section, T1-weighted image FSE fat-saturated contrast-enhanced. (C) Axial section, T1-weighted image FSE fat-saturated contrast-enhanced. FSE, fast spin-echo.
Faced with refractory response to CYP, rituximab (RTX) intravenous infusion 750 mg/once weekly was initiated (375 mg/m2/week for 4 weeks). The patient received four doses of RTX, again without significant improvement of orbital swelling, proptosis or ocular motility (figures 5A, B). Progressive oral prednisolone tapering to 20 mg/day was possible, but the therapy could not be stopped altogether.
Figure 5.

Orbital granulomatosis with polyangiitis. (A) Anterior view. (B) Lateral view.
The patient is currently taking oral CYP 175 mg/day (2 mg/kg/day, body weight of 92 kg) and prednisolone 30 mg/day. A new orbital MRI performed 2 months after starting this treatment, again revealed no improvement.
Despite locally aggressive orbital disease, this patient never presented with irreversible visual loss related to the disease itself or to systemic GPA.
She developed typical steroid-related Cushing's facies, bilateral posterior subcapsular cataracts with transient visual acuity impairment and ocular hypertension controlled with timolol maleate ophthalmic solution. Phacoemulsification surgery was performed, with rapid visual recovery (her best corrected visual acuity is now 10/10 in both eyes).
Discussion
A localised form of GPA involving the head and neck region has been recognised as a subtype of the disease, without renal involvement and with less frequent progression to systemic disease. This limited form of disease is usually associated with better prognosis, but there are a few cases refractory to classical treatments and associated with poor prognosis, as this patient illustrates. Orbital disease occurs in 45% of patients with GPA and can be associated with significant morbidity, including visual loss and facial deformity. Limited orbital GPA is uncommon and its diagnosis may be delayed, especially in patients with isolated lacrimal involvement as the initial presentation, because clinical manifestations are non-specific and may mimic other less significant illnesses, and systemic diagnostic criteria are not adaptable. The incidence of immunocompetent patients presenting with purely adnexal and orbital GPA, suggests that advanced localised orbital disease may occur over the years, without systemic involvement.10 Orbital GPA may be the result of extension from adjacent sinuses or nasopharynx (contiguous disease), or primary inflammation (focal disease). Sinonasal involvement, such as purulent or sanguineous nasal discharge, chronic nasal congestion, epistaxis and sinus pain, is present in up to 69% of patients with orbital and adnexal GPA.11 12
Orbital GPA differential diagnosis includes bacterial or fungal (especially Aspergillus or Mucor spp) orbital inflammatory infiltration, sarcoidosis, EGPA, Graves’ orbitopathy, lymphoma of the orbit, IOI and IgG4-related disease. Polyarteritis nodosa and Kawasaki disease have also, rarely, been seen in orbital biopsies.9 13
Orbital disease can involve the lacrimal gland (inflammatory dacryoadenitis), extraocular muscles (orbital myositis) and soft tissues (inflammatory orbital pseudotumour). Orbital inflammation usually has a relatively sudden onset (within days) of ocular pain, epiphora, injection and proptosis. Diplopia and vision loss can arise in severe orbital disease and occurs in 20–50% of patients as a direct consequence of mass effect, optic nerve compression due to adjacent inflammation, optic neuropathy secondary to vasculitis, exposure keratopathy, corneal ulceration or eyelid destruction. Proptosis resulting from orbital involvement is the most common finding, affecting 15–20% of patients with GPA. One-third of patients with proptosis develop orbital socket contraction, defined as enophthalmos, and fibrotic orbital changes, leading to restrictive ophthalmopathy, optic nerve injury and chronic orbital pain that may be unresponsive to immunosuppressive drugs. Extensive orbital disease may lead to intranasal bone erosion, particularly loss of the nasal septum, orbital fistula and infectious orbital abscess.8 9 11 12 14 Lacrimal gland involvement as the initial presentation of orbital GPA is uncommon and usually unilateral, and it may predict locally aggressive disease. There are previous case reports detailing unilateral and bilateral dacryoadenitis at the early stages of GPA.12 15–18
An early complete diagnostic work up is important to distinguish GPA from other infiltrative lesions of the lacrimal gland and orbit. A high index of suspicion is important, especially in young adults, with repeated ANCA testing, biopsy and imaging to evaluate progression of disease, and to identify diagnostic signals that may not exist at presentation.12 c-ANCAs have 91% of sensitivity and 99% of specificity for active GPA involving lungs and kidneys, although they are negative in up to 50% of cases of limited disease to the sinuses and orbit. c-ANCA titres are dependent on the extent and activity of the disease and, comprehensibly, they can be low in limited orbital GPA. Moreover, the probability of a definitive diagnosis from biopsy of a tissue such as of the sinus or orbit is very low, with the diagnostic pathological triad of parenchymal necrosis, vasculitis and granulomatous inflammation being demonstrated in only 50% of patients, in part because medium-sized vessels are not typically biopsied. Mid-facial bone erosion and destruction of the nasal septum are independent of ANCA or systemic involvement, so they are good predictors of GPA in patients with orbital inflammatory symptoms and signs.10 11 13 19 Radiological findings are helpful in discriminating between GPA and other aetiologies of orbital inflammation. Imaging investigation of orbital disease in patients with suspected GPA is performed with conventional CT and MRI. Sinonasal involvement and bony changes on imaging are highly suggestive of GPA, but they are often absent at the initial stage.12 An advantage of CT is its ability to better characterise sinus structure disorders and osseous invasion. MRI is more useful in identifying granulomas and delineating mucosal changes in sinuses, the nasal cavity and orbits. Orbital masses are slightly hyperdense relative to nasal mucosa on contrast-enhanced CT; they are hypointense relative to orbital fat in both T1 and T2 modalities on MRI, and enhance with iv gadolinium contrast.11 13 20 21 All of these clinical and imagiological characteristics are non-specific and may occur in other orbital inflammatory diseases. In these cases, biopsy of the orbital inflammatory mass is useful to reach a definitive histopathological diagnosis when orbital GPA is considered. Biopsy immunohistochemical stains are helpful in the diagnosis of lymphoma of the orbit. As with other vasculitides, correlation of clinical, laboratorial, imaging and histological findings is the optimal strategy for a confident diagnosis of GPA.8 9
The orbit may be the only affected site but it can also be the first presenting feature of GPA before progression to multisystem involvement despite systemic therapy. Thus, the clinician should perform an investigation to determine the extent of disease, including assessment of lung and renal involvement, with chest CT scans and urinalysis (detection of microscopic haematuria and cellular casts, indicating glomerulonephritis).9 11
GPA limited to orbital and adnexal structures may progress over the years, without systemic involvement. Making an early correct diagnosis despite the absence of systemic progression is extremely important because in some of these patients the disease is locally destructive, with irreversible visual and functional loss, and it can be refractory to systemic corticosteroids and conventional immunosuppressive drugs including CYP. This ophthalmic morbidity highlights the importance of identifying atypical clinical presentations of GPA, for timely institution of adequate aggressive immunosuppression to reach remission and prevent visual loss.10 12 Recent advances in immunosuppressive therapy, with a combination of corticosteroids with either MTX or azathioprine, or CYP or RTX, have radically changed the prognosis of GPA, leading to disease remission and prolonged survival rates of 95% at 5-year follow-up and 80% after 10 years. Despite this, GPA remains an incurable disease and treatment side effects are a major concern, boosting investigation of alternative therapies, including biological agents.11 12 Immunosuppressive therapy should be considered in all patients with active GPA. Induction therapy with a corticosteroid and an immunosuppressive drug has the finality of achieving remission (absence of active disease), and maintenance treatment is then given to sustain the remission. The main induction drug used is CYP, a cytotoxic alkylating agent capable of killing B-cells and T-cells.22 An effective treatment with control of the disease in severe forms of active GPA can be achieved with 3–4 months of daily CYP and corticosteroids in a high percentage of patients. CYP can be given orally in a daily scheme or using iv pulses. Both regimens have similar efficacy at controlling disease activity, but pulse CYP is associated with a lower cumulative dose, thus exposing the patient to a higher risk of relapse while at the same time lowering toxicity. When using iv CYP as a first line route, oral CYP can still be successful if iv pulses did not control the disease or in the case of relapse within the first 6 months of treatment. For milder cases of GPA, chronic CYP therapy may not be necessary and using azathioprine or MTX is often as effective as CYP for induction of remission. Prednisolone 1 mg/kg/day is usually tapered and discontinued over 6–9 months when there is favourable clinical evolution with remission maintenance.11 22–24 Patients treated with azathioprine or MTX who are non-responders, have progressive disease or lateral effects, should be treated with a regimen consisting of corticosteroids in combination with either CYP (oral or intravenous) or RTX. If there is high risk for malignancy or concerns about fertility, RTX should be preferred.10 12 RTX is a chimeric monoclonal antibody that leads to the depletion of peripheral B-cells, but not plasma cells, by targeting the B-cell–specific CD20 calcium channel. It has been approved for the induction treatment of ANCA-associated vasculitides as an alternative to CYP, but is not yet approved in all countries and is much more expensive. In open-label trials, the complete remission rates were higher than 75%. Once RTX induces a remission, maintenance treatment must be initiated. A controlled trial achieved by the French Vasculitis Study Group has corroborated the usefulness of RTX for maintenance therapy, but the use of RTX for the maintenance of long-term remission needs further investigation.22 Two randomised controlled trials compared RTX to CYP for induction, in combination with corticosteroids. Short-term safety profiles were identical and complete remission rates were not substantially different.25 26 RTX had higher remission rates when relapses occurred after CYP therapy.26 RTX retreatment decisions can be individualised based on combined disease activity scores, B-cell recovery and ANCA reascension.27 Infections and a few cases of progressive multifocal leukoencephalopathy due to the JC virus induced by severe immunosuppression have been reported as RTX side effects. Symptoms of serum-like sickness, neutropaenia or vasculitis may also occur.22 28 Anti-CD20 agents are promising treatment options for ANCA-associated vasculitides, but long-term studies of their efficacy and safety profiles are necessary to upgrade our knowledge in these chronic and often recurrent diseases.22 In over 90% of patients with small-vessel vasculitis, remission should be attained by 6 months with standard induction therapy. Patients with persistent active vasculitis, despite at least 6 months of treatment with CYP or RTX in combination with corticosteroids, have treatment-resistant disease.25 26 29 Mycophenolate mofetil is used in patients who could not achieve remission with CYP and RTX. One option is the regimen used in the IMPROVE study, starting at 2000 mg/day, followed by a reduction to 1500 and 1000 mg/day after 12 and 18 months, respectively.30 Open-label trials and one controlled trial presented promising results and good safety profile of intravenous immunoglobulin use in relapsing or naïve ANCA-associated vasculitides. However, relapses at treatment discontinuation were documented and limited their use. This therapy may be useful in periods of active vasculitis with severe immunodepression. The total dosage is 2 g/kg, either in two doses or over 5 days.22 31
This patient is the paradigm of an unusual form of progressive limited orbital GPA, refractory to classical treatments that induce remission. She presented with bilateral inflammatory dacryoadenitis, evolving later to locally aggressive bilateral orbital pseudotumour leading to bilateral proptosis, extraocular myositis, diplopia and medial deviation of the right lamina papyracea of the ethmoid bone. Despite this, she had no systemic manifestations, no significant elevation of inflammatory markers and no c-ANCA titre. Her disease remained reasonably controlled during 2 years of azathioprine and corticosteroid therapy, although subsequently there was local progression of orbital disease, refractory to MTX, and to intravenous and oral CYP and RTX.
In conclusion, limited orbital GPA is a challenging ‘simulator’ disease that may be persistently active and unresponsive to immunosuppressive therapies. Control of active disease should be performed through a collaborative multidisciplinary approach according to the clinical state, and may be achieved using a range of therapies, depending on how rapidly and aggressively the treatment is required. The choice should be based on evidence, but interpreted for the individual, to minimise damage and always taking into account comorbidities.29
Learning points.
Limited orbital granulomatosis with polyangiitis (GPA) is an uncommon and challenging disease the diagnosis of which may be delayed, especially in patients with isolated lacrimal gland involvement as the initial presentation, because clinical manifestations are non-specific and may mimic other less significant illnesses, and systemic diagnostic criteria are not applicable.
Making an early correct diagnosis of GPA limited to orbital structures despite the absence of systemic progression is extremely important because it can be locally destructive, with irreversible visual and functional loss.
Correlation of clinical, laboratorial, imaging and histological findings is the optimal strategy for a confident diagnosis of limited GPA to the sinuses and orbit because cytoplasmic antineutrophil cytoplasmatic antibody titres are negative in up to 50% of cases and the probability of a definitive diagnosis from biopsy is very low, with the diagnostic pathological triad of parenchymal necrosis, vasculitis and granulomatous inflammation occurring in only 50% of patients.
Limited orbital GPA may be persistently active, and unresponsive to systemic corticosteroids and conventional immunosuppressive therapies. Our patient is a paradigm of this type of presentation.
Acknowledgments
The authors wish to express their gratitude to Dr Miguel Baptista of the Neuroradiology Department of Hospital Pedro Hispano, for his solicitude and extreme helpfulness in the discussion of the MRI images of this clinical case.
Footnotes
Contributors: All the authors were responsible for the patient's management and follow-up. RLC and SM collected all significant clinical information. RLC drafted the manuscript. RM and RC reviewed, redrafted and made a significant contribution to the final version. All the authors gave final approval of this version.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Falk RJ, Gross WL, Guillevin L et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Ann Rheum Dis 2011;70:704 10.1136/ard.2011.150714 [DOI] [PubMed] [Google Scholar]
- 2.Leavitt RY, Fauci AS, Bloch DA et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum 1990;33:1101–7. 10.1002/art.1780330807 [DOI] [PubMed] [Google Scholar]
- 3.Lutalo PM, D'Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 2014;48–49:94–8. 10.1016/j.jaut.2014.01.028 [DOI] [PubMed] [Google Scholar]
- 4.Comarmond C, Cacoub P. Granulomatosis with polyangiitis (Wegener): clinical aspects and treatment. Autoimmun Rev 2014;13:1121–5. 10.1016/j.autrev.2014.08.017 [DOI] [PubMed] [Google Scholar]
- 5.Cartin-Ceba R, Peikert T, Specks U. Pathogenesis of ANCA-associated vasculitis. Curr Rheumatol Rep 2012;14:481–93. 10.1007/s11926-012-0286-y [DOI] [PubMed] [Google Scholar]
- 6.Gao Y, Zhao MH. Review article: drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrol (Carlton) 2009;14:33–41. 10.1111/j.1440-1797.2009.01100.x [DOI] [PubMed] [Google Scholar]
- 7.Lyons PA, Rayner TF, Trivedi S et al. Genetically distinct subsets within ANCA-associated vasculitis . N Engl J Med 2012;367:214–23. 10.1056/NEJMoa1108735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pakrou N, Selva D, Leibovitch I. Wegener's granulomatosis: ophthalmic manifestations and management. Semin Arthritis Rheum 2006;35:284–92. 10.1016/j.semarthrit.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 9.Muller K, Lin JH. Orbital granulomatosis with polyangiitis (Wegener granulomatosis): clinical and pathologic findings. Arch Pathol Lab Med 2014;138:1110–14. 10.5858/arpa.2013-0006-RS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan LT, Davagnanam I, Isa H et al. Clinical and Imaging Features Predictive of Orbital Granulomatosis with Polyangiitis and the Risk of Systemic Involvement. Ophthalmology 2014;121:1304–9. 10.1016/j.ophtha.2013.12.003 [DOI] [PubMed] [Google Scholar]
- 11.Tarabishy AB, Schulte M, Papaliodis GN et al. Wegener's granulomatosis: clinical manifestations, differential diagnosis, and management of ocular and systemic disease. Surv Ophthalmol 2010;55:429–44. 10.1016/j.survophthal.2009.12.003 [DOI] [PubMed] [Google Scholar]
- 12.Tan LT, Davagnanam I, Isa H et al. Clinical and Imaging Features of Lacrimal Gland Involvement in Granulomatosis with Polyangiitis. Ophthalmology 2015;122:2125–9. 10.1016/j.ophtha.2015.06.026 [DOI] [PubMed] [Google Scholar]
- 13.Vischio JA, McCrary CT. Orbital Wegener's granulomatosis: a case report and review of the literature. Clin Rheumatol 2008;27:1333–6. 10.1007/s10067-008-0949-2 [DOI] [PubMed] [Google Scholar]
- 14.Garrity JA. Ocular manifestations of small-vessel vasculitis. Cleve Clin J Med 2012;79(Suppl 3):S31–3. 10.3949/ccjm.79.s3.07 [DOI] [PubMed] [Google Scholar]
- 15.Woo TL, Francis IC, Wilcsek GA et al. Australasian orbital and adnexal Wegener's granulomatosis. Ophthalmology 2001;108:1535–43. 10.1016/S0161-6420(01)00655-8 [DOI] [PubMed] [Google Scholar]
- 16.Leavitt JA, Butrus SI. Wegener's granulomatosis presenting as dacryoadenitis. Cornea 1991;10:542–5. 10.1097/00003226-199111000-00015 [DOI] [PubMed] [Google Scholar]
- 17.Soheilian M, Bagheri A, Aletaha M. Dacryoadenitis as the earliest presenting manifestation of systemic Wegener's granulomatosis. Eur J Ophthalmol 2002;12:241–3. [DOI] [PubMed] [Google Scholar]
- 18.Kiratli H, Sekeroğlu MA, Soylemezoğlu F. Unilateral dacryoadenitis as the sole presenting sign of Wegener's granulomatosis. Orbit 2008;27:157–60. 10.1080/01676830701523863 [DOI] [PubMed] [Google Scholar]
- 19.Rosenbaum JT, Choi D, Wilson DJ et al. Molecular diagnosis of orbital inflammatory disease. Exp Mol Pathol 2015;98:225–9. 10.1016/j.yexmp.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bitik B, Kılıç L, Küçükşahin O et al. Retro-orbital granuloma associated with granulomatosis with polyangiitis: a series of nine cases. Rheumatol Int 2015;35:1083–92. 10.1007/s00296-014-3179-8 [DOI] [PubMed] [Google Scholar]
- 21.Muhle C, Reinhold-Keller E, Richter C et al. MRI of the nasal cavity, the paranasal sinuses, and orbits in Wegener's granulomatosis. Eur Radiol 1997;7:566–70. 10.1007/s003300050206 [DOI] [PubMed] [Google Scholar]
- 22.Puéchal X, Guillevin L. Therapeutic immunomodulation in systemic vasculitis: taking stock. Joint Bone Spine 2013;80:374–9. 10.1016/j.jbspin.2012.10.023 [DOI] [PubMed] [Google Scholar]
- 23.De Groot K, Rasmussen N, Bacon PA et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461–9. 10.1002/art.21142 [DOI] [PubMed] [Google Scholar]
- 24.López-González R, Martínez-González O, Martín-Luquero Ibáñez M et al. Red eye as the primary manifestation of Wegener's granulomatosis. Reumatol Clin 2014;10:193–4. 10.1016/j.reuma.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 25.Jones RB, Cohen Tervaert JW, Hauser T et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010;363:211–20. 10.1056/NEJMoa0909169 [DOI] [PubMed] [Google Scholar]
- 26.Stone JH, Merkel PA, Spiera R et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363:221–32. 10.1056/NEJMoa0909905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cartin-Ceba R, Golbin JM, Keogh KA et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener's): ten-year experience at a single center. Arthritis Rheum 2012;64:3770–8. 10.1002/art.34584 [DOI] [PubMed] [Google Scholar]
- 28.Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy associated with immunosuppressive therapy in rheumatic diseases: evolving role of biologic therapies. Arthritis Rheum 2012;64:3043–51. 10.1002/art.34468 [DOI] [PubMed] [Google Scholar]
- 29.Luqmani RA. State of the art in the treatment of systemic vasculitides. Front Immunol 2014;5:1–9. 10.3389/fimmu.2014.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miloslavsky EM, Specks U, Merkel PA et al. Rituximab for the treatment of relapses in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol 2014;66:3151–9. 10.1002/art.38788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez V, Cohen P, Pagnoux C et al. Intravenous immunoglobulins for relapses of systemic vasculitides associated with antineutrophil cytoplasmic autoantibodies: results of a multicenter, prospective, open-label study of twenty-two patients. Arthritis Rheum 2008;58:308–17. 10.1002/art.23147 [DOI] [PubMed] [Google Scholar]