

Abstract
Malaria is a critical public health issue in the tropical world, causing extensive morbidity and mortality. Infection by unicellular, obligate intracellular Plasmodium parasites causes malaria. The emergence of resistance to current antimalarial drugs necessitates the development of novel therapeutics. A potential novel drug target is the purine import transporter. Because Plasmodium parasites are purine auxotrophic, they must import purines from their host to fulfill metabolic requirements. They import purines via equilibrative nucleoside transporter 1 (ENT1) homologs. Recently, we used a yeast-based high-throughput screen to identify inhibitors of the P. falciparum ENT1 (PfENT1) that kill P. falciparum parasites in culture. P. berghei infection of mice is an animal model for human malaria. Because P. berghei ENT1 (PbENT1) shares only 60% amino acid sequence identity with PfENT1, we sought to characterize PbENT1 and its sensitivity to our PfENT1 inhibitors. We expressed PbENT1 in purine auxotrophic yeast and used radiolabeled substrate uptake to characterize its function. We showed that PbENT1 transports both purines and pyrimidines. It preferred nucleosides compared with nucleobases. Inosine (IC50 = 3.7 µM) and guanosine (IC50 = 21.3 µM) had the highest affinities. Our recently discovered PfENT1 inhibitors were equally effective against both PbENT1- and PfENT1-mediated purine uptake. The PfENT1 inhibitors are at least 10-fold more potent against PfENT1 than human hENT1. They kill P. berghei parasites in 24-hour ex vivo culture. Thus, the P. berghei murine malaria model may be useful to evaluate the efficacy of PfENT1 inhibitors in vivo and their therapeutic potential for treatment of malaria.
Introduction
Every year, approximately 500,000 deaths, mostly children aged younger than 5 years, result from nearly 200 million cases of malaria (World Health Organization, 2014). In Southeast Asia, Plasmodium falciparum parasites have developed resistance to current first-line artemisinin-based combination therapies (ACTs) (Uhlemann and Fidock, 2012; Ariey et al., 2014; Ashley et al., 2014; Burrows et al., 2014; Straimer et al., 2015). Therefore, it is essential to identify new drug targets to facilitate the development of novel antimalarial drugs.
The Plasmodium parasite purine import pathway is one potential target, because the parasites are purine auxotrophic and require imported purines to synthesize RNA and replicate DNA during proliferation in the human host. The parasites import purines via equilibrative nucleoside transporters (Baldwin et al., 2004; Downie et al., 2008; Riegelhaupt et al., 2010; Frame et al., 2015a). In P. falciparum, knockout of the primary purine transporter, P. falciparum equilibrative nucleoside transporter 1 (PfENT1), is conditionally lethal at purine concentrations found in human blood (<10 μM) (Traut, 1994; El Bissati et al., 2006; Frame et al., 2015b). Thus, we and others hypothesized that small molecule inhibitors of the parasite purine transporter would kill malaria parasites (El Bissati et al., 2006, 2008; Baldwin et al., 2007; Frame et al., 2015a). We developed a robust, yeast-based high-throughput screen (HTS) to identify PfENT1 inhibitors (Frame et al., 2015b). The basis for the HTS was that 5-flurouridine (5-FUrd) is cytotoxic for PfENT1-expressing fui1Δ yeast due to 5-FUrd entry via PfENT1. With 5-FUrd in the growth media, PfENT1-expressing fui1Δ yeast will only grow if a PfENT1 inhibitor is present in the media. We screened 64,500 compounds, identified 171 hits, and characterized nine of the top compounds. The nine compounds, representing six distinct chemical scaffolds, inhibit tritiated adenosine uptake into red blood cell (RBC) free parasites with IC50 values in the 5- to 50-nM range (Frame et al., 2015b). They kill P. falciparum parasites in culture with 5- to 50-μM IC50 values. Furthermore, the compounds inhibit the P. vivax equilibrative nucleoside transporter 1 (PvENT1) transporter and PvENT1 nonsynonymous single nucleotide polymorphic variants identified in field isolates (Deniskin et al., 2015). Collectively, these findings support the hypothesis that PfENT1 inhibitors may be developed into novel antimalarial drugs.
P. berghei causes malaria in mice. It is currently the best animal model to study the human disease, although significant differences have been noted between human P. falciparum malaria and the P. berghei mouse model (Chisholm et al., 2016). PbENT1 is 60% amino acid sequence identical with PfENT1 (Supplemental Fig. 1). Mice infected with PbENT1-knockout P. berghei ANKA parasites develop a nonfatal, high parasitemia without cerebral malaria symptoms (Niikura et al., 2013). However, knockouts of other P. berghei genes that reduce parasite viability also cause loss of the ability to induce cerebral malaria (Chisholm et al., 2016). Thus, the inability to induce cerebral malaria may not be related to the PbENT1 knockout, but rather could reflect the loss of parasite fitness. Loss of fitness after equilibrative nucleoside transporter 1 (ENT1) knockout is also seen in mice infected with ENT1-disrupted P. yoelii, another murine malaria species. They do not develop obvious infection but develop sterilizing immunity against subsequent infection with wild-type (WT) P. yoelii (Aly et al., 2010). The limited viability of ENT1-knockout murine-infective Plasmodia indicates that they have a secondary purine import pathway. This pathway plus higher mouse blood purine concentrations presumably allows the ENT1-knockout parasites to remain viable during in vivo infection, albeit with reduced pathogenicity.
Adenosine uptake into human RBCs is mediated by human equilibrative nucleoside transporter 1 (hENT1) (Domin et al., 1988; Griffiths et al., 1997). hENT1 is 17%–18% amino acid sequence identical to PbENT1 and PfENT1. It is inhibited by nitrobenzylmercaptopurineriboside and the Food and Drug Administration–approved drug, dipyridamole, at nanomolar concentrations (Domin et al., 1988; Griffiths et al., 1997). Neither nitrobenzylmercaptopurineriboside nor dipyridamole inhibits PfENT1 at concentration up to 20 μM (Carter et al., 2000a; Parker et al., 2000; Riegelhaupt et al., 2010). Thus, it may be feasible to develop inhibitors with high specificity for malaria ENT1s. The existence of known nanomolar potency hENT1 inhibitors indicates that the ENTs are druggable targets.
In this study, we expressed PbENT1 in Saccharomyces cerevisiae and characterized its substrate specificity profile. We investigated whether our PfENT1 inhibitors block PbENT1-mediated transport and their effects on the human RBC ENT1 homolog. We show that PbENT1 is a purine and pyrimidine transporter. We found that the PfENT1 inhibitors inhibit PbENT1 with similar nanomolar affinities, but with 10- to 1000-fold lower affinity for hENT1. Thus, after the development of more potent PfENT1 inhibitors, the in vivo mouse malaria model may be useful to test their efficacy.
Materials and Methods
Yeast DNA Construct.
We purchased a yeast codon-optimized gene of PbENT1 with a C-terminal HA epitope tag (pbent1-HA-CO) for expression in S. cerevisiae (DNA 2.0, Newark, CA) (Supplemental Fig. 2). The construct was cloned into a Gateway entry vector pENTR using a pENTR/D-TOPO cloning kit (Life Technologies, Waltham, MA). From there, pbent1-HA-CO was cloned into a modified pYES2 destination vector using LR clonase enzyme (Life Technologies). The construct contains an upstream GAL1 promoter and a downstream CYC1 terminator. The construct also contains the Ura3 gene, to allow for positive selection of yeast carrying the plasmid when using media lacking uracil.
Yeast Growth Media.
Purine auxotrophic yeast were maintained on synthetic defined media (SDM) that contained 2% (w/v) galactose, 1% (w/v) raffinose, 0.5% (w/v) ammonium sulfate, 0.17% yeast nitrogen base (US Biologicals, Salem, MA), 0.02% (w/v) yeast dropout mix lacking uracil, adenine, histidine, and tryptophan (US Biologicals), 40 mg/l tryptophan, and 40 mg/l histidine. Media were supplemented with 300 µM adenine for ade2Δ-empty vector (EV) yeast or 1 mM adenosine for PbENT1-HA–expressing yeast. Solid media plates contained 2% agar.
Yeast Strains and Transformation.
DNA constructs were transformed into purine auxotrophic yeast as previously described (Frame et al., 2015b). Briefly, S. cerevisiae BY4741 with FUI1 and ADE2 gene deletions were used as the WT (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0; fui1Δ::KanMX4; ade2Δ::hphNT1). Hereafter, this yeast strain is referred to as purine auxotrophic yeast. Yeast were transformed using the lithium acetate/dimethylsulfoxide (DMSO) method (Hill et al., 1991). WT yeast were grown in 10 ml yeast extract peptone dextrose media to a cell density of 2 × 107 cells/ml. Cells were pelleted and washed with 20 ml cold lithium acetate (LiOAc; 100 mM LiOAc, 10 mM Tris, pH 8). Pellet was resuspended in 100 µl LiOAc, and 10 µl salmon-sperm DNA (2 mg/ml) and 1 µg DNA construct were added. After incubation at room temperature for 5 minutes, 280 µl 50% polyethylene glycol 3350 (% w/v) was added. Final solution was incubated at 30°C for 45 minutes. DMSO was added [8% (v/v)] and the sample was heat shocked at 42°C for 15 minutes. Cells were pelleted and resuspended in 5 ml yeast extract peptone dextrose and allowed to recover for 3 hours. Cells were pelleted and plated on SDM/agar.
Yeast Growth Assays.
Growth of PbENT1-HA–expressing and EV-transformed purine auxotrophic yeast was assessed in media where adenine or adenosine was the sole purine source. First, EV yeast and PbENT1-HA–expressing yeast were grown overnight in SDM containing adenine to mid-log phase. Yeast cell density was determined by measuring optical density at 600 nm (Benchmark Plus; Bio-Rad, Hercules, CA). Cells were pelleted and washed three times with sterile water. Final cell pellet was diluted to 4 × 106 cells/ml in 2× SDM lacking purine. A 96-well plate was preloaded with 100 µl serially diluted adenosine in sterile water. One-hundred microliters of cells was added and incubated for 30 hours at 30°C. EC50 values were calculated using Prism 6 software (GraphPad Software, La Jolla, CA). All experiments were repeated at least three times on different days.
Radiolabel Uptake Experiments with PbENT1-HA–Expressing Purine Auxotrophic Yeast.
PbENT1-HA–expressing purine auxotrophic yeast were grown to mid-log phase in SDM containing 1 mM adenosine. Cells were pelleted and washed three times in phosphate-buffered saline (PBS) supplemented with galactose (150 mM NaCl, 10 mM KH2PO4, 40 mM K2HPO4, and 11 mM galactose, pH 7.4). Cells were resuspended in PBS to a concentration of 2 × 108 cells/ml. For uptake time course experiments, a 96-well plate was preloaded with 100 µl 100 nM [3H]adenosine ([2,8-3H]adenosine, 35 Ci/mmol; Moravek Biochemicals, Brea, CA) or [3H]uridine ([2,8-3H]uridine, 22 Ci/mmol; Moravek Biochemicals). One-hundred microliters of suspended yeast was added at the appropriate time points. For purine/pyrimidine uptake competition, a 96-well plate was preloaded with 50 µl serially diluted purines or pyrimidines. Fifty microliters of 200 nM [3H]adenosine was added to pyrimidines and 50 µl 1 mM [3H]uridine was added to purines. One-hundred microliters of resuspended yeast was added and incubated for 15 minutes. For all experiments, uptake was terminated by harvesting cells onto glass fiber filtermats (Filtermat, GF/C; Perkin Elmer, Waltham, MA) using a Tomtec 96-well cell harvester (96-3-469; Tomtec, Hamden, CT). Filtermats were dried for > 1 hour and sealed in plastic bags containing 5 ml Betaplate Scint LSC (Perkin Elmer). Filtermats were counted using 1450 Microbeta Trilux (Perkin Elmer). IC50 values were calculated using Prism 6 software (GraphPad Software). All experiments were repeated at least three times on different days.
Inhibition of PbENT1 by PfENT1 Inhibitors.
PbENT1-HA–expressing purine auxotrophic yeast were grown to mid-log phase in SDM plus 1 mM adenosine. Cells were washed and resuspended to a concentration of 2 × 108 cells/ml as above. A 96-well plate was preloaded with 100 µl 100 nM [3H]adenosine. One-half microliter of compound, serially diluted in DMSO, was added to each well. One-hundred microliters of cells was added and cells were harvested after 15 minutes, as above. Compounds were also tested on PfENT1-expressing purine auxotrophic yeast as described in Frame et al. (2015b). PfENT1 inhibitors were purchased from Chembridge Corp. (San Diego, CA). Chembridge catalog numbers for the PfENT1 inhibitors are as follows: compounds 1 (9001893), 2 (6718896), 3 (6946484), 4 (6081106), 5 (9039333), 6 (9011026), 7 (6736283), 13 (6517398), and 19 (9011680); structures are provided in Table 2. Compound names are listed in Frame et al., (2015b). Chemical structure and composition of PfENT1 inhibitors was validated previously by nuclear magnetic resonance and mass spectrometry (Frame et al., 2015b). All experiments were repeated at least three times on different days.
TABLE 2.
IC50 values for inhibition of uptake of 50 nM [3H]adenosine into PbENT1-expressing and PfENT1-expressing yeast by the PfENT1 inhibitors identified by Frame et al. (2015b)
IC50 values are means ± S.D. from three experiments. Compounds are numbered as in Frame et al. (2015b), with rank-order number from the HTS. Chembridge catalog numbers of the compounds are in the Materials and Methods. The compound names are in Frame et al., (2015b).
| Compound | Structure | PbENT1 IC50 | PfENT1 IC50 | Pb:Pf |
|---|---|---|---|---|
| nM | nM | |||
| 1 | 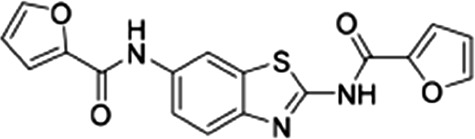 |
3.4 ± 0.7 | 4.8 ± 0.5 | 0.7 |
| 2 | 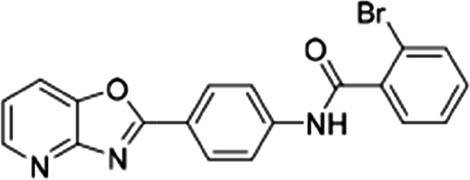 |
16 ± 7.2 | 33.5 ± 6.5 | 0.47 |
| 3 | 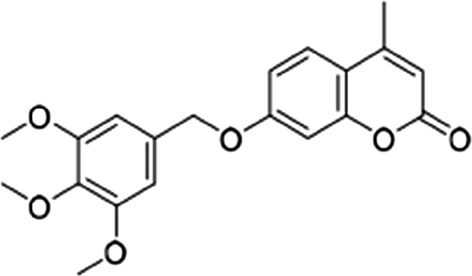 |
5.4 ± 2.3 | 7 ± 0.4 | 0.77 |
| 4 | 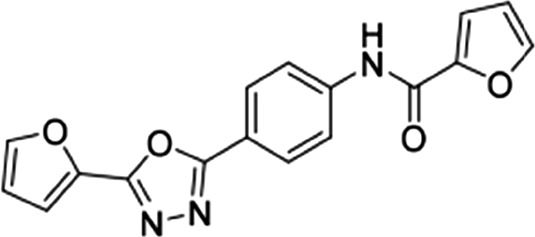 |
21.6 ± 2.3 | 23.1 ± 3.5 | 0.93 |
| 5 | 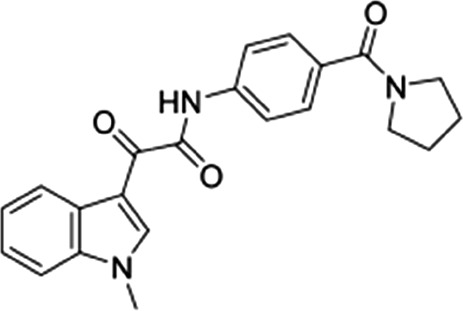 |
56 ± 14 | 54.5 ± 7.2 | 1.02 |
| 6 | 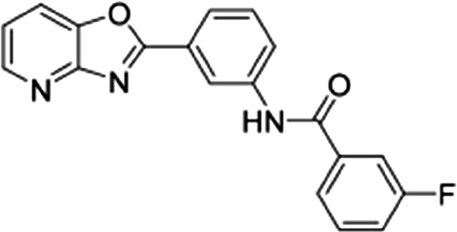 |
27 ± 7 | 14 ± 2.7 | 1.93 |
| 7 | 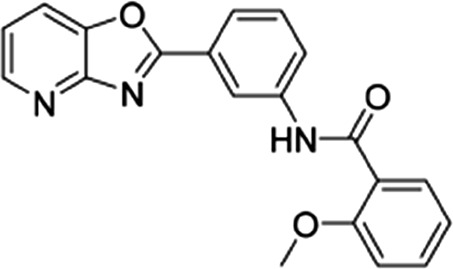 |
12.3 ± 3.2 | 15 ± 2.3 | 0.82 |
| 13 | 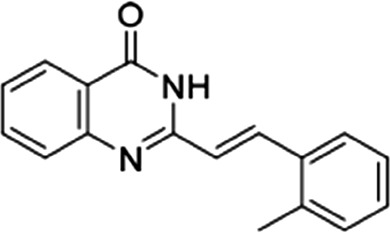 |
11.6 ± 3.1 | 10.3 ± 2.4 | 1.12 |
| 19 | 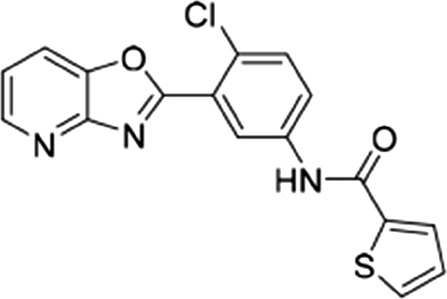 |
17 ± 3.6 | 42.5 ± 2.5 | 0.4 |
Ex vivo parasite drug susceptibility assay
Donor outbred CD1 mice (female, aged 6–8 weeks; Charles River Laboratories, Stone Ridge, NY) were infected with 1 × 107 P. berghei parasites (strain ANKA 676m1cl1). This strain was obtained from the BEI Resources Repository (National Institutes of Health National Institute of Allergy and Infectious Diseases, Bethesda, MD) as item MRA-868, contributed by Chris J. Janse and Andrew P. Waters. Once the parasitemia reached 5%–7%, the parasites were harvested by cardiac puncture. One milliliter of the harvested blood was incubated in 50 ml RPMI-1640 malaria culture medium (supplemented with 25% fetal bovine serum) with gentle shaking and incubated at 36.8°C for 23 hours (Janse et al., 2006). The parasites were then pelleted, washed in physiologic saline solution, and reinjected (in saline solution, total volume 0.5 ml) into another mouse by tail vein injection. After 1 day (the duration of one intraerythrocytic developmental cycle), we harvested ring-stage parasites by cardiac puncture and diluted these in 10 ml malaria culture medium per 100-µl packed blood cell pellet. One-half milliliter of this parasite suspension (at approximately 3%–4% parasitemia) was mixed with 0.5 ml compound across a range of ten 2-fold dilutions in 24-well plates. Plates were incubated for 24 hours at 36.8°C. Parasitemias were determined by microscopic analysis of Giemsa-stained thin blood smears, and IC50 values were extrapolated by nonlinear regression analysis. Experiments were performed on two separate occasions in duplicate. As a positive control, we included amodiaquine, which yielded IC50 values of 3.9 ± 0.3 nM, consistent with earlier reports of its activity against P. berghei parasites assayed ex vivo (Orjuela-Sánchez et al., 2012). All animal experiments were conducted under a protocol approved by the Columbia University Institutional Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals.
PfENT1 Inhibitor Effects on the Human RBC hENT1 Transporter.
The PfENT1 inhibitors were evaluated for their ability to inhibit uptake of 50 nM [3H]adenosine ([2,8-3H]adenosine, 35 Ci/mmol; Moravek Biochemicals) into human RBCs. Uninfected red blood cells (uRBCs) were washed with a Ringer solution (122.5 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 0.8 mM MgCl2, 11 mM d-glucose, 25 mM HEPES, and 1 mM Na2HPO4, pH 7.4) and resuspended at 3% hematocrit in prewarmed (37°C) Ringer solution. The PfENT1 inhibitors were serially diluted 1:4 in DMSO and mixed with radiolabeled purine in Ringer solution (final DMSO concentration was 0.5%). One-hundred microliters of uRBCs was mixed with 100 μl radioisotope/PfENT1-inhibitor solution and incubated at room temperature for 15 minutes (final hematocrit was 1.5%, approximately 18 million cells). Discs punched from fiberglass filters (Filtermat A) were inserted into 1-ml filter tips (catalog number 1182-1830; USA Scientific, Ocala, FL) and mounted on a vacuum suction manifold. At the end of the incubation, the samples were transferred to the filter tips and vacuum-filtered onto the filtermats and each sample was washed with 4 ml ice-cold Ringer solution. Filtermats were transferred to scintillation vials and allowed to dry for 1 hour. RBCs were solubilized with 200 μl 5% SDS for 30 minutes and then mixed with 3 ml scintillation fluid (UltimaGold; Perkin Elmer). Samples were counted for 1 minute each using a Wallac TriCarb Liquid Scintillation Counter (Perkin Elmer). Experiments were repeated at least three times on different days.
We also used a second method to measure the concentration dependence of compound inhibition of [3H]adenosine uptake into uRBCs, which gave similar IC50 values to the method described above. Values from the two methods were pooled in calculating the average values. We preloaded 96-well plates (clear flat-bottom 96-well polystyrene, nonsterile plate; Fisher Scientific) with 0.75 μl compound (serially diluted 4-fold in DMSO from a 25-mM stock) and 100 μl 100 nM [3H]adenosine ([2,8-3H]adenosine,23 Ci/mmol; Moravek Biochemicals) in PBS solution (137 mM NaCl, 2.7 mM KCl, 10 mM KH2PO4, and 10 mM Na2HPO4, pH 7.4). RBCs acquired from healthy blood donors were washed five times with PBS solution and resuspended at 4% hematocrit in PBS. One-hundred microliters of RBCs (4% hematocrit) was added to each well containing compound/radiolabel, resuspended, and incubated at room temperature for 15 minutes. At the end of the time course, cells were harvested onto glass fiber filter mats (Filtermat A, GF/C; Perkin Elmer) using a Tomtec 96-well cell harvester system (number 96-3-469). Filtermats were dried and sealed in plastic bags with Betaplate Scint (Perkin Elmer) scintillation fluid. Counts were measured using a microplate scintillation counter (1450 Microbeta Trilux; Perkin Elmer).
Results
PbENT1 shares 60% amino acid identity with PfENT1 (Supplemental Fig. 1), so we hypothesized that PbENT1 should have a similar substrate transport profile. Because Plasmodium genes are often A-T rich, heterologous expression is often difficult. To eliminate effects of codon-usage bias on expression, we used a yeast codon-optimized version of PbENT1 (Supplemental Fig. 2). The expression vector was transformed into yeast lacking the endogenous uridine transporter, FUI1, and one of the enzymes in the de novo purine synthesis pathway, ADE2. Growth of these purine auxotrophic yeast can be rescued in media containing adenine, which can enter via the endogenous yeast FCY2 transporter (Weber et al., 1990). However, yeast lack an endogenous adenosine transporter. Thus, growth in media containing adenosine as the sole purine source can only be rescued if there is heterologous expression of an adenosine transporter. As expected, PbENT1-expressing yeast grew in the presence of adenosine (EC50 542 ± 59 µM, mean ± S.D.), but yeast transformed with EV did not (Fig. 1).
Fig. 1.

PbENT1-HA-CO expression in purine auxotrophic yeast. Concentration-dependent growth of yeast containing PbENT1-HA construct or EV in the presence of adenosine, after 30 hours. Adenosine growth EC50 of PbENT1-expressing yeast is 542 ± 59 µM. Data shown are the average of three independent experiments. OD600, optical density at 600 nm.
Next, we examined the uptake time course of radiolabeled substrate into PbENT1-expressing yeast. PbENT1-HA–expressing yeast take up [3H]adenosine and [3H]uridine in a time-dependent, linear fashion over a 1-hour period (Fig. 2). Uptake was significantly greater for PbENT1-HA–expressing yeast than for EV-transformed yeast. Subsequent uptake experiments were done at 15 minutes, within the linear uptake range.
Fig. 2.

Time course of uptake of [3H]adenosine and [3H]uridine into PbENT1-HA–expressing purine auxotrophic yeast. (A and B) Uptake of 50 nM [3H]adenosine (A) and 250 nM [3H]uridine (B) into PbENT1-HA–expressing yeast. Open boxes represent yeast transformed with EV. Filled boxes represent yeast expressing PbENT1-HA. Uptake is measured in counts per minute (CPM) per million cells. Note differing y-axis scales in the two panels. Data shown are the average of three independent experiments.
To determine the substrate specificity of PbENT1, we measured the ability of unlabeled purines and pyrimidines to inhibit the uptake of radiolabeled tracer into PbENT1-expressing yeast. To ensure that the radiolabel and test substrate were competing at the transporter, and not at a downstream metabolic enzyme, we used [3H]adenosine to test pyrimidine substrates and [3H]uridine to test purine substrates. Thus, the radiolabel and test substrate would not share metabolic enzymes and competition could only occur at the transporter. We determined the IC50 values for four purine nucleosides and their equivalent nucleobases (Fig. 3). All of the tested purines inhibited [3H]uridine uptake. Inosine and guanosine showed the highest affinity, with IC50 values of 3.7 µM and 21.3 µM, respectively (Table 1). IC50 values for nucleobases were 1.6- to 50-fold higher than for the corresponding nucleosides (Table 1).
Fig. 3.

Inhibition of [3H]uridine uptake into PbENT1-HA–expressing yeast by various purine nucleosides and nucleobases. (A–D) Uptake inhibition of 250 nM [3H]uridine into PbENT1-HA– expressing yeast by adenosine, adenine (A); inosine, hypoxanthine (B); guanosine, guanine (C); and xanthosine, xanthine (D). Uptake is normalized to remove background and is shown as the percentage of maximum uptake. All tested substrates fully inhibited [3H]uridine uptake, with the exception of the nucleobases xanthine and guanine and the nucleoside xanthosine. IC50 values are shown in Table 1. Data from a representative experiment are shown.
TABLE 1.
IC50 values for inhibition of radiolabel substrate uptake by purine and pyrimidine substrates
IC50 values are means ± S.D. for ≥3 experiments. Pyrimidines were competed with 50 nM [3H]adenosine; purines were competed with 250 nM [3H]uridine.
| Uptake Inhibition IC50 Values | |||
|---|---|---|---|
| Pyrimidine | IC50 Versus [3H]Adenosine | Purine | IC50 Versus [3H]Uridine |
| µM | µM | ||
| Uracil | 2833 ± 243 | Adenine | 306 ± 19 |
| Uridine | 400 ± 152 | Adenosine | 190 ± 2.5 |
| Thymine | 1142 ± 185 | Hypoxanthine | 53 ± 1.65 |
| Thymidine | 91.3 ± 38 | Inosine | 3.7 ± 0.1 |
| Cytosine | 9697 ± 455 | Guanine | 1142 ± 44 |
| Cytidine | 26726 ± 4116 | Guanosine | 21.3 ± 0.6 |
| Xanthine | 979 ± 13 | ||
| Xanthosine | 626 ± 17 | ||
The uptake of [3H]uridine showed that PbENT1 was able to transport a pyrimidine (Fig. 2). We tested the ability of six pyrimidine nucleosides and their equivalent nucleobases to inhibit [3H]adenosine uptake (Fig. 4). The nucleosides thymidine and uridine showed the highest affinity, with IC50 values of 91.3 μM and 400 µM, respectively (Table 1). The nucleobase cytosine and nucleoside cytidine were unable to completely inhibit radiolabel uptake, even at the maximum concentration tested, 12.5 mM (Fig. 4).
Fig. 4.

Inhibition of [3H]adenosine uptake into PbENT1-HA–expressing yeast by various pyrimidine nucleobases and nucleosides. (A and B) Uptake inhibition of 50 nM [3H]adenosine into PbENT1-HA-CO–expressing yeast by pyrimidine nucleobases (A) and nucleosides (B). Uptake is normalized to remove background and is shown as the percentage of maximum uptake. Of the tested substrates, only uridine and thymidine showed complete inhibition. IC50 values are shown in Table 1. Data from a representative experiment are shown.
We recently identified and characterized nine small molecule inhibitors of PfENT1 (Table 2) (Frame et al., 2015b). We showed that these compounds inhibit [3H]adenosine uptake into erythrocyte-free P. falciparum parasites and kill parasites in culture (Frame et al., 2015b). In this study, we tested their ability to inhibit [3H]adenosine uptake into PbENT1-HA–expressing yeast (Fig. 5). All nine compounds inhibited PbENT1 with IC50 values in the 3- to 60-nM range (Table 2). The ratios of PbENT1 IC50 to PfENT1 IC50 for the different compounds were all within a factor of 2 (Table 2). Thus, the compounds have similar efficacy against both transporters despite the 40% amino acid sequence differences.
Fig. 5.

Concentration-dependent inhibition of PbENT1 function by PfENT1 inhibitors. Concentration-dependent inhibition of 50 nM [3H]adenosine uptake in the presence of nine PfENT1 inhibitor compounds. Uptake is normalized to remove background and is shown as the percentage of maximum uptake. Single experiment inhibition curves are shown, representative of three independent trials. IC50 values are shown in Table 2.
Based on the ability of the PfENT1 inhibitors to block PbENT1, we sought to test the hypothesis that they would inhibit proliferation of P. berghei parasites. It should be noted that the compounds are not cytotoxic to yeast at concentrations up to 125 μM, the highest concentration tested (Frame et al., 2015b). We tested the effect of three of the inhibitors (compounds 3, 4, and 13) on P. berghei parasite proliferation in 24-hour ex vivo culture. The compounds inhibited parasite proliferation, with IC50 values between 5 and 25 μM (Table 3). Amodiaquine, a 4-aminoquinoline compound similar to chloroquine, was included as a positive control (Table 3). Similar IC50 values were obtained for inhibition of P. falciparum parasite proliferation in culture (Table 3).
TABLE 3.
IC50 values for inhibition of P. berghei parasite proliferation in 24-hour ex vivo culture
Compound numbers refer to PfENT1 inhibitors as per Table 2 and from Frame et al. (2015b). Data are means ± S.E.M. from two independent experiments.
| Compound | IC50 P. berghei Parasites Ex Vivo Culture | IC50 P. falciparum 3D7 Parasitesa |
|---|---|---|
| 3, (μM) | 6.5 ± 0.6 | 19.2 ± 4.3 |
| 4, (μM) | 23.4 ± 1.0 | 15.0 ± 1.5 |
| 13, (μM) | 5.6 ± 0.4 | 6.9 ± 0.4 |
| Amodiaquine, (μM) | 3.9 ± 0.4 |
From Frame et al. (2015b).
We also tested the specificity of the PfENT1 inhibitors relative to the human erythrocyte hENT1 purine transporter. We assessed their effect using uninfected human RBCs in which hENT1 function was assayed by [3H]adenosine uptake (Fig. 6A). Compound 7 displayed the lowest selectivity: it had 27 times higher affinity for PfENT1 than for hENT1 (Fig. 6B). Compound 1 had the highest selectivity: it inhibited hENT1 at a concentration 1200 times higher than the concentration at which it inhibited PfENT1 (Fig. 6B). These results confirmed that the compounds displayed significant specificity for PfENT1 over the human erythrocyte hENT1 transporter.
Fig. 6.

Specificity of the PfENT1 inhibitors for the malaria transporter compared with the human RBC hENT1 transporter. (A) Inhibition of 50 nM [3H]adenosine uptake into RBC-free parasites (filled symbols) and uninfected human RBCs (open symbols) by compounds 5 and 7. Representative experiments are shown. (B) Ratio of IC50 values for inhibition of radiolabeled purine uptake via hENT1 compared with the IC50 values for inhibition of uptake into RBC-free 3D7 strain P. falciparum parasites. IC50 values used to calculate the ratios were the average of at least three separate experiments. Activity of hENT1 measured using [3H]adenosine uptake into uninfected RBC. IC50 values for inhibition of [3H]adenosine uptake into RBC-free P. falciparum parasites are from Frame et al. (2015b). Note that the y-axis is a log scale.
Discussion
The long-term goal of this project is to develop antimalarial drugs against a novel target, the primary purine import transporter. As a step toward that goal, in this work we sought to determine the feasibility of using the mouse malaria model. To establish the feasibility of using the mouse model, we characterized the functional properties of PbENT1 and determined whether the best hits from our HTS for PfENT1 inhibitors would also work on the P. berghei homolog, PbENT1. PbENT1, like its homologs in P. falciparum and P. vivax (Riegelhaupt et al., 2010; Deniskin et al., 2015), transports both purines and pyrimidines (Fig. 2; Table 1). PbENT1 has higher affinity for purines compared with pyrimidines and higher affinity for nucleosides compared with nucleobases (Table 1). Inosine and guanosine showed the highest affinities (Table 1), much like the Leishmania LdNT2 nucleoside transporter (Carter et al., 2000b). For P. falciparum, adenosine and hypoxanthine are the preferred substrates for purine import, because these purines are present at the highest concentration in human plasma and are also present in human erythrocytes (Möser et al., 1989; Traut, 1994). The inhibition constants (Ki) for PfENT1 transport of adenosine and hypoxanthine are around 650 µM and 300 µM, respectively (Riegelhaupt et al., 2010). In contrast, the IC50 values for PbENT1 were 4- to 6-fold lower for these substrates (Table 1). The different affinities for various purines are presumably due to amino acid differences between PfENT1 and PbENT1, because they are only 60% sequence identical. Whether these differences in substrate affinity are physiologically significant is currently unknown. Of note, differences exist in the purine import pathways of Plasmodium species that infect primates and rodents. Genome sequencing reveals that P. vivax and P. falciparum encode four ENT homologs (ENT1–ENT4), whereas the species that infect rodents, P. berghei and P. yoelii, lack an ENT3 ortholog (Frame et al., 2012, 2015a). The substrate specificity and functional role of PfENT3 remains to be determined. However, its presence in Plasmodium species that infect humans and not in those that infect rodents suggests that there may be differences in purine transport and metabolism between rodents and humans that may be important for proliferation of the respective Plasmodium species.
Efforts to combat malaria have been hampered by the development of resistance to antimalarial drugs (Sá et al., 2011). Thus, it is important to have a robust pipeline of new therapeutics that target novel aspects of Plasmodium parasite biology to replace current drugs as they become less effective (Burrows et al., 2014). One potential target is the purine import and salvage pathway, which is essential for parasite survival (Ducati et al., 2013; Frame et al., 2015a). Using a yeast-based HTS, we previously identified compounds that inhibit PfENT1 at concentrations in the nanomolar range (Frame et al., 2015b). We characterized nine of the hits in a series of secondary assays and showed that they inhibit P. falciparum parasite proliferation in culture. In this article, we show that despite the 40% amino acid sequence differences between PbENT1 and PfENT1, these nine inhibitors also block PbENT1-mediated purine transport with IC50 values comparable to those for PfENT1 (Table 2).
Ideally, an antimalarial drug would not inhibit hENT1. However, dipyridamole, a Food and Drug Administration–approved drug that inhibits hENT1, has been used safely in patients (Griffiths et al., 1997). Thus, avoiding interactions with hENT1 may not be essential for a viable antimalarial drug targeting PfENT1. Nonetheless, because hENT1 is only 17% amino acid sequence identical with PfENT1 and dipyridamole does not block PfENT1 (Riegelhaupt et al., 2010), it may be feasible to identify inhibitors that display specificity for PfENT1 over hENT1. In fact, the PfENT1 inhibitors that we have identified have lower potency against the human RBC purine transporters, hENT1 (Fig. 6). They are 27- to 1200-fold more potent against the Plasmodium ENT1 homologs relative to the human hENT1 (Fig. 6B). Thus, it may be possible to maintain selectivity for the Plasmodium ENTs during the hit-to-lead medicinal chemistry process that will be necessary to develop our current compounds into antimalarial drugs.
Based on the assumption that PbENT1-mediated purine import is essential for P. berghei parasite proliferation, we expected that the PfENT1 inhibitors would kill P. berghei parasites. Because these compounds are HTS hits that need to be optimized through medicinal chemistry, we did not think it worthwhile to determine the mouse pharmacokinetics for these compounds at this time. Thus, we did not test the efficacy of the inhibitors in P. berghei–infected mice. To assess their efficacy on parasite proliferation, we tested the effect of three of the inhibitors on P. berghei parasite proliferation in ex vivo culture (Table 3). The inhibitors blocked parasite proliferation with IC50 values similar to their efficacy against P. falciparum parasites in culture (Frame et al., 2015b). This suggests that with improved potency through medicinal chemistry efforts, Plasmodium ENT1 inhibitors will display efficacy in the in vivo mouse malaria model.
One strategy to reduce the development of resistance to antimalarial drugs has been to pair drugs with different targets. This was the rationale behind pairing artemisinin derivatives with other drugs in ACTs. Unfortunately, because the partner drugs for the artemisinins already had been widely used, resistance to the partner drugs was already present in the P. falciparum parasite population. This has contributed to the development of resistance to the artemisinin component of the ACTs (Uhlemann and Fidock, 2012; Ariey et al., 2014; Ashley et al., 2014; Burrows et al., 2014; Straimer et al., 2015). With our PfENT1 inhibitors, we were surprised that PfENT1-knockout parasites grown in high purine concentrations were also killed by the PfENT1 inhibitors with 2- to 4-fold higher IC50 values (Frame et al., 2015b). Killing of PfENT1-knockout parasites showed a delayed-death phenotype not observed in the killing of the WT parasites (Frame et al., 2015b). This indicates that the secondary target causing death in the PfENT1-knockout parasites is distinct from the primary target in WT parasites. The existence of two targets with similar affinities may reduce the likelihood of parasites developing resistance to these compounds. Whether the affinity for both targets can be improved simultaneously during medicinal chemistry optimization is uncertain. An alternative strategy might be to pair a PfENT1 inhibitor with another drug that acts elsewhere in the purine salvage pathway. Schramm and coworkers have shown that transition state analogue inhibitors of the purine salvage pathway enzyme, purine nucleoside phosphorylase, kill malaria parasites (Cassera et al., 2011; Ducati et al., 2013). Targeting two points in the purine metabolic pathway might lead to synergistic effects. Future experiments will be necessary to test this hypothesis. In summary, we showed that the PfENT1 inhibitors were active against PbENT1 and that they were able to inhibit the proliferation of P. berghei parasites in ex vivo culture. This indicates that we will be able to use the mouse malaria model to test the utility of inhibition of purine uptake as a strategy for development of novel antimalarial drugs.
Supplementary Material
Acknowledgments
The authors thank I.J. Frame for advice in initiating this project and David Pierce for expert technical assistance.
Abbreviations
- ACT
artemesinin-based combination therapy
- DMSO
dimethylsulfoxide
- ENT
equilibrative nucleoside transporter
- EV
empty vector
- 5-FUrd
5-flurouridine
- HTS
high-throughput screen
- LiOAc
lithium acetate
- PBS
phosphate-buffered saline
- PbENT1
P. berghei equilibrative nucleoside transporter 1
- PfENT1
P. falciparum equilibrative nucleoside transporter 1
- PvENT1
P. vivax equilibrative nucleoside transporter 1
- RBC
red blood cell
- SDM
synthetic defined media
- uRBC
uninfected red blood cell
- WT
wild type
Authorship Contributions
Participated in research design: Arora, Fidock, Akabas.
Conducted experiments: Arora, Deniskin, Sosa, Nishtala, Henrich, Kumar.
Contributed new reagents or analytic tools: Arora.
Performed data analysis: Arora, Deniskin, Sosa, Nishtala, Henrich, Kumar, Fidock, Akabas.
Wrote or contributed to the writing of the manuscript: Arora, Deniskin, Sosa, Nishtala, Henrich, Kumar, Fidock, Akabas.
Footnotes
This research was supported by the Albert Einstein College of Medicine, the National Institutes of Health National Institute of Allergy and Infectious Diseases [Grant R01AI116665 (to M.H.A.)], and in part by the National Institutes of Health National Institute of General Medical Sciences [Medical Scientist Training Program Grant T32GM007288 (A.A. and R.D.)].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Aly AS, Downie MJ, Mamoun CB, Kappe SH. (2010) Subpatent infection with nucleoside transporter 1-deficient Plasmodium blood stage parasites confers sterile protection against lethal malaria in mice. Cell Microbiol 12:930–938. [DOI] [PubMed] [Google Scholar]
- Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, et al. (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, et al. Tracking Resistance to Artemisinin Collaboration (TRAC) (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. (2004) The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 447:735–743. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, McConkey GA, Cass CE, Young JD. (2007) Nucleoside transport as a potential target for chemotherapy in malaria. Curr Pharm Des 13:569–580. [DOI] [PubMed] [Google Scholar]
- Burrows JN, Burlot E, Campo B, Cherbuin S, Jeanneret S, Leroy D, Spangenberg T, Waterson D, Wells TN, Willis P. (2014) Antimalarial drug discovery - the path towards eradication. Parasitology 141:128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter NS, Ben Mamoun C, Liu W, Silva EO, Landfear SM, Goldberg DE, Ullman B. (2000a) Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J Biol Chem 275:10683–10691. [DOI] [PubMed] [Google Scholar]
- Carter NS, Drew ME, Sanchez M, Vasudevan G, Landfear SM, Ullman B. (2000b) Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J Biol Chem 275:20935–20941. [DOI] [PubMed] [Google Scholar]
- Cassera MB, Hazleton KZ, Merino EF, Obaldia N, 3rd, Ho MC, Murkin AS, DePinto R, Gutierrez JA, Almo SC, Evans GB, et al. (2011) Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS One 6:e26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm SA, McHugh E, Lundie R, Dixon MW, Ghosh S, O’Keefe M, Tilley L, Kalanon M, de Koning-Ward TF. (2016) Contrasting inducible knockdown of the auxiliary PTEX component PTEX88 in P. falciparum and P. berghei unmasks a role in parasite virulence. PLoS One 11:e0149296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniskin R, Frame IJ, Sosa Y, Akabas MH. (2015) Targeting the Plasmodium vivax equilibrative nucleoside transporter 1 (PvENT1) for antimalarial drug development. Int J Parasitol Drugs Drug Resist 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domin BA, Mahony WB, Zimmerman TP. (1988) Purine nucleobase transport in human erythrocytes. Reinvestigation with a novel “inhibitor-stop” assay. J Biol Chem 263:9276–9284. [PubMed] [Google Scholar]
- Downie MJ, Saliba KJ, Bröer S, Howitt SM, Kirk K. (2008) Purine nucleobase transport in the intraerythrocytic malaria parasite. Int J Parasitol 38:203–209. [DOI] [PubMed] [Google Scholar]
- Ducati RG, Namanja-Magliano HA, Schramm VL. (2013) Transition-state inhibitors of purine salvage and other prospective enzyme targets in malaria. Future Med Chem 5:1341–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bissati K, Downie MJ, Kim SK, Horowitz M, Carter N, Ullman B, Ben Mamoun C. (2008) Genetic evidence for the essential role of PfNT1 in the transport and utilization of xanthine, guanine, guanosine and adenine by Plasmodium falciparum. Mol Biochem Parasitol 161:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bissati K, Zufferey R, Witola WH, Carter NS, Ullman B, Ben Mamoun C. (2006) The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc Natl Acad Sci USA 103:9286–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame IJ, Deniskin R, Arora A, Akabas MH. (2015a) Purine import into malaria parasites as a target for antimalarial drug development. Ann N Y Acad Sci 1342:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame IJ, Deniskin R, Rinderspacher A, Katz F, Deng SX, Moir RD, Adjalley SH, Coburn-Flynn O, Fidock DA, Willis IM, et al. (2015b) Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites. ACS Chem Biol 10:775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame IJ, Merino EF, Schramm VL, Cassera MB, Akabas MH. (2012) Malaria parasite type 4 equilibrative nucleoside transporters (ENT4) are purine transporters with distinct substrate specificity. Biochem J 446:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths M, Beaumont N, Yao SY, Sundaram M, Boumah CE, Davies A, Kwong FY, Coe I, Cass CE, Young JD, et al. (1997) Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med 3:89–93. [DOI] [PubMed] [Google Scholar]
- Hill J, Donald KA, Griffiths DE. (1991) DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res 19:5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse CJ, Ramesar J, Waters AP. (2006) High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 1:346–356. [DOI] [PubMed] [Google Scholar]
- Möser GH, Schrader J, Deussen A. (1989) Turnover of adenosine in plasma of human and dog blood. Am J Physiol 256:C799–C806. [DOI] [PubMed] [Google Scholar]
- Niikura M, Inoue S, Mineo S, Yamada Y, Kaneko I, Iwanaga S, Yuda M, Kobayashi F. (2013) Experimental cerebral malaria is suppressed by disruption of nucleoside transporter 1 but not purine nucleoside phosphorylase. Biochem Biophys Res Commun 432:504–508. [DOI] [PubMed] [Google Scholar]
- Orjuela-Sánchez P, Duggan E, Nolan J, Frangos JA, Carvalho LJ. (2012) A lactate dehydrogenase ELISA-based assay for the in vitro determination of Plasmodium berghei sensitivity to anti-malarial drugs. Malar J 11:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MD, Hyde RJ, Yao SY, McRobert L, Cass CE, Young JD, McConkey GA, Baldwin SA. (2000) Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem J 349:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegelhaupt PM, Cassera MB, Fröhlich RF, Hazleton KZ, Hefter JJ, Schramm VL, Akabas MH. (2010) Transport of purines and purine salvage pathway inhibitors by the Plasmodium falciparum equilibrative nucleoside transporter PfENT1. Mol Biochem Parasitol 169:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sá JM, Chong JL, Wellems TE. (2011) Malaria drug resistance: new observations and developments. Essays Biochem 51:137–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, et al. (2015) Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. (1994) Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140:1–22. [DOI] [PubMed] [Google Scholar]
- Uhlemann AC, Fidock DA. (2012) Loss of malarial susceptibility to artemisinin in Thailand. Lancet 379:1928–1930. [DOI] [PubMed] [Google Scholar]
- Weber E, Rodriguez C, Chevallier MR, Jund R. (1990) The purine-cytosine permease gene of Saccharomyces cerevisiae: primary structure and deduced protein sequence of the FCY2 gene product. Mol Microbiol 4:585–596. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2014) WHO Global Malaria Programme: World Malaria Report 2014, World Health Organization, Geneva, Switzerland. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.