

Abstract
Drug-induced toxicity is often mediated by electrophilic metabolites, such as bioactivation of acetaminophen (APAP) to N-acetyl-p-benzoquinone imine (NAPQI). We have shown that APAP hepatotoxicity can be prevented by 2-acetylcyclopentanone (2-ACP). This 1,3-dicarbonyl compound ionizes to form an enolate nucleophile that scavenges NAPQI and other electrophilic intermediates. In this study, we expanded our investigation of enolate-forming compounds to include analyses of the phloretin pharmacophores, 2′,4′,6′-trihydroxyacetophenone (THA) and phloroglucinol (PG). Studies in a mouse model of APAP overdose showed that THA provided hepatoprotection when given either by intraperitoneal injection or oral administration, whereas PG was hepatoprotective only when given intraperitoneally. Corroborative research characterized the molecular pharmacology (efficacy, potency) of 2-ACP, THA, and PG in APAP-exposed isolated mouse hepatocytes. For comparative purposes, N-acetylcysteine (NAC) cytoprotection was also evaluated. Measurements of multiple cell parameters (e.g., cell viability, mitochondrial membrane depolarization) indicated that THA and, to a lesser extent, PG provided concentration-dependent protection against APAP toxicity, which exceeded that of 2-ACP or NAC. The enolate-forming compounds and NAC truncated ongoing APAP exposure and thereby returned intoxicated hepatocytes toward normal viability. The superior ability of THA to protect is related to multifaceted modes of action that include metal ion chelation, free radical trapping, and scavenging of NAPQI and other soft electrophiles involved in oxidative stress. The rank order of potency for the tested cytoprotectants was consistent with that determined in a parallel mouse model. These data suggest that THA or a derivative might be useful in treating drug-induced toxicities and other conditions that involve electrophile-mediated pathogenesis.
Introduction
The therapeutic benefits of certain clinically important drugs can be limited by biotransformation of the parent compounds to reactive metabolites that subsequently produce toxicity. These drug-induced toxicities are often caused by electrophilic metabolites (e.g., cyclophosphamide biotransformation to acrolein) that form irreversible covalent adducts with nucleophilic sulfhydryl groups on cysteine residues. Cytotoxicity is mediated by the resulting depletion of cellular glutathione (GSH), protein inactivation and mitochondrial dysfunction (Stachulski et al., 2013; Kalgutkar and Dalvie, 2015). Some of these reactive metabolites also function as oxidants that cause toxicity by oxidizing sulfhydryl groups on cysteine residues of GSH and proteins (Yuan and Kaplowitz, 2013). Acetaminophen (APAP; 4′-hydroxyacetanilide) is considered to be prototypical among marketed drugs (e.g., clozapine, atorvastatin) that produce toxicity mediated by a reactive metabolite (Erve, 2006; Kalgutkar and Dalvie, 2015). Specifically, excess APAP is metabolized via the liver cytochrome P450 system to N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive metabolite that causes hepatotoxicity through both electrophilic and oxidant mechanisms (Hinson et al., 2010; Lancaster et al., 2015). Drug-induced toxicities are a notable therapy-limiting problem. For example, there are 300,000 hospitalizations per year in the United States for APAP-induced hepatotoxicity (Blieden et al., 2014); therefore, development of effective pharmacological interventions would have significant clinical implications.
One approach to mitigating the risk of an adverse drug reaction is through the use of nucleophilic compounds [e.g., N-acetylcysteine (NAC), diphenyl diselenide] that scavenge toxic electrophiles (Carvalho et al., 2013; Chughlay et al., 2015). In this regard, we have shown that 2-acetylcyclopentanone (2-ACP; Fig. 1A) prevented lethality in a mouse model of APAP overdose. This protection was associated with inhibition of APAP-induced derangement of histologic (e.g., hepatocyte necrosis) and biochemical (e.g., GSH depletion; plasma appearance of liver enzymes) indices of hepatotoxicity (Zhang et al., 2013). 2-ACP is a 1,3-dicarbonyl (β-diketone) compound that can ionize in solution (Table 1) to form a nucleophilic enolate that binds and thereby detoxifies pathogenic electrophiles (e.g., NAPQI, 4-hydroxy-2-nonenal). The enolate can also chelate metal ions such as those involved in free-radical generating Fenton chemistry (LoPachin et al., 2011; Zhang et al., 2013). That enolate-forming 1,3-dicarbonyl compounds might be cytoprotective stems from the recognition that the central bridge of curcumin contains a 1,3-dicarbonyl group (Fig. 1A) that plays a critical role in preventing oxidative stress (Vajragupta et al., 2005; Weber et al., 2006; Begum et al., 2008; reviewed in LoPachin et al., 2012). Whereas the ability of curcumin and other phytopolyphenols to function as free radical traps has been the focus of much mechanistic research, this bridge exists predominately as a keto-enol tautomer (Balasubramanian, 2006; Payton et al., 2007) that can ionize (pKa = 7.8) to form an enolate nucleophile. The chemistry of the dicarbonyl enolates is well understood (Loudon, 2002; Bug and Mayr, 2003; Eames, 2009) and corroborative mechanistic research has demonstrated the role of this chemistry in cytoprotection (Awasthi et al., 1996; Bernabé-Pineda et al., 2004; Vajragupta et al., 2005; Weber et al., 2006; Begum et al., 2008; Eames, 2009; LoPachin et al., 2011).
Fig. 1.

(A) Color-coded line drawing of curcumin and the 1,3-dicarbonyl derivative, 2-ACP. Functionally important shared structural moieties are in red. (B) Color-coded line drawing of phloretin and pharmacophores, THA and PG. Functionally important shared structural moieties are in blue (PG) or both black and blue (phloretin, THA).
TABLE 1.
Line structures and physicochemical characteristics of selected nucleophiles
| Compounda |
Structure (Ionization) |
Nucleophile |
ω−b,c |
σb |
pKa |
|---|---|---|---|---|---|
| 2-ACP (partial) | 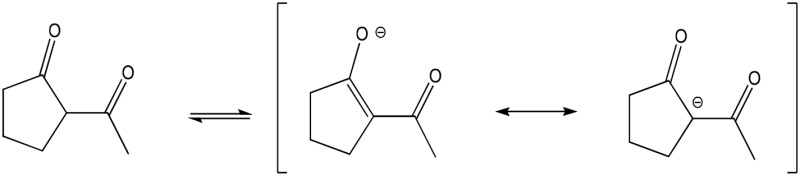 |
Enolate | 485 (204) | 418 | 7.8 |
| Phloretin (toxic) | 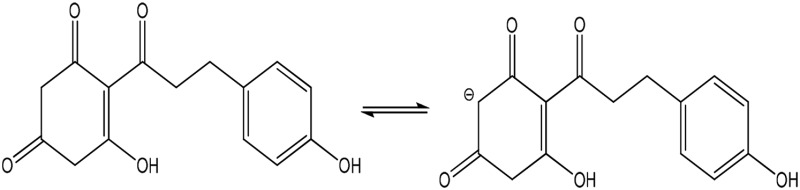 |
Enolate | 221 (105) | 494 | 7.3 |
| PG (full) | 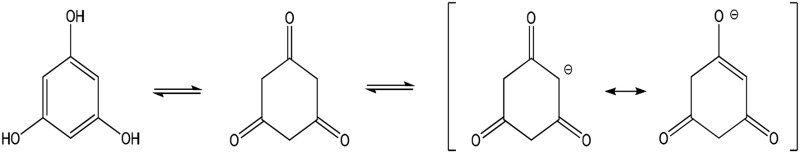 |
Enolate | 366 (133) | 540 | 8.5 |
| THA (full) | 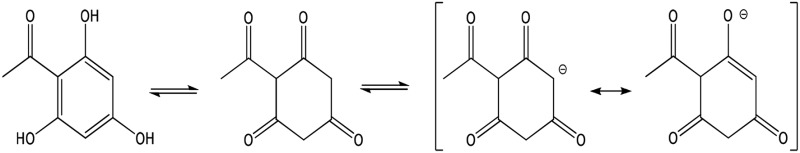 |
Enolate | 325 (114) | 485 | 7.7 |
| NAC (partial) | 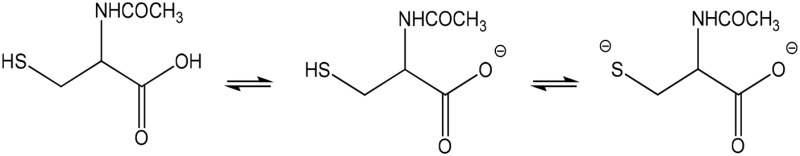 |
Thiolate | 667 (316) | 367 | 9.5 |
| GSH (partial) | 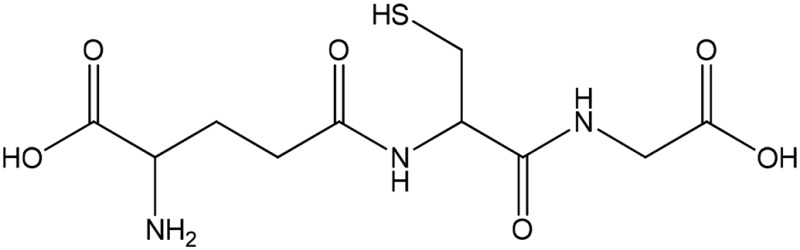 |
Thiolate | 548 (239) | 427 | 8.7 |
| N-acetyl-l-lysine (none) | 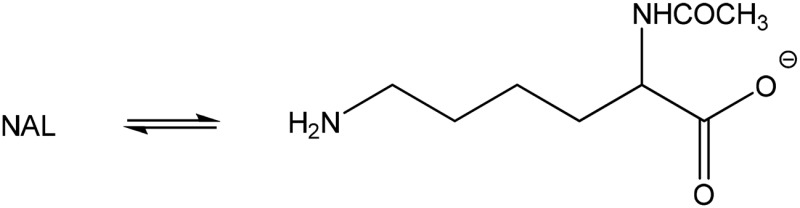 |
Amine | 387 (162) | 292 | 10 |
| Carnosine (none) | 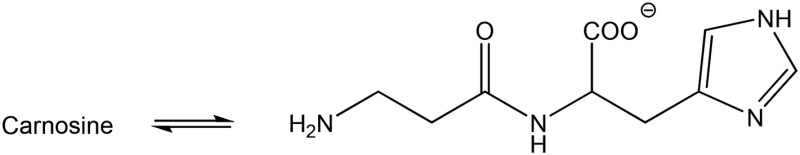 |
Amine | 389 (155) | 307 | 9.5 |
Data in parentdeses indicate level of cytoprotection.
Values are ×10−3 eV.
Values for reaction witd NAPQI (values in parentdeses are for reaction witd acrolein).
The three phenolic groups on the A ring of phloretin are also enols that can ionize to form potentially cytoprotective enolate sites (Table 1). Phloretin is a flavanoid that has cytoprotective potential based on an ability to trap free radicals and scavenge unsaturated aldehyde electrophiles that are involved in oxidative stress (Calliste et al., 2001; Zhu et al., 2011). In addition, two phloretin pharmacophores, 2′,4′,6′-trihydroxyacetophenone (THA; Fig. 1B) and phloroglucinol (PG; Fig. 1B), are polyphenolic compounds that can form nucleophilic enolates. We have proposed that enolate formation is an important platform for development of efficacious cytoprotectants (LoPachin et al., 2011, 2012). Therefore, to extend our analysis of enolate-based cytoprotection, we determined the relative abilities of the phloretin pharmacophores to provide hepatoprotection in a mouse model of acute APAP overdose. In corroborative studies, the cellular pharmacological properties of efficacy and potency were determined for 2-ACP, THA, and PG in APAP-exposed freshly isolated mouse hepatocytes (Reid et al., 2005; Burke et al., 2010a). For comparative purposes, the relative protective abilities of several other nucleophilic compounds, including the thiol-based nucleophile NAC, were evaluated in this hepatocyte model. To define the chemical basis of enolate cytoprotection further, we determined the role of acidity (pKa) and several other physicochemical parameters (softness/hardness, nucleophilicity) in preventing electrophile (NAPQI, acrolein)–induced GSH depletion in an in chemico model. The isolated hepatocyte system is a relevant experimental model of APAP hepatotoxicity (Burke et al., 2010b), which avoids extrahepatic pharmacokinetic influences that complicate the interpretation of mechanism. The results of this study offer a comprehensive view of enolate protection in an experimental drug-induced toxicity.
Materials and Methods
Reagents.
APAP, 2-ACP, β-alanyl-l-histidine (carnosine), N-acetyl-l-lysine, dithiothreitol, N-acetyl-l-cysteine, THA, 1,3,5,-trihydroxybenzene (phloroglucinol), GSH, 3-(4-hydroxyphenyl)-1-(2,4,6-trihydroxyphenyl)-propan-1-one (phloretin), HEPES, porcine heparin sodium salt, penicillin G sodium salt, dimethylsulfoxide, RPMI-1640 modified media without sodium bicarbonate or phenol red, Percoll, and trypan blue were purchased from Sigma-Aldrich (St. Louis, MO). Tetramethylrhodamine ethyl ester (TMRE) mitochondrial membrane potential kits were purchased from Abcam (Cambridge, MA). Prestoblue was purchased from Life Technologies (Grand Island, NY). Collagenase A from Clostridium histolyticum was purchased from Roche Laboratories (Nutley, NJ). Regardless of the source, all chemicals were of the highest grade commercially available.
Animals and Treatments.
All aspects of animal use in this study were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Montefiore Medical Center Animal Care Committee. Three-month-old male C57BL/6N mice (mean weight 21 g) were purchased from Charles River Laboratory (Wilmington, MA). Mice were housed individually in polycarbonate boxes. Filtered drinking water and Purina Rodent Laboratory Chow (Purina Mills, Inc., St. Louis, MO) were available ad libitum. The animal room was maintained at approximately 22°C and 50% humidity with a 12-hour light/dark cycle. Prior to drug administration, mice were fasted overnight and treatments began at 8:00 AM the following morning. Food was returned 1 hour post-treatment. APAP and all experimental compounds were administered in phosphate-buffered polyethylene glycol. Previous studies demonstrated that this vehicle did not affect the experimental outcome (Zhang et al., 2013). As a general protocol, groups of mice (n = 10–15) were pretreated by intraperitoneal injection (10 ml/kg; THA, 0.2–0.8 mmol/kg; PG, 0.4–2.4 mmol/kg) or oral administration (6 ml/kg; THA, 0.8–2.4 mmol/kg; PG, 0.8 and 2.4 mmol/kg) of putative hepatoprotectant followed 20 minutes later by oral (4 ml/kg) APAP (500 mg/kg). A separate group of animals were given vehicle by intraperitoneal injection or oral administration followed 20 minutes later by oral APAP. To define the temporal limitations of hepatoprotection, groups of mice were treated with oral APAP (500 mg/kg) followed 60 minutes later by intraperitoneal or oral administration of putative protectant (Zhang et al., 2013). Control mice received an intraperitoneal/oral sequence of vehicle injections. An assessment of general toxicity was conducted at 2-day intervals over a 7-day postintoxication period. Specifically, animals were weighed and assessed by a blinded observer for changes in grooming, nest building, open field behavior, recumbency, and gait (Zhang et al., 2013). Kaplan–Meier survival curves were used to illustrate the cumulative percent daily lethality of mice in different experimental groups and were generated in Prism 6.0 software (GraphPad Software Inc., La Jolla, CA; see Zhang et al., 2013).
Hepatocyte Isolation Procedures and Incubations.
Freshly isolated mouse hepatocytes were used in all experiments. Hepatocytes for individual experiments were derived from a single anesthetized mouse (isoflurane inhalation) by collagenase perfusion according to a modification of the Burke et al. (2010a) method. Briefly, to separate dead cells, isolated hepatocytes were centrifuged (140g × 8 minutes) in a Percoll gradient and then washed in media (140g × 3 minutes) to remove Percoll. The isolation procedure yielded approximately 30–40 million cells with 80%–90% viability as determined by trypan blue exclusion. Hepatocytes (100,000 cells/ml) were incubated in covered 35-mm plastic dishes containing RPMI-1640 media supplemented with 25 mM HEPES, heat-inactivated horse serum, 10 IU/ml heparin, and 500 IU/ml penicillin G at 37°C in a humidified atmosphere of 95% O2/5% CO2. In general, the concentration-dependent (0.01–3.0 mM) abilities of putative cytoprotectants from different chemical classes (e.g., polyphenols, 1,3-dicarbonyl enols, cysteine, and nitrogen-based compounds) were determined in isolated hepatocytes coexposed to APAP (1 mM × 4-hour incubation). Vehicle (0.1% dimethylsulfoxide in media), vehicle plus APAP (1 mM × 4-hour incubation), and cytoprotectant (3.0 mM × 4 hours) controls were included in these analyses. In corroborative studies, the cytoprotectant concentration was fixed (0.25 or 1.0 mM) and the APAP content was varied (0.25–5.0 mM). To evaluate the relative abilities of the different cytoprotectants to reverse (rescue) the injury process, test compounds (0.25–1.0 mM) or vehicle were added 2 hours after APAP (1 mM) exposure and hepatocyte toxicity was measured after an additional 2 hours of incubation. The concentration-response data were fitted by nonlinear regression analyses (LoPachin et al., 2009a). In all studies, hepatocyte viability and other toxic measures (see ahead) were determined in at least six to eight independent experiments conducted in triplicate after 2–4 hours of APAP or vehicle control exposure.
Cytotoxicity Parameters.
Hepatocyte viability was measured using PrestoBlue, which contains an oxidized cell-permeant compound (blue resazurin) that is converted exclusively in viable cells to a reduced (red resorufin) form. Cell viability assays were performed according to the manufacturer’s specifications. Preliminary studies were conducted to establish experimental conditions such as Prestoblue concentration, cell number, and incubation times. Isolated hepatocytes were stained by adding Prestoblue (50 nM final concentration) directly to individual samples followed by incubation for 30 minutes at 37°C in an atmosphere of 95% O2/5% CO2. At the completion of sample incubation, absorbance was determined in a SpectraMax (Sunnyvale, CA) M5 plate reader at a wavelength of 570 nm (reduced form) and 600 nm (oxidized form). A relative decrease in absorbance ratio (570 nm/600 nm) represented a corresponding degree of cell lethality.
As a direct measure of hepatocyte damage, the appearance of liver-specific enzymes, alanine aminotransferase and aspartate aminotransferase, was measured in the incubation media. In addition, we determined serum levels of lactate dehydrogenase as a measure of general cell damage. Cells were pelleted (14,000g for 5 minutes) and aliquots (700 µl) were collected from the supernatant. Media samples were analyzed by an automated analyzer (Hitachi Modular Automated Clinical Chemistry Analyzer; Roche Diagnostics, Indianapolis, IN) and are expressed as mean media U/l ± S.E.M.
Mitochondrial membrane potential (ΔΨm) was measured using a TMRE–Mitochondrial Membrane Potential Assay Kit (Abcam) according to the manufacturer’s specifications. TMRE reagent is a cell-permeable positively charged dye that accumulates in viable polarized mitochondria that are negatively charged. TMRE was added to hepatocyte suspensions (50 nM final concentration) and cells were incubated for 20 minutes at 37°C in a humidified atmosphere of 95% O2/5% CO2. In parallel suspensions, background fluorescence was determined by the addition of carbonyl cyanide 4-trifluoromethoxy/pehylhydrazone (20 µm), an ionophore uncoupler of oxidative phosphorylation, 10 minutes prior to adding TMRE. After incubation, cells were pelleted (140g × 3 minutes), washed twice in 0.2% bovine serum albumin in phosphate-buffered saline (PBS), and then resuspended in PBS (100 µl). Fluorescence was measured in 96-well plates using a SpectraMax M5 plate reader at the TMRE excitation/emission fluorescence of 549 nm/575 nm.
In Chemico Studies: Effects of Cytoprotectants on Electrophile-Induced GSH Loss.
Graded concentrations of NAPQI (2–128 μM) or acrolein (2–256 μM) were preincubated (15 minutes) in PBS (pH 7.4, 25°C) with selected cytoprotectants (50 μM) from different chemical classes or vehicle. After preincubation, GSH (30 μM) was added and the remaining sulfhydryl content was measured after 15 minutes by the DTNB (5,5′-Dithiobis(2′-nitrobenzoic acid) method of LoPachin et al. (2009a). For each hepatoprotectant, respective sulfhydryl data were fitted by nonlinear regression analyses (r2 for all curves ≥ 0.90) and electrophile concentrations that produced 50% thiol loss (IC50 values) and their 95% confidence intervals were calculated by the Cheng–Prusoff equation (LoPachin et al., 2007).
Hard And Soft, Acids and Bases Parameters: Quantum Mechanical Calculations.
To calculate hard and soft, acids and bases (HSAB) parameters for the electrophiles and nucleophiles involved in this study, the respective energies of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO) were derived using Spartan14 software (version 1.1.8; Wavefunction Inc., Irvine, CA). For each chemical structure, ground-state equilibrium geometries were calculated with Density Functional B3LYP 6-31G* in water starting from 6-31G* geometries. Global (whole-molecule) hardness (η) was calculated as η = (ELUMO − EHOMO)/2 and softness (σ) was calculated as the inverse of hardness or σ = 1/η. The nucleophilicity index (ω−) was calculated as ω− = ηA(µA − µB)2/2(ηA + ηB)2, where µ = (ELUMO + EHOMO)/2, A = reacting nucleophile, and B = NAPQI (η = 2.005 eV and μ = −5.235 eV) or acrolein (η = 2.695 eV and μ = −4.535 eV; see LoPachin et al., 2012).
Statistical Analyses.
All statistical analyses were conducted using Prism 6.0 software (GraphPad Software Inc.) with significance set at the 0.05 level of probability. In studies evaluating the relative abilities of potential hepatoprotectants to modify APAP lethality, the Mantel–Cox log-rank test was used to compare survival rates among the experimental groups. For analyses of cytoprotective and toxicity indices, statistically significant differences between group mean data were determined by a Bonferroni test for multiple comparisons.
Results
Intraperitoneal Hepatoprotectant Administration.
Intraperitoneal injection of either THA (Fig. 2A) or PG (Fig. 2B) provided dose-dependent hepatoprotection when administered 20 minutes before oral APAP intoxication. However, the dose range for THA hepatoprotection (Fig. 2A; 0.20–0.80 mmol/kg) was substantially lower than that of PG (Fig. 2B; 0.40–2.40 mmol/kg). In addition, intraperitoneal THA was fully protective at 0.80 mmol/kg (Fig. 2A), whereas the same dose of PG provided only 65% protection (Fig. 2B). Administration of either pharmacophore at the highest effective dose did not produce toxicity or lethality over a 7-day observation period (data not shown). Neither THA, PG, nor NAC (2.40 mmol/kg) produced hepatoprotection when given by intraperitoneal injection 60 minutes after oral APAP (data not shown). This is consistent with our previous findings (Zhang et al., 2013).
Fig. 2.

(A) Kaplan–Meier survival curves illustrating the effects of intraperitoneal THA (0.20–0.80 mmol/kg) on oral APAP (500 mg/kg)–induced lethality in mice (n = 10–15/group). Joining lines indicate statistically significant differences in treatment groups. (B) Effects of PG (0.40–2.40 mmol/kg) administered by intraperitoneal injection on oral APAP (500 mg/kg)–induced lethality in mice (n = 10–15/group). Joining lines indicate statistically significant differences in treatment groups. **P < 0.01; *** P < 0.001.
Oral Hepatoprotectant Administration.
THA (Fig. 3A) produced dose-dependent hepatoprotection when administered orally, whereas similar administration of PG (Fig. 3B) was completely ineffective over a broad dose range (0.80–2.40 mmol/kg). Although neither oral THA nor PG (2.40 mmol/kg) was effective when given 60 minutes after APAP, similar administration of equimolar NAC was hepatoprotective (data not shown; see Corcoran et al., 1985a,b).
Fig. 3.

(A) Kaplan–Meier survival curves illustrating the effects of oral THA (0.80–2.40 mmol/kg) on oral APAP (500 mg/kg)–induced lethality in mice (n = 10–15/group). Joining line indicates statistically significant differences in treatment groups. **P < 0.01. (B) Effects of oral PG (0.80–2.40 mmol/kg) on oral APAP (500 mg/kg)–induced lethality in mice (n = 10–15/group). Statistically significant differences were not determined among experimental groups.
Cytoprotective Properties of Phloretin Pharmacophores.
The relative cytoprotective abilities of the enolate-forming analogs (2-ACP, THA, and PG) were determined and compared with that of the thiolate-forming nucleophile NAC. Hepatocytes were exposed to APAP (1.0 mM) and incubated (4 hours) with graded concentrations of putative protectant (0.010–5.0 mM). Initial determinations showed that the cytoprotectants were not toxic in these experimental conditions. Results (Fig. 4) indicated that exposure of hepatocytes to APAP (1.0 mM × 4 hours) caused a decrease in mean (± S.E.M.) cell viability of 52% ± 7%. The lowest concentration (0.010 mM) of all cytoprotectants used in this study did not alter the APAP baseline effect (i.e., 48% ± 5%; see ordinate of Fig. 4). At higher concentrations, all compounds provided concentration-dependent protection against APAP-induced hepatocyte toxicity. As evidenced by the respective concentration profiles, THA and PG were substantially more potent than either 2-ACP or NAC. For example, THA produced measurable cytoprotection at 0.050 mM (Fig. 4, arrow), whereas the lowest concentrations of 2-ACP and NAC to provide protection were both 1.0 mM (Fig. 4, respective arrows). Although the cytoprotectants differed in potency, all compounds tested ultimately produced maximal cytoprotection (Fig. 4).
Fig. 4.

Effects of graded protectant (0.010–5.0 mM) on viability of APAP-exposed (1.0 mM × 4 hours) isolated mouse hepatocytes. Data are expressed as mean percent of control ± S.E.M. cell viability (n = 6–8). Arrows indicate the first cytoprotectant concentration at which the mean viability data are significantly different from the APAP-alone data. Sequentially higher protectant concentrations were statistically significant. P < 0.05 (level of significance relative to APAP-alone data).
To obtain a more complete definition of cytoprotective ability, isolated hepatocytes were exposed to protectant (either 0.25 or 1.0 mM) and graded concentrations of APAP (0.25–5.0 mM × 4 hours). Results show that APAP exposure caused progressive concentration-dependent loss of hepatocyte viability (Fig. 5) that was associated with substantial derangement of both hepatocyte enzyme concentrations (alanine aminotransferase, aspartate aminotransferase, and lactate dehydrogenase) in media and mitochondrial membrane potential (Fig. 6). At a lower cytoprotectant concentration (0.25 mM; Fig. 5A), neither 2-ACP nor NAC provided cytoprotection beyond the lowest APAP concentration (0.25 mM). In contrast, both THA and PG were fully protective up to the 1.0 mM APAP concentration (Fig. 5B). This differential cytoprotection among the compounds tested is reflected in the corresponding changes in the toxicity indices (Fig. 6). At the highest APAP concentration (5.0 mM), THA (0.25 mM) preserved cell viability, whereas PG (0.25 mM) provided only partial cytoprotection as evidenced by significantly reduced cell viability (Fig. 5A) and corresponding changes in toxicity parameters (Fig. 6, A and C). These data are consistent with the previous rank order of concentration dependent cytoprotection (Fig. 4). When the concentration of cytoprotectant was increased to 1.0 mM (Fig. 5B), THA or PG provided full hepatocyte protection irrespective of APAP concentration. Protection was characterized by normal (THA) to near-normal (PG) toxicity measures (Fig. 6, B and D). When APAP-exposed hepatocytes were incubated with either 2-ACP or NAC at 1.0 mM, the partial cytoprotection provided (Fig. 5B) was accompanied by a return toward normalization of the toxicity indices (Fig. 6, B and D). Together, these findings identify THA as a highly potent antagonist of APAP toxicity in isolated hepatocytes.
Fig. 5.

Effects of 0.25 mM (A) or 1.0 mM (B) protectant on viability of isolated mouse hepatocytes exposed to graded APAP concentrations (0.25–5.0 mM × 4 hours). Data are expressed as mean percent of control ± S.E.M. (n = 6–8) cell viability. *P < 0.05 (level of significance relative to the APAP-alone control data).
Fig. 6.

This figure is a continuation of analyses presented in Fig. 5. Here we describe the respective effects of 0.25 mM or 1.0 mM protectant on ALT appearance in media (A and B) and changes in mitochondrial membrane potential (ΔΨm; C and D) in isolated mouse hepatocytes exposed to 1.0 mM APAP (× 4 hours) as per Fig. 5. ALT data are expressed as mean IU/mg protein ± S.E.M. (n = 6–8) and changes in ΔΨm are expressed as mean percent of control ± SEM (n = 6–8). Also measured were AST and LDH media levels that showed mean changes equivalent to those of ALT (data not shown). *P < 0.05; **P < 0.01 (levels of significance relative to the APAP-alone control data). ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase.
Physicochemical Characteristics related to Cytoprotection.
According to the HSAB theory of Pearson, substances can be classified as either relatively soft (polarizable) or hard (nonpolarizable) depending on the respective orbital energy values. A governing theorem of the HSAB concept is that electrophiles react preferentially with nucleophilic targets of similar softness or hardness. Softness (σ) and hardness (η) are ordinarily determined using quantum mechanical models and the values obtained can be combined with chemical activity parameters to produce indices of electrophilic (ω) and nucleophilic (ω-) reactivity. The application of HSAB concepts and parameters to environmental, pharmaceutical, and endogenous electrophiles has been extensively reviewed by LoPachin et al. (2008, 2009b, 2012) and LoPachin and Gavin (2014). We have suggested that NAPQI, acrolein, and other soft electrophiles (Table 2) involved in APAP hepatotoxicity rapidly form adducts with soft nucleophilic thiolate sites on proteins or the soft carbanionic enolate nucleophiles of 2-ACP or THA (Zhang et al., 2013). If the proposed soft–soft interactions govern the surrogate activity of the cytoprotectants, then hard nucleophiles should be less effective. The nitrogen atoms on the ε-amino group of lysine and imidazole moiety of histidine are harder nucleophiles than the enolate or thiolate nucleophiles. Using respective analogs of these potential amino acid targets, N-acetyl lysine and carnosine, we found that neither compound provided protection against APAP cytotoxicity in our isolated hepatocyte model (Table 1). These data, in conjunction with our previous studies (LoPachin et al., 2007, 2009b; Zhang et al., 2013) suggest that APAP hepatotoxicity involves toxic soft electrophiles (Table 2) that can be scavenged by soft enolate or thiolate nucleophiles.
TABLE 2.
Softness, electrophilicity, and IC50 values for quinone and α,β-unsaturated carbonyl derivatives
Softness (σ) and electrophilicity (ω) for the selected compounds were calculated as described in the Materials and Methods. The respective IC50 values represent in vitro electrophile concentrations that produce 50% thiol loss and reflect the relative electrophilic potency of each chemical (Zhang et al., 2013).
| Electrophile | Softness (σ)a | Electrophilicity (ω) | IC50 |
|---|---|---|---|
| eV | μM | ||
| NAPQI | 499 | 6.83 | 8.4 |
| Benzoquinone | 524 | 7.78 | 11.6 |
| Acrolein | 371 | 3.82 | 52.5 |
| 4-Hydroxy-2-nonenal | 377 | 3.80 | 398b |
Values are ×10−3 eV−1.
The IC50 value for HNE is higher than expected based on corresponding electrophilicity (ω). However, the second-order reaction rate for reaction with sulfhydryl groups is affected by the relatively larger size and shape of this molecule (i.e., the reaction rate is slower due to steric hindrance; see LoPachin et al., 2009b).
Although PG and THA are weaker nucleophiles than 2-ACP (Table 1), our studies (Figs. 4 and 5; Table 1) showed that 2-ACP was a less potent protectant than either THA or PG. As illustrated in Table 1, the enolate protectants vary in softness (σ), nucleophilicity (ω−), and acidity (pKa values). To determine how these physicochemical attributes might influence electrophile scavenging and therefore cytoprotection, we conducted in chemico experiments that determined the relative abilities of the nucleophiles to prevent NAPQI depletion of GSH (Fig. 7A). For comparative purposes, we included analyses of cytoprotectant modulation of acrolein-induced thiol loss (Fig. 7B).
Fig. 7.

In chemico effects of selected cytoprotectants on NAPQI-induced (A) or acrolein-induced (B) loss of GSH sulfhydryl groups. Graded concentrations of toxicant were incubated with selected test hepatoprotective compounds. Data are expressed as mean percent control ± S.E.M. (n = 3 to 4 experiments) and calculated IC50 values are provided in parentheses. 2-ACP, 2-acetylcyclopentanone; Phl, phloretin.
Consistent with electrophilic reactivity (Table 2), the IC50 for NAPQI (ω = 6.83 eV) was 10 μM (Fig. 7A), whereas the IC50 for the less powerful electrophile, acrolein (ω = 3.82 eV), was 21 μM (Fig. 7B). The more basic pKa of PG (8.5; Table 1) indicates that only 7% of this substance will be in the nucleophilic carbanion state at physiologic pH, which significantly limits NAPQI scavenging (i.e., IC50 = 20 µM; Fig. 7A). Alternatively, phloretin significantly shifted the NAPQI concentration-response curve rightward, as reflected by the increased IC50 for GSH loss (IC50 = 59 μM). In this regard, phloretin is relatively acidic (pKa value = 7.3; Table 1); therefore, at cellular pH, a large proportion of this polyphenol (55%) exists as the ionized NAPQI-scavenging nucleophilic enolate. THA and 2-ACP are equivalent in acid strength (pKa ≈ 7.7; Table 1). Accordingly, 30% of the corresponding cellular concentrations will be in the ionized nucleophilic enolate state at physiologic conditions. However, even though 2-ACP is a more powerful nucleophile in reaction with NAPQI (ω−ACP = 0.485 eV versus ω−THA = 0.325 eV; Table 1), THA provided significantly greater GSH protection (i.e., IC50 = 73 µM for THA, whereas IC50 = 25 µM for 2-ACP; Fig. 7A).
When acrolein was used as the reacting electrophile (Fig. 7B; Table 2), the effectiveness of the protectant was related to a combination of acidity (tendency to ionize) and the nucleophilicity of the corresponding anion. Phloretin, THA, and 2-ACP have roughly equivalent pKa values (range, 7.3–7.8; Table 1), indicating that each will be highly ionized (30%–60%) and thus provide a significant concentration of the protecting anionic species at cellular conditions. However, 2-ACP is a much better nucleophile in reaction with acrolein (ω−ACP = 0.204 eV, ω−THA = 0.114 eV, ω-Phl = 0.105 eV; Table 1) and, as the data in Fig. 7B demonstrate, provides superior GSH protection. It should be noted that although PG is a good nucleophile (ω−PG = 0.133 eV; Table 1), the relatively poor ionization capacity (pKa = 8.5) results in a low anion concentration (approximately 7%) and, consequently, reduced ability to scavenge electrophiles. The rank order of this protection (2-ACP > THA > PG; Fig. 7B) is, therefore, consistent with the respective ω− values, suggesting that relative nucleophilicity is the determining factor for GSH protection when acrolein is the reacting electrophile. However, this does not appear to be the case for NAPQI-mediated GSH depletion. Instead, our results suggest that THA, and perhaps PG as well, act via additional mechanisms to prevent NAPQI toxicity (see Discussion).
Hepatocyte Recovery.
In this study, we determined the relative abilities of the cytoprotectants to arrest or otherwise modify the ongoing development of APAP hepatocyte toxicity. After an initial 2 hours of incubation with APAP (1.0 mM) alone, there was a 31.5% ± 7.0% reduction in mean (± S.E.M.) cell viability (Fig. 8) associated with increases in the media content of hepatocyte enzymes (Fig. 9, A–C) and depolarization of mitochondrial membranes (Fig. 9, D–F). Hepatocyte toxicity continued to progress over an additional 2 hours of APAP incubation [i.e., mean cell viability decreased to 49.5% ± 9.0% after 4 hours (total) of incubation; Fig. 8]. This loss of viability was associated with further derangement of the toxicity parameters (Fig. 9). However, addition of cytoprotectant 2 hours after APAP exposure modified subsequent progression of cytotoxicity in a concentration-dependent manner. Thus, although 2-ACP and NAC were ineffective at the lowest concentration tested (0.25 mM; Fig. 8A), both compounds truncated the rapid decline in viability at 1.0 mM (Fig. 8B). At 5 mM (Fig. 8C), 2-ACP and NAC promoted recovery of APAP-exposed hepatocytes. The concentration-dependent restoration of cell viability mediated by 2-ACP and NAC was correlated with significant improvements in corresponding toxicity indices (Fig. 9). THA provided partial recovery of viability at the lowest concentration (0.25 mM; Fig. 8A), whereas PG arrested the progression of APAP-induced toxicity (Fig. 8A). At the 1.0-mM concentration, PG and THA promoted complete restoration of viability in APAP-exposed hepatocytes (Fig. 8B). The return to control viability was accompanied by normalization of the toxicity indices (Fig. 9).
Fig. 8.

Effects of graded cytoprotectant concentrations (0.25–5.0 mM) on temporal loss of viability induced by APAP exposure (1.0 mM). After 2 hours of APAP incubation, cytoprotectant was added and viability was determined after additional 2 hours. Note: Fig. 8C only pertains to the representative effects of 2-ACP and NAC. Data are expressed as mean percent of control ± S.E.M. (n = 6–8) cell viability. *P < 0.05 (level of significance relative to the APAP alone control data after 4 hours of incubation).
Fig. 9.

This figure is a continuation of our analysis presented in Fig. 8. Here we describe the effects of graded protectant concentrations (0.25–5.0 mM) added 2 hours after APAP exposure on ALT appearance in media (A–C) and changes in mitochondrial membrane potential (ΔΨm; D–F) in isolated mouse hepatocytes exposed to 1.0 mM APAP. Parameters were measured at 4 hours after APAP exposure as per Fig. 8. ALT data are expressed as mean IU/mg protein ± S.E.M. (n = 6–8) and changes in ΔΨm are expressed as mean percent of control ± S.E.M. Also measured were AST and LDH media levels that showed mean changes equivalent to those of ALT (data not shown). *P < 0.05; **P < 0.01 (levels of significance relative to the APAP-alone control data). ALT, alanine aminotransferase.
Discussion
Many drug-induced toxicities involve liver cell injury mediated by electrophilic metabolites (Table 2) that form covalent adducts with cysteine sulfhydryl groups on GSH and proteins (Erve, 2006; Kalgutkar and Dalvie, 2015). Some of these toxic metabolites can also act as oxidants that convert sulfhydryl groups to the corresponding disulfide (Hinson et al., 2010; Kalgutkar and Dalvie, 2015). One approach to preventing metabolite-induced toxicity is the use of nucleophilic compounds that act as surrogate targets for the electrophilic metabolites (e.g., see LoPachin et al., 2012; Chughlay et al., 2015; Mansour et al., 2015). Adduction of these electrophiles can prevent or reduce subsequent development of cytotoxicity. We previously showed that 2-ACP could prevent experimental APAP hepatotoxicity through formation of a carbon-based enolate nucleophile that scavenges electrophilic NAPQI (Zhang et al., 2013). APAP hepatotoxicity is considered to be a prototypical example of drug-induced toxicity and, consequently, enolate-forming compounds might be useful in treating or preventing this and other adverse drug outcomes. Therefore, we expanded our investigation of these compounds to include analyses of the phloretin pharmacophores, THA and PG, in a mouse model of APAP overdose. Corroborative studies in an isolated mouse hepatocyte system were conducted to advance our understanding of enolate pharmacology and its corresponding role in remediation of drug-induced toxicity.
Our studies in the APAP overdose animal model showed that THA and PG were hepatoprotective and were nontoxic at the doses tested. THA was effective regardless of administration route and, on a comparative basis (intraperitoneal injection), was more potent than PG. THA was also more potent than either 2-ACP or NAC when administered intraperitoneally (Zhang et al., 2013). Earlier studies of NAC showed that this thiol protectant was effective only when given orally over a dose range similar to that of 2-ACP or PG. It is possible that hepatoprotection afforded by 2-ACP, THA, and PG involved inhibition of cytochrome P450 activation of APAP or changes in APAP tissue distribution (Corcoran et al., 1985b). However, Zhang et al. (2013) provided evidence that APAP metabolism was not disrupted in mice treated with 2-ACP, since early transient (2–4 hours after APAP exposure) changes in liver injury biomarkers (e.g., thiol depletion) were evident, indicating parallel NAPQI-mediated hepatotoxicity. In addition, the observation that THA/PG provided cytoprotection in an isolated cell system argues against the possibility that corresponding cytoprotection involved changes in APAP tissue disposition (e.g., see Corcoran et al., 1985a,b).
Results from the isolated hepatocyte model suggest that differences among cytoprotectants in our animal model are based on characteristic molecular and physicochemical traits that affect potency and mechanism. Although the test compounds exhibited similar efficacy (Fig. 4), THA and PG were more potent as cytoprotectants in APAP-exposed hepatocytes than NAC or 2-ACP (Figs. 4 and 5). Like 2-ACP, THA and PG undergo ionization (Table 1) and the resulting soft enolate nucleophile can scavenge NAPQI (Fig. 7A), acrolein (Fig. 7B), and other soft electrophiles involved in secondary oxidative stress. However, the lower acidity of PG (pKa = 8.5) greatly diminishes corresponding enolate-based electrophile scavenging at physiologic conditions. In contrast, THA and 2-ACP are more acidic and, consequently, the enolates are present at higher a concentration, which facilitates electrophile scavenging. However, HSAB calculations of the respective nucleophilic indices (ω−; Table 1) revealed that the 2-ACP enolate was more reactive with NAPQI than either the PG or THA enolates. This is inconsistent with the rank order of GSH protection (THA > 2-ACP > PG) demonstrated in chemico (Fig. 7A). The discrepancy between nucleophilicity and cytoprotective abilities suggests that, in addition to electrophile adduction, other mechanisms of cytoprotection must be considered for THA and PG (see ahead).
For comparative purposes, our investigation also included evaluation of NAC cytoprotection in APAP-exposed isolated hepatocytes. NAC is the clinical antidote for APAP poisoning (Mucomyst; Bristol-Myers Squibb, Princeton, NJ) and, when ionized, is a thiolate nucleophile. In isolated hepatocyte studies, NAC was virtually equivalent to 2-ACP with respect to cytoprotective potency and efficacy. HSAB calculations indicated that, although NAC is a substantial nucleophile (ω− = 0.667 eV; Table 1), it is a relatively weak acid (pKa = 9.5) and very little (<1.0%) of the cysteine sulfhydryl group will exist in the nucleophilic anionic thiolate state at cellular pH. Therefore, it is unlikely that NAC will effectively scavenge NAPQI and other involved electrophiles (see Massey and Racz, 1981; Lauterburg et al., 1983; Corcoran and Wong, 1986). Furthermore, NAC is not known to form bidentate coordination complexes with metal ions but does have modest antioxidant abilities (Jaeschke et al., 2003; LoPachin et al., 2011). Thus, neither electrophile scavenging nor metal ion chelation appears to play a significant role in NAC hepatoprotection. Based on these findings, this thiol operates through an alternative, likely indirect, mechanism of protection [e.g., substrate for mitochondrial GSH synthesis (Lauterburg et al., 1983) and/or bioenergetic processes (Saito et al., 2010)].
Our animal studies showed that oral NAC administered 1 hour after APAP significantly reduced lethality, whereas similar administration of THA (this study) and 2-ACP (Zhang et al., 2013) failed to provide such delayed protection. Nonetheless, in a cell model of delayed protection, THA, PG, and 2-ACP were capable of rescuing isolated hepatocytes from ongoing APAP-induced injury (Figs. 8 and 9). Provided APAP-induced hepatotoxicity follows a similar pathophysiological sequence in both whole-animal and isolated hepatocytes, these findings suggest that delayed enolate-based hepatoprotection is possible. Therefore, the lack of such protection observed in mice likely indicates that intervening variables specific to whole-animal pharmacokinetics (e.g., half-life, tissue distribution) might compromise the expression of delayed hepatoprotection.
The relative abilities of the enolate-forming compounds to rescue APAP-exposed isolated hepatocytes also provided molecular insight into the cytoprotective mechanisms. Specifically, THA and PG were significantly more effective than either 2-ACP or NAC at rescuing hepatocytes from ongoing APAP-induced injury (Figs. 8 and 9). Clearly, the isolated hepatocytes were not irreversibly damaged at 2 hours after APAP exposure (Fig. 8), which suggests that a hepatotoxic mechanism involving reversible changes in sulfhydryl oxidation status might be more important at this time point than irreversible formation of covalent adducts (Yang et al., 2012, 2013). Nonetheless, the noted potency of THA and PG in this model of delayed cytoprotection could be related to their ability to trap free radicals, since generation of reactive oxygen/nitrogen species appears to mediate the oxidative phase of APAP injury (Reid et al., 2005). THA and, to a lesser extent, PG are free radical trapping agents that exhibit antioxidant activity (Fig. 8; Mathiesen et al., 1997; Rezk et al., 2002; Kim and Kim, 2010; Bentes et al., 2011; So and Cho, 2014). It is likely THA also has redox properties that are operational at cellular conditions and, like GSH (Rosen et al., 1984; Albano et al., 1985), might decrease the effective NAPQI concentration via reduction to APAP. In addition, the flexible structure of THA allows the formation of bidentate complexes with iron and copper ions (Shao et al., 2008; Mammino, 2013), thereby reducing free radical generation via the metal-catalyzed Fenton reaction (Loudon, 2002; Eames, 2009; LoPachin et al., 2011). PG cannot, however, chelate metal ions due to a more rigid molecular architecture. Together, these data suggest that the primary cytoprotective mechanism of PG is free radical trapping, since both enolate-mediated electrophile scavenging and metal chelation are limited. In contrast, the multifaceted cytoprotective properties of THA likely explain the superior ability to prevent APAP toxicity (Fig. 10).
Fig. 10.

This figure presents a schematic diagram of a typical drug-induced toxicity pathway mediated by a soft electrophile metabolite. The illustration also highlights the possible molecular sites of intervention by cytoprotective enolate-forming compounds. As per our current understanding of drug-induced toxicities, the electrophilic metabolite (e.g., NAPQI) will form adducts with nucleophilic cysteine residues on GSH and proteins. Some of these electrophilic metabolites also function as oxidants. The resulting GSH depletion, protein inactivation, and mitochondrial disruption promote cellular oxidative stress by initiating the generation of superoxide anion (O2−·) and hydrogen peroxide (H2O2). Through metal-catalyzed Fenton reactions, these free radicals generate highly reactive hydroxyl radicals (OH·) that can cause direct macromolecular damage. In addition, the hydroxyl and superoxide radicals can initiate peroxidation of polyunsaturated fatty acids to yield α,β-unsaturated aldehydes (e.g., acrolein, 4-hydroxy-2-nonenal). As soft electrophiles, these aldehyde toxicants contribute to the cellular electrophile burden and can thereby augment cytotoxicity. Enolate-forming cytoprotectants such as THA can act at several steps in this cascade to truncate development of toxicity. As soft nucleophiles, these compounds can scavenge the initiating metabolite and can bind membrane lipid-derived aldehyde electrophiles. The ability of these chemicals to chelate transition metal ions can inhibit the Fenton reaction and subsequent generation of free radicals. The radicals that nonetheless develop can be trapped by aromatic enolate-forming compounds (e.g., THA, PG). Therefore, the multifunctional enol cytoprotectants represent a platform for development of effective compounds that can treat drug-induced toxicities and pathogenic conditions involving cellular oxidative stress. HNE, 4-hydroxy-2-nonenal; RXN, reaction.
In this study, we have shown that the 1,3,5-trihydroxy aromatic derivative, THA, was a substantially more potent cytoprotectant against APAP-induced isolated hepatocyte toxicity than PG, 2-ACP, or NAC. These findings are generally consistent with parallel animal-based research, which supports their corresponding in vivo relevance. THA is characterized by several cytoprotective properties, such as electrophile scavenging, metal chelation, and free radical trapping, which enable this polyphenol to interrupt electrophile initiation and several succeeding stages of the drug-induced toxicity cascade (Fig. 10). We have also described the soft nucleophilicity of the carbanionic enolates and the fact that, in accordance with HSAB principles, these cytoprotectants react preferentially with soft electrophiles that mediate many drug-induced toxicities (see also LoPachin et al., 2011; Zhang et al., 2013). Critical to pharmacotherapeutic development is the understanding that hard nucleophilic candidates, such as those tested in our study (Table 1; N-acetyl lysine, carnosine), react more slowly with soft electrophiles and are therefore less effective cytoprotectants. The soft nucleophilic compounds tested in this research are pharmacophores of the phytopolyphenols, curcumin (2-ACP) and phloretin (THA, PG). However, unlike the parent compounds, the pharmacophores were less toxic, more stable, and relatively water soluble and had larger volumes of distribution (Ballantyne and Cawley, 2001; LoPachin et al., 2011; Zhang et al., 2013). That enolate-forming compounds can be developed for pharmaceutical use is evidenced by the fact that PG is marketed as the antispasmodic, Spasfon (Cephalon, Maisons-Alfort, France). The efficacy and potency demonstrated in our study, in conjunction with favorable pharmacokinetics, suggest that THA (or more likely an analog) could be coformulated with, for example, Tylenol (Johnson & Johnson, Fort Washington, PA) as an “on-board” protectant against drug-induced toxicities. In this regard, NAC would be less effective since the cytoprotective mechanism does not involve direct electrophile (e.g., NAPQI) scavenging, which is necessary to truncate the onset of acute hepatotoxicity. These findings support our enolate theory of cytoprotection (LoPachin et al., 2011, 2012) and provide proof-of-principle evidence that multifunctional enolate-forming compounds might represent a developmental platform for cytoprotectants that prevent drug-induced toxicity mediated by electrophilic metabolites.
Acknowledgments
The authors thank Irene Ostrovsky (Chemistry Laboratory, Department of Pathology) and Tewolde Yimer and staff members Anie George, Faina Kremerman, Don Kutsyk, Kseniya Vayner, and Aleksandr Yakubov for analysis of hepatocyte media biochemistry. The authors also thank Dr. J.A. Hinson and staff for helpful guidance regarding the mouse hepatocyte isolation procedure.
Abbreviations
- APAP
acetaminophen
- EHOMO
highest occupied molecular orbital energy
- ELUMO
lowest unoccupied molecular orbital energy
- GSH
glutathione
- HSAB
hard and soft, acids and bases
- NAC
N-acetylcysteine
- NAPQI
N-acetyl-p-benzoquinonimine
- PBS
phosphate-buffered saline
- THA
2′,4′,6′-trihydroxyacetophenone
- TMRE
tetramethylrhodamine ethyl ester
- 2-ACP
2-acetylcyclopentanone
Authorship Contributions
Participated in research design: Geohagan, Gavin, LoPachin.
Conducted experiments: Geohagen, Vydyanathan, Kosharskyy.
Performed data analysis: Geohagen, Vydyanathan, Kosharskyy, Gavin.
Wrote or contributed to the writing of the manuscript: Shaparin, Gavin, LoPachin.
Footnotes
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant R01ES00383028].
References
- Albano E, Rundgren M, Harvison PJ, Nelson SD, Moldéus P. (1985) Mechanisms of N-acetyl-p-benzoquinone imine cytotoxicity. Mol Pharmacol 28:306–311. [PubMed] [Google Scholar]
- Awasthi S, Srivatava SK, Piper JT, Singhal SS, Chaubey M, Awasthi YC. (1996) Curcumin protects against 4-hydroxy-2-trans-nonenal-induced cataract formation in rat lenses. Am J Clin Nutr 64:761–766. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K. (2006) Molecular orbital basis for yellow curry spice curcumin’s prevention of Alzheimer’s disease. J Agric Food Chem 54:3512–3520. [DOI] [PubMed] [Google Scholar]
- Ballantyne B, Cawley TJ. (2001) 2,4-Pentanedione. J Appl Toxicol 21:165–171. [DOI] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, et al. (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentes ALA, Borges RS, Monteiro WR, de Macedo LGM, Alves CN. (2011) Structure of dihydrochalcones and related derivatives and their scavenging and antioxidant activity against oxygen and nitrogen radical species. Molecules 16:1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabé-Pineda M, Ramírez-Silva MT, Romero-Romo MA, González-Vergara E, Rojas-Hernández A. (2004) Spectrophotometric and electrochemical determination of the formation constants of the complexes Curcumin-Fe(III)-water and Curcumin-Fe(II)-water. Spectrochim Acta A Mol Biomol Spectrosc 60:1105–1113. [DOI] [PubMed] [Google Scholar]
- Blieden M, Paramore LC, Shah D, Ben-Joseph R. (2014) A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert Rev Clin Pharmacol 7:341–348. [DOI] [PubMed] [Google Scholar]
- Bug T, Mayr H. (2003) Nucleophilic reactivities of carbanions in water: the unique behavior of the malodinitrile anion. J Am Chem Soc 125:12980–12986. [DOI] [PubMed] [Google Scholar]
- Burke AS, MacMillan-Crow LA, Hinson JA. (2010a) Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol 23:1286–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke AS, MacMillan-Crow LA, Hinson JA. (2010b) The hepatocyte suspension assay is superior to the cultured hepatocyte assay for determining mechanisms of acetaminophen hepatotoxicity relevant to in vivo toxicity. Chem Res Toxicol 23:1855–1858. [Google Scholar]
- Calliste CA, Le Bail JC, Trouillas P, Pouget C, Habrioux G, Chulia AJ, Duroux JL. (2001) Chalcones: structural requirements for antioxidant, estrogenic and antiproliferative activities. Anticancer Res 21 (6A):3949–3956. [PubMed] [Google Scholar]
- Carvalho NR, da Rosa EF, da Silva MH, Tassi CC, Dalla Corte CL, Carbajo-Pescador S, Mauriz JL, González-Gallego J, Soares FA. (2013) New therapeutic approach: diphenyl diselenide reduces mitochondrial dysfunction in acetaminophen-induced acute liver failure. PLoS One 8:e81961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chughlay MF, Kramer N, Werfalli M, Spearman W, Engel ME, Cohen K. (2015) N-acetylcysteine for non-paracetamol drug-induced liver injury: a systematic review protocol. Syst Rev 4:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran GB, Racz WJ, Smith CV, Mitchell JR. (1985a) Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. J Pharmacol Exp Ther 232:864–872. [PubMed] [Google Scholar]
- Corcoran GB, Todd EL, Racz WJ, Hughes H, Smith CV, Mitchell JR. (1985b) Effects of N-acetylcysteine on the disposition and metabolism of acetaminophen in mice. J Pharmacol Exp Ther 232:857–863. [PubMed] [Google Scholar]
- Corcoran GB, Wong BK. (1986) Role of glutathione in prevention of acetaminophen-induced hepatotoxicity by N-acetyl-L-cysteine in vivo: studies with N-acetyl-D-cysteine in mice. J Pharmacol Exp Ther 238:54–61. [PubMed] [Google Scholar]
- Eames J. (2009) Acid-base properties of enols and enolates, in The Chemistry of Metal Enolates (Zablicky J. ed) pp 411–460, John Wiley & Sons, West Sussex, UK. [Google Scholar]
- Erve JCL. (2006) Chemical toxicology: reactive intermediates and their role in pharmacology and toxicology. Expert Opin Drug Metab Toxicol 2:923–946. [DOI] [PubMed] [Google Scholar]
- Hinson JA, Roberts DW, James LP. (2010) Mechanisms of acetaminophen-induced liver necrosis. Handbook Exp Pharmacol 196:369–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Knight TR, Bajt ML. (2003) The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett 144:279–288. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Dalvie D. (2015) Predicting toxicities of reactive metabolite-positive drug candidates. Annu Rev Pharmacol Toxicol 55:35–54. [DOI] [PubMed] [Google Scholar]
- Kim MM, Kim SK. (2010) Effect of phloroglucinol on oxidative stress and inflammation. Food Chem Toxicol 48:2925–2933. [DOI] [PubMed] [Google Scholar]
- Lancaster EM, Hiatt JR, Zarrinpar A. (2015) Acetaminophen hepatotoxicity: an updated review. Arch Toxicol 89:193–199. [DOI] [PubMed] [Google Scholar]
- Lauterburg BH, Corcoran GB, Mitchell JR. (1983) Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 71:980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Barber DS, Gavin T. (2008) Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Toxicol Sci 104:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T. (2014) Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol 27:1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Decaprio A, Barber DS. (2012) Application of the Hard and Soft, Acids and Bases (HSAB) theory to toxicant--target interactions. Chem Res Toxicol 25:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Geohagen BC, Das S. (2007) Neurotoxic mechanisms of electrophilic type-2 alkenes: soft soft interactions described by quantum mechanical parameters. Toxicol Sci 98:561–570. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Geohagen BC, Zhang L, Casper D, Lekhraj R, Barber DS. (2011) β-dicarbonyl enolates: a new class of neuroprotectants. J Neurochem 116:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. (2009b) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol 22:1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Geohagen BC, Gavin T. (2009a) Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Toxicol Sci 107:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudon GM. (2002) Chemistry of enolate ions, enols and α,β-unsaturated carbonyl compounds, in Organic Chemistry, 4th ed, pp 997–1068, Oxford University Press, New York. [Google Scholar]
- Mammino L. (2013) Investigation of the antioxidant properties of hyperjovinol A through its Cu(II) coordination ability. J Mol Model 19:2127–2142. [DOI] [PubMed] [Google Scholar]
- Mansour HH, El Kiki SM, and Hasan HF (2015) Protective effect of N-acetylcysteine on cyclophosphamide-induced cardiotoxicity in rats. Environ Toxicol Pharmacol 40:417–422. [DOI] [PubMed] [Google Scholar]
- Massey TE, Racz WJ. (1981) Effects of N-acetylcysteine on metabolism, covalent binding, and toxicity of acetaminophen in isolated mouse hepatocytes. Toxicol Appl Pharmacol 60:220–228. [DOI] [PubMed] [Google Scholar]
- Mathiesen L, Malterud KE, Sund RB. (1997) Hydrogen bond formation as basis for radical scavenging activity: a structure-activity study of C-methylated dihydrochalcones from Myrica gale and structurally related acetophenones. Free Radic Biol Med 22:307–311. [DOI] [PubMed] [Google Scholar]
- Payton F, Sandusky P, Alworth WL. (2007) NMR study of the solution structure of curcumin. J Nat Prod 70:143–146. [DOI] [PubMed] [Google Scholar]
- Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA, Hinson JA. (2005) Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther 312:509–516. [DOI] [PubMed] [Google Scholar]
- Rezk BM, Haenen GRMM, van der Vijgh WJF, Bast A. (2002) The antioxidant activity of phloretin: the disclosure of a new antioxidant pharmacophore in flavonoids. Biochem Biophys Res Commun 295:9–13. [DOI] [PubMed] [Google Scholar]
- Rosen GM, Rauckman EJ, Ellington SP, Dahlin DC, Christie JL, Nelson SD. (1984) Reduction and glutathione conjugation reactions of N-acetyl-p-benzoquinone imine and two dimethylated analogues. Mol Pharmacol 25:151–157. [PubMed] [Google Scholar]
- Saito C, Zwingmann C, Jaeschke H. (2010) Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Bai N, He K, Ho CT, Yang CS, Sang S. (2008) Apple polyphenols, phloretin and phloridzin: new trapping agents of reactive dicarbonyl species. Chem Res Toxicol 21:2042–2050. [DOI] [PubMed] [Google Scholar]
- So MJ, Cho EJ. (2014) Phloroglucinol attenuates free radical-induced oxidative stress. Prev Nutr Food Sci 19:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachulski AV, Baillie TA, Park BK, Obach RS, Dalvie DK, Williams DP, Srivastava A, Regan SL, Antoine DF, Goldring CEP, et al. (2013) The generation, detection, and effects of reactive drug metabolites. Med Res Rev 33:985–1080. [DOI] [PubMed] [Google Scholar]
- Vajragupta O, Boonchoong P, Morris GM, Olson AJ. (2005) Active site binding modes of curcumin in HIV-1 protease and integrase. Bioorg Med Chem Lett 15:3364–3368. [DOI] [PubMed] [Google Scholar]
- Weber WM, Hunsaker LA, Gonzales AM, Heynekamp JJ, Orlando RA, Deck LM, Vander Jagt DL. (2006) TPA-induced up-regulation of activator protein-1 can be inhibited or enhanced by analogs of the natural product curcumin. Biochem Pharmacol 72:928–940. [DOI] [PubMed] [Google Scholar]
- Yang X, Greenhaw J, Ali A, Shi Q, Roberts DW, Hinson JA, Muskhelishvili L, Beger R, Pence LM, Ando Y, et al. (2012) Changes in mouse liver protein glutathionylation after acetaminophen exposure. J Pharmacol Exp Ther 340:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Greenhaw J, Shi Q, Roberts DW, Hinson JA, Muskhelishvili L, Davis K, Salminen WF. (2013) Mouse liver protein sulfhydryl depletion after acetaminophen exposure. J Pharmacol Exp Ther 344:286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Kaplowitz N. (2013) Mechanisms of drug-induced liver injury. Clin Liver Dis 17:507–518, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Gavin T, Geohagen BC, Liu Q, Downey KJ, LoPachin RM. (2013) Protective properties of 2-acetylcyclopentanone in a mouse model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther 346:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Sun Z, Jiang Y, Chen F, Wang M. (2011) Acrolein scavengers: reactivity, mechanism and impact on health. Mol Nutr Food Res 55:1375–1390. [DOI] [PubMed] [Google Scholar]