

Abstract
Gonadal dysgenesis and Mayer-Rokitansky-Küster-Hauser syndrome (MRKHS) are the most common causes of primary amenorrhoea. Patients with gonadal dysgenesis present with primary amenorrhoea and lack of secondary sexual characteristics, which, in contrast, are present in patients with MRKHS. The coexistence of the 2 syndromes has been reported in only a few studies so far. We describe a case of a 15-year-old girl who presented with short stature and primary amenorrhoea. Investigations revealed hypergonadotropic hypogonadism, and absence of the uterus, and upper two-thirds of the vagina, with presence of the rudimentary lower third of the vagina and non-visualised bilateral ovaries on imaging. Karyotyping obtained by lymphocyte culture GTG banding revealed 45X/46XX. The patient was diagnosed as having a rare case of gonadal dysgenesis with MRKH. She was started on growth hormone therapy. The association of these syndromes is uncommon, and has further implications on fertility and pregnancy, affecting the quality of life.
Background
Gonadal dysgenesis (Turner syndrome) in females is defined as absent or insufficient development of the ovaries. These individuals can have varied karyotypes in the form of 46XX, 45XO, mosaicism or deletion of a certain part of the X chromosome.1 Patients usually present with primary amenorrhoea and lack of development of secondary sexual characteristics due to inability of the ovaries to produce sex steroids. The overall incidence is about 1:2500 live birth females.
Mayer-Rokitansky-Küster-Hauser syndrome (MRKHS) is characterised by an absent or hypoplastic uterus and upper two-thirds of the vagina in phenotypically and karyotypically normal females, with incidence of ∼1 case per 5000 newborn females.2 A patient with MRKHS has normal secondary sexual characteristics due to normally functioning ovaries.
The association of gonadal dysgenesis and MRKHS, though reported, remains rare.
Case presentation
We report a case of a 15-year-old girl who presented to the outpatient department, with reports of inability to gain height after 10 years of age. She was a full term normal vaginal delivery born out of a non-consanguineous marriage. The patient had normal mentation with no behavioural abnormalities and had not attained menarche. There was no history of antenatal stress events, or any chronic illness in the mother or the patient. There was no family history of genetic disease. The patient had been fully vaccinated. Her elder sister and younger brother were healthy and taller than the patient (figure 1).
Figure 1.

Patient (middle) is shorter than younger and older siblings (from left to right).
On examination, her height was 125 cm, weight 38 kg and mid-parental height was 154 cm. She had a short fourth metacarpal of the right hand (figure 2). There was presence of a shield chest with widely placed nipples (figure 3), however, no webbing of the neck and no wide carrying angle at the elbow were present. Her pubic hair and breast development were Tanner stage 1. Her blood pressure was 116/70 mm Hg with no difference in upper and lower limbs, and pulse was 72/min. Pulmonary, cardiovascular and abdominal examination was normal.
Figure 2.
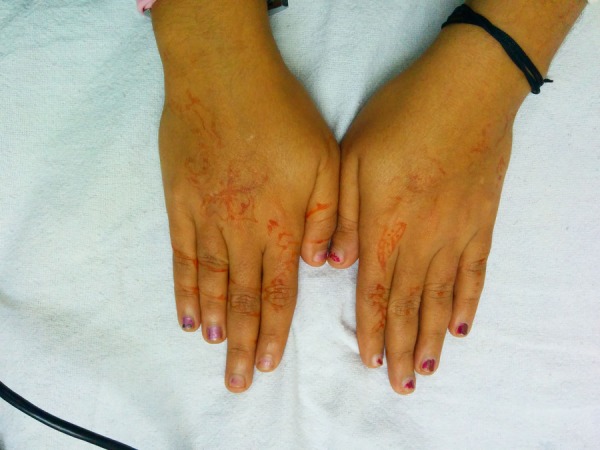
Short fourth metacarpal of the right hand.
Figure 3.
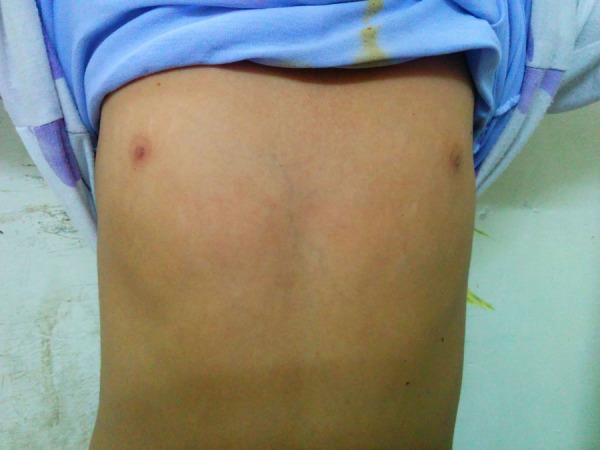
Shield chest.
Investigations
Investigations revealed haemoglobin of 13.3 g%, total leucocyte count 8900 mm3, platelet count 2.44 mm3 and erythrocyte sedimentation rate 12 mm/hour. Lipid profile, liver function and kidney function tests were normal. Her calcium was 9.4 mg/dL (8.7–10.2 mg/dL), phosphorus 5.0 mg/dL (2.5–4.3 mg/dL), alkaline phosphatase of 296 U/L (33–96 U/L) and bone alkaline phosphatase >140 U/L. Hormonal profile showed suppressed intact parathyroid hormone levels of 2.8 pg/mL (8–52 pg/mL), elevated luteinising hormone 33.9 mIU/mL (2–15 mIU/mL) and follicle-stimulating hormone 72.9 mIU/mL (3–20 mIU/mL). Thyroid function tests were normal. Bone age was estimated radiologically to be 8.5–12 years. Ultrasound of the pelvis did not show the uterus and ovaries. MRI of the pelvis revealed absence of the uterus, and upper two-thirds of the vagina, with presence of the rudimentary lower third of the vagina and non-visualised bilateral ovaries (figures 4 and 5). The kidneys on abdomen MRI were normal in size, shape and signal intensity (figure 6). Karyotyping obtained by lymphocyte culture GTG banding revealed 45XO/46XX.
Figure 4.
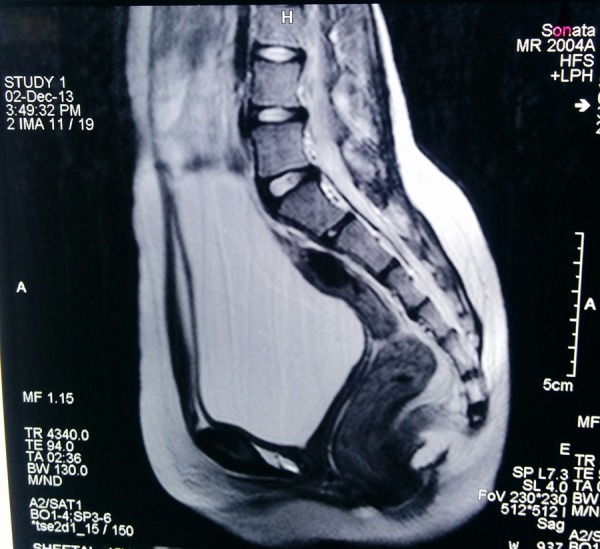
Mid-sagittal T2-weighted image showing absence of uterine tissue.
Figure 5.
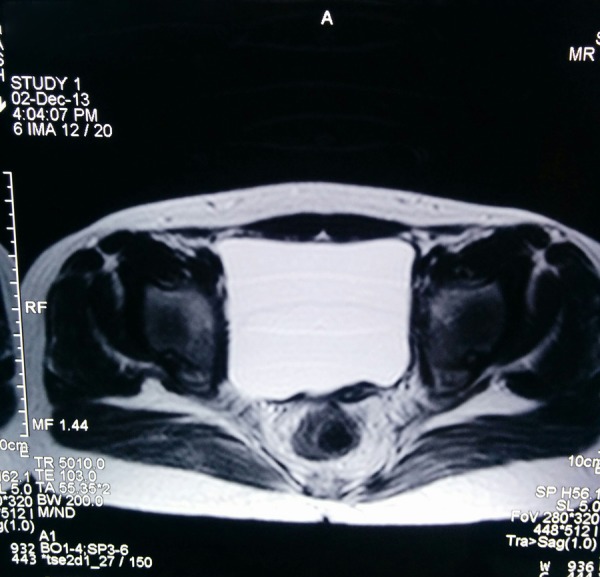
Axial T2-weighted image shows absence of uterine and ovarian tissue.
Figure 6.
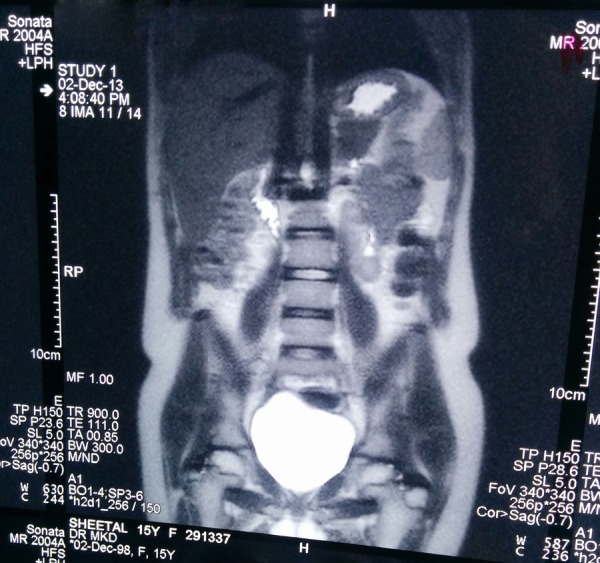
Both kidneys are normal in size, position and signal intensity.
Treatment
Following the diagnosis, the patient was started on replacement therapy with recombinant growth hormone (GH) (0.05 mg/kg/day subcutaneously) after endocrinology consultation. She has currently been on therapy for 12 months and is doing well.
Outcome and follow-up
The patient has been on GH therapy for 12 months and has achieved a growth velocity of 10 cm/year, without showing signs of complications from treatment.
Discussion
Gonadal dysgenesis is the most common cause of primary amenorrhoea and absent secondary sexual characteristics.3 This may arise from an early defect in primordial follicle formation or a defect in differentiation of the ovaries. Patients usually present with short stature and have streak or immature ovaries. Other features classically described include webbed neck, low set ears, shield-like chest, broadly placed nipples, cubitus valgus, lymphoedema of hands and feet, nail dysplasia, genu valgum, etc. There is no clear association with any established risk factors, maternal age, or familial inheritance pattern. Diagnosis is established on the basis of karyotype analysis. Deletion of the whole (45X) or part of one X chromosome will invariably be the basis of the definitive diagnosis. GH therapy should be considered in all girls with Turner syndrome whose height is below the 95th centile and who do not exhibit significant catch-up on growth charts. Oestrogen therapy in these patients can help in sexual development and bone maturation. In vitro fertilisation using donor oocytes has been successfully employed with good pregnancy outcomes for these patients.
Müllerian agenesis, on the other hand, presents with normal secondary sexual characters and developmental anomalies of the female genital tract including an absent/malformed uterus and upper two-thirds of the vagina (MRKHS syndrome). However, due to different embryonic origin, the lower third of the vagina is always present. It is also associated with urogenital, skeletal or dental defects and other dysmorphias. These individuals have normal female genotype 46 and XX, and they ovulate normally. The most common presentation is primary amenorrhoea. The levels of luteinising hormone and follicle-stimulating hormone are normal due to normally functioning ovaries. Diagnosis is established on the basis of clinical findings, MRI of the pelvis and/or laparoscopy and karyotype analysis. However, MRI of the pelvis is preferred over diagnostic laparoscopy as it is non-invasive, gives better soft tissue contrast resolution and multiplanar capability, and is cost effective. With appropriate reconstructive surgeries (creating a neovagina), these patients can have a normal sex life.
This rare association of the two syndromes has been described in 25 published case reports so far.2–25 A brief summary of presentation is shown in table 1.
Table 1.
Review of published cases showing association of Turner syndrome and Mayer-Rokitansky-Küster-Hauser syndrome
| Author (year) | Age of presentation (years) | Karyotype | Ovaries | Uterus | Fallopian tubes | Consanguinity | |
|---|---|---|---|---|---|---|---|
| 1 | Bhandari and Chaudhary (2015)4 | 17 | 46XX | Agenetic | Absent | NR | Absent |
| 2 | Białka et al (2016)5 | 17 | 46,X/X(q10) | Dysgenetic | Hypoplasia | NR | Absent |
| 3 | Kebaili et al (2013)6 | 21 | 46XX | Agenetic | Absent | Absent | Absent |
| 4 | Viral et al (2013)7 | 21 | 46XX | Agenetic | Absent | Absent | Absent |
| 5 | Bousfiha et al (2010)3 | 19 | 46XX | Dysgenetic | Absent | Absent | Absent |
| 6 | Tatar et al (2009)2 | 2 sister (34 and 23) | 46XX | Agenetic | Hypoplasia | Hypoplasia | Present |
| 7 | Zaman and Nisar (2009)8 | 2 sisters (22 and 13) | 46XX | Dysgenetic | One absent The other, rudimentary |
Hypoplasia | Present |
| 8 | Güvan et al (2008)9 | 17 | 45,X/46,X delX (p11.21) | Agenetic | Absent | NR | Absent |
| 9 | Kumar et al (2007)10 | 18 | 46XX | Right side, Agenetic | Absent | NR | Absent |
| 10 | Colombani et al (2007)11 | 15 | 46XX | Dysgenetic | Absent | N | Present |
| 11 | Marrakchi et al (2004)12 | 19 | 46XX | Dysgenetic | Absent | N | Absent |
| 12 | Plevraki et al (2004)13 | 6 patients | 46XX with testis specific protein 1-Y linked gene (in Patient 1 and 4) | Patient 1: left side, Agenetic Patient 6: Agenetic |
Patient 1: hypoplastic uterus with symmetrical uterine buds, with no endometrium Patient 6: uterus, symmetrical hypoplastic |
Patient 1: left fallopian tube, absent Patient 6: both fallopian tubes were symmetric, but hypoplastic |
Absent |
| 13 | Kaya et al (2003)14 | 17 | 46XX | Left Agenetic | Absent | Right, normal Left, hypoplastic |
Absent |
| 14 | Aydos et al (2003)15 | 19 | Agenetic | Rudimentary | NR | Absent | |
| 15 | Mégarbané et al (2003)16 | 2 sisters | 46XX | Dysgenetic | Hypoplasia | Hypoplastic | Present |
| 16 | Gorgojo et al (2002)17 | 17 | 46XX | Agenetic | Absent | Absent | Absent |
| 17 | Ting and Chang (2002)18 | 22 | 45x/46x, del(X) (p22.22) | Dysgenetic | Absent | Rudimentary | Absent |
| 18 | Güitrón-Cantú et al (1999)19 | 19 | 45,X/46,Xdic(X) | Agenetic | Absent | N | Absent |
| 19 | Güitrón-Cantú et al (1999)19 | 19 | 45,X/46,Xdic(X) | Agenetic | Absent | N | Absent |
| 20 | Oyer et al (1994)20 | Neonate | 46XX | Agenetic | Defects in Müllerian derivatives | NA | Absent |
| 21 | Aughton (1993)21 | NA | 46XX | Dysgenetic | Absent | Absent | Absent |
| 22 | Alper et al (1985)22 | 16 | NA | Dysgenetic | Absent | NA | Absent |
| 23 | Al-Awadi et al (1985)23 | 2 sisters (18 and 16) | 46XX | One Agenetic The other, dysgenetic |
Hypoplastic | One, absent, The other, hypoplastic |
Present |
| 24 | De Leon et al (1984)24 | NA | 46,X,i(Xq) | Agenetic | Absent | NR | Absent |
| 25 | Levinson et al (1976)25 | 17 | 46XX | Agenetic | Absent | Absent | Absent |
N, normal; NA, not available; NR, not reported.
It is difficult to postulate a common theory for coexistence of these two anomalies in the same patient. Mutations of the gene encoding the anti-Müllerian hormone receptor (AMH-R) and the lack of oestrogen receptors during embryonic development have been hypothesised to cause MRKH syndrome.17 However, mutation in neither AMH nor AMH-R has been found. This combination of Turner syndrome and MRKH is a double blow to the patient, with very poor chances of conception. Hormone substitution therapy remains the only therapeutic option; it aims at triggering the development of secondary sexual characters and preventing osteoporosis. However, the unsolved problem of infertility remains.12
Learning points.
It is important to consider Turner syndrome in all cases of short stature and primary amenorrhoea.
Early diagnosis of the condition might help in sensitising caregivers to understand the importance of instituting hormonal therapy for feminisation and bone health during adult life to improve the quality of life.
Coexisting Müllerian agenesis in these patients can jeopardise the chances of future pregnancy as they are associated with structural abnormalities of the uterus and vagina.
Footnotes
Contributors: AM was the chief author and investigator and takes full responsibility for the publication. MKD supervised the writing and proofread the manuscript. RD provided input on the radiological images.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Bhuyan AK, Sarma D, Saikia UK. Contemporary issues in primary amenorrhea: an experience from a tertiary care center. Indian J Endocrinol Metab 2012;16:S387–8. 10.4103/2230-8210.104103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tatar A, Ocak Z, Tatar A et al. Primary hypogonadism, partial alopecia, and Mullerian hypoplasia: report of a third family and review. Am J Med Genet A 2009;149A:501–4. 10.1002/ajmg.a.32645 [DOI] [PubMed] [Google Scholar]
- 3.Bousfiha N, Errarhay S, Saadi H et al. Gonadal dysgenesis 46, XX Associated with Mayer-Rokitansky-Kuster-Hauser Syndrome: one case report. Obstet Gynecol Int 2010;2010:847370 10.1155/2010/847370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhandari B, Chaudhary BK. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster- Hauser syndrome in a girl with a 46, XX karyotype: a case report. Int J Contemp Pediatr 2015;2:246–8. [Google Scholar]
- 5.Białka A, Gawlik A, Drosdzol-Cop A et al. Coexistence of Mayer-Rokitansky- Küster-Hauser Syndrome and Turner's Syndrome: a case report. J Paediatr Adolesc Gynaecol 2016;29:e35–8. 10.1016/j.jpag.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 6.Kebaili S, Chaabane K, Mnif MF et al. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser Syndrome in a girl with a 46, XX karyotype: a case report and review of literature. Indian J Endocrinol Metab 2013;17:505–8. 10.4103/2230-8210.111663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viral S, Parth G, Rajni P et al. Coexistence of gonadal dysgenesis and Mayer- Rokitansky-Kuster-Hauser syndrome in 46, XX female: a case report and review of literature. Indian J Endocrinol Metab 2013;17(Suppl 1):S274–7. 10.4103/2230-8210.119605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaman SM, Nisar M. Primary hypergonadotrophic hypogonadism, alopecia totalis, and müllerian hypoplasia: a clinical study. J Pak Med Assoc 2009;59:571–3. [PubMed] [Google Scholar]
- 9.Güven A, Kara N, Saðlam Y et al. The Mayer-Rokitansky-Kuster-Hauser and gonadal dysgenesis anomaly in a girl with 45, X/46, X, del (X) (p11.21). Am J Med Genet A 2008;146A:128–31. 10.1002/ajmg.a.32048 [DOI] [PubMed] [Google Scholar]
- 10.Kumar A, Mishra S, Dogra PN. Management of an unusual case of atypical Mayer-Rokitansky-Kuster-Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J Pelvic Floor Dysfunct 2007;18:823–5. 10.1007/s00192-006-0238-z [DOI] [PubMed] [Google Scholar]
- 11.Colombani M, Cau D, Sapin E et al. Fourth case of uterine aplasia, ovarian dysgenesis, amenorrhea and impuberism: a variant of Mayer-Rokitansky-Kuster-Hauser syndrome. Acta Paediatr 2007;96:1371–2. 10.1111/j.1651-2227.2007.00404.x [DOI] [PubMed] [Google Scholar]
- 12.Marrakchi A, Gharbi MH, Kadiri A. Gonadal dysgenesis associated with Mayer-Rokitansky-Küster-Hauser syndrome: a case report. Ann Endocrinol (Paris) 2004;65:466–8. 10.1016/S0003-4266(04)95953-7 [DOI] [PubMed] [Google Scholar]
- 13.Plevraki E, Kita M, Goulis DG et al. Bilateral ovarian agenesis and the presence of the testis-specific protein 1-Y-linked gene: two new features of Mayer-Rokitansky-Küster- Hauser syndrome. Fertil Steril 2004;81:689–92. 10.1016/j.fertnstert.2003.07.029 [DOI] [PubMed] [Google Scholar]
- 14.Kaya H, Sezik M, Ozkaya O et al. Mayer-Rokitansky-Küster-Hauser syndrome associated with unilateral gonadal agenesis. A case report. J Reprod Med 2003;48:902–4. [PubMed] [Google Scholar]
- 15.Aydos S, Tükün A, Bökesoy I. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster- Hauser syndrome in a girl with 46, X, del (X)(pter—>q22:). Arch Gynecol Obstet 2003;267:173–4. 10.1007/s00404-001-0274-3 [DOI] [PubMed] [Google Scholar]
- 16.Mégarbané A, Gannagé-Yared MH, Khalifé AA et al. Primary hypergonadotropic hypogonadism, partial alopecia, and müllerian hypoplasia: report of a second family with additional findings. Am J Med Genet A 2003; 119A:214–17. [DOI] [PubMed] [Google Scholar]
- 17.Gorgojo JJ, Almodóvar F, López E et al. Gonadal agenesis 46,XX associated with the atypical form of Rokitansky syndrome. Fertil Steril 2002;77:185–7. [DOI] [PubMed] [Google Scholar]
- 18.Ting TC, Chang SP. Coexistence of gonadal dysgenesis and Mullerian agenesis with two mosaic cell lines 45, X/46, X, del (X) (p22.2). Zhonghua Yi Xue Za Zhi (Taipei) 2002;65:450–2. [PubMed] [Google Scholar]
- 19.Güitrón-Cantú A, López-Vera E, Forsbach-Sánchez G et al. Gonadal dysgenesis and Rokitansky syndrome. A case report. J Reprod Med 1999;44:891–3. [PubMed] [Google Scholar]
- 20.Oyer CE, Ramos D, Shoji T et al. 46, XX gonadal agenesis in a neonate with multiple congenital anomalies: case report and review of the literature. Pediatr Pathol 1994;14:967–72. [DOI] [PubMed] [Google Scholar]
- 21.Aughton DJ. Müllerian duct abnormalities and galactosaemia heterozygosity: report of a family. Clin Dysmorphol 1993;2:55–61. [PubMed] [Google Scholar]
- 22.Alper MM, Garner PR, Spence JE. Coexistence of gonadal dysgenesis and uterine aplasia. A case report. J Reprod Med 1985;30:232–4. [PubMed] [Google Scholar]
- 23.Al-Awadi SA, Farag TI, Teebi AS et al. Primary hypogonadism and partial alopecia in three sibs with müllerian hypoplasia in the affected females. Am J Med Genet 1985;22:619–22. 10.1002/ajmg.1320220322 [DOI] [PubMed] [Google Scholar]
- 24.De Leon FD, Hersh JH, Sanfilippo JS et al. Gonadal and müllerian duct agenesis in a girl with 46, X, i (Xq). Obstet Gynecol 1984;63:81S–3 S. [PubMed] [Google Scholar]
- 25.Levinson G, Zárate A, Guzmán-Toledano R et al. An XX female with sexual infantilism, absent gonads, and lack of Müllerian ducts. J Med Genet 1976;13:68–9. [DOI] [PMC free article] [PubMed] [Google Scholar]