

Abstract:
Background:
Diabetes aggravates brain injury after cerebral ischemia/reperfusion (I/R).
Objective:
To investigate whether limb I/R causes cerebral injury in a rat diabetes model and whether glycogen synthase kinase-3β (GSK-3β) is involved.
Methods:
Male adult Sprague-Dawley rats were assigned into streptozotocin-induced diabetes (n = 30; blood glucose ≥16.7 mmol/L) or control (n = 20) groups, further subdivided into diabetes I/R (3-hour femoral artery/vein clamping), diabetes-I/R + TDZD-8 (I/R plus GSK-3β inhibitor), diabetes-sham, control-sham and control-I/R groups (n = 10 each). Cortical and hippocampal morphology (hematoxylin/eosin); hippocampal CA1 apoptosis (TUNEL assay); cleaved caspase-3 (apoptosis), and Iba1 (microglial activation) protein expression (immunohistochemistry); phosphorylated/total GSK-3β and nuclear factor-κB (NF-κB) protein levels (Western blotting); and serum and brain tissue tumor necrosis factor (TNF)-α levels (enzyme-linked immunosorbent assay) were analyzed.
Results:
The diabetes-I/R group showed greater cortical and hippocampal injury, apoptosis, cleaved caspase-3 expression and Iba1 expression than the control-I/R group; TDZD-8 reduced injury/apoptosis and cleaved caspase-3/Iba1 expressions. The diabetes-I/R group had lower p-GSK-3β and p-NF-κBp65 expression than the control-I/R group (P < 0.05); TDZD-8 increased p-GSK-3β expression but decreased p-NF-κBp65 expression (P < 0.05). The diabetes-I/R group showed higher elevation of serum and brain tissue TNF-α than the control-I/R group (P < 0.05); TDZD-8 reduced TNF-α production.
Conclusions:
Diabetes exacerbates limb I/R-induced cerebral damage and activates NF-κB and GSK-3β.
Key Indexing Terms: Glycogen synthase kinase-3β (GSK-3β), Diabetes mellitus, Ischemia/reperfusion, Cerebral injury, Nuclear factor-κB (NF-κB), Tumor necrosis factor-α (TNF-α)
Diabetes is a systemic disease that can cause chronic and progressive destruction of many organs, including the eyes, kidneys, cardiovascular system and peripheral nervous system. More recently, increasing attention has been paid to the central nervous system complications of diabetes,1 which include hippocampal damage,2 impairments in learning and memory3,4 and an enhanced risk of age-related neurodegeneration.5 A variety of factors have been implicated in the cognitive deficits associated with diabetes.1 Interestingly, diabetes has also been reported to aggravate brain injury after cerebral ischemia/reperfusion (I/R), which is characterized by neuronal cell apoptosis.6
Lower limb ischemia is a common scenario encountered in clinic practice that can have a variety of underlying causes (including peripheral blood vessel embolism or damage, severe injury to the lower limb or inappropriate use of a tourniquet). Although restoration of the blood supply is the premise for rescuing the ischemic limb, the reperfusion can itself induce I/R injury, a metabolic disorder associated with structural destruction of the ischemic tissues. Clinical and animal studies have shown that severe I/R injury not only damages the limb skeletal muscle but can also induce systemic inflammatory response syndrome and even remote multiple organ dysfunction syndrome7 affecting the lungs, heart, liver and intestine. The brain is also sensitive to I/R injury, and there is evidence in an animal model that hindlimb I/R can cause a systemic inflammatory response that results in microglial cell activation and neuronal apoptosis.8
Glycogen synthase kinase-3β (GSK-3β), one of the rate-limiting enzymes in glycogen synthesis, is a serine/threonine protein kinase that is highly expressed in the central nervous system, especially in the hippocampus and neocortex. GSK-3β has almost 50 substrates, including plasmosin and nuclear transcription factors and can modulate transduction in multiple cell signaling pathways closely related to diabetes, I/R, inflammation, neurodegenerative disease and cancer.9 GSK-3β has been shown to have critical functions in regulating the balance between cell survival and apoptosis.10 An important feature of GSK-3β is that it has intracellular catalytic activity in the absence of any stimuli, and that its kinase activity can be enhanced by phosphorylation at Thr-216. Under normal conditions, the activity of GSK-3β is downregulated by cell survival signals, including insulin and growth factors, which act through the phosphoinositide 3-kinase (PI3K)/Akt pathway. Hence, excessive activation or inadequate suppression of GSK-3β can induce apoptosis.
GSK-3β has been implicated in the cognitive changes observed in models of diabetes and indeed has been proposed to be a molecular link between insulin, diabetes and Alzheimer's disease.11 In streptozotocin (STZ)- or pancreatectomy-induced rat models of diabetes, the activity of hippocampal GSK-3β is significantly higher than that in normal rats,12 whereas learning and memory functions are decreased.13 Furthermore, treatment with a GSK-3β inhibitor has been reported to improve learning and memory in rats with diabetes.14 Inflammation induced by I/R injury can aggravate the destruction of neurons and GSK-3β may be relevant to this process, as it exerts critical functions in modulating the balance between proinflammatory and anti-inflammatory cytokine production in the central nervous system.15 For example, GSK-3β, nuclear factor-κB (NF-κB) and tumor necrosis factor-α (TNF-α) have been suggested to play a role in inflammation and apoptosis of neurons and microglia.16,17 Furthermore, inhibiting GSK-3β in microglial cells can block the inflammatory response stimulated by activation of NF-κB and production of TNF-α and enhance the levels of anti-inflammatory cytokines.18
Many patients with diabetes will require surgery and anesthesia, and limb I/R injury is not rare. The authors have observed that patients with diabetes have a lower tolerance to surgery and anesthesia and a poorer prognosis than patients without diabetes. Insulin resistance has been shown to exacerbate cortical injury during ischemia,19 and importantly, GSK-3β, NF-κB and TNF-α have been implicated as having a role.20 Furthermore, Albadawi et al21 have reported that mice with diet-induced obesity have an exaggerated acute response to hindlimb I/R, with an enhanced metabolic deficit, fat accumulation and defective functional recovery during the regenerative phase of I/R. However, it is not currently known whether the detrimental effects of lower limb ischemia on the brain are exacerbated if diabetes is also present, and if so, which mechanisms contribute to this phenomenon.
The aim of this study was to investigate whether limb I/R can cause cerebral injury in a rat model of diabetes and to explore the potential mechanisms that may be involved with a focus on GSK-3β signaling.
METHODS
Animals and Grouping
Fifty male Sprague-Dawley rats, weighing 300 to 350 g, were obtained from the Experimental Animal Center of Guangdong Province, China. All rats were housed under specific pathogen-free conditions in the Medical Laboratory Animal Center, Guangzhou University of Traditional Chinese Medicine, Guangdong, China. The animals were kept in a quiet environment with a 12:12 hour light-dark cycle, under temperature- (21–24°C) and humidity- (40%–70%) controlled conditions.
The rats were divided randomly into 2 groups: a T1DM group (n = 30) and a control group (n = 20). The rats in the T1DM group were divided randomly into 3 subgroups (n = 10 for each): a sham group, an I/R group and an I/R + TDZD-8 group, whereas rats in the control group were divided randomly into 2 subgroups (n = 10 for each): a sham group and an I/R group. Rats in the T1DM group were treated with STZ to induce diabetes, whereas rats in the control group were not treated with STZ. Animals in the I/R groups (both control and T1DM) were exposed to surgery (limb I/R model) to induce experimental I/R, whereas animals in the I/R + TDZD-8 group were injected intravenously with 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8; a selective GSK-3β inhibitor) 5 minutes before the surgery. A sham operation was performed in the sham group of rats.
Establishment of a Murine Model of T1DM
All rats were acclimatized to their environment for 1 week before any experiments were undertaken. The health condition of the rats was monitored before treatment with STZ was initiated, and only rats maintaining a normal diet, no body weight loss and a normal blood glucose level for 1 week were used for establishment of the murine model of T1DM.
During the 1st week of the experimental procedure, the rats in the T1DM and control groups were fasted for 12 hours and their body weights recorded; the T1DM group animals were then given a single intraperitoneal injection of 1% STZ (30 mg/kg; Sigma, Sigma-Aldrich, St. Louis, MO) diluted in sodium citrate–hydrochloric acid buffer solution (pH = 4.5; Sigma), whereas the control group received the vehicle, sodium citrate–hydrochloric acid buffer solution (pH = 4.5; 3 mL/kg; Sigma). One week later, animals in the T1DM group were given a single intraperitoneal injection of 1% STZ (50 mg/kg), whereas the control group received the vehicle, sodium citrate–hydrochloric acid buffer solution (pH = 4.5; 5 mL/kg).
Blood Glucose Measurements and Criteria for Successful Establishment of the T1DM Model
Blood glucose levels were measured using a blood glucose monitor (Onetouch Ultra blood glucose meter; Johnson, TN) from blood obtained by tail nicking on day 3, week 1, week 2 and week 3 after the 2nd injection of STZ or buffer. Water intake, food intake, urine volume and body weight were monitored. Hyperglycemia was defined as a blood glucose level ≥16.7 mmol/L.
Limb I/R Model
Before surgery, the rats were fasted for 12 hours with free access to water. The animals were anesthetized with chloral hydrate (3 mL/kg, intraperitoneal). For rats in the I/R group, the femoral artery and vein were 1st exposed and ligated for 3 hours; the vascular clamps were then removed to restore the limb blood supply. A similar surgical procedure was used for rats in the sham group except that the vasculature was not ligated.
Therapy
Rats in the I/R + TDZD-8 group were injected intravenously with TDZD-8 (1 mg/kg, dissolved in 10% DMSO, 1 mg/mL) 5 minutes before the I/R surgery was performed; rats in the other groups received intravenous injection of 10% DMSO solution (1 mL/kg).
Histological Examination
Rats were deeply anesthetized with an intraperitoneal injection of 10% chloral hydrate (3 mL/kg) 4 hours after the limb I/R event. A median sternotomy was performed, blood collected from the right ventricle and the aorta was perfused with 400 mL heparinized saline, followed by cold 4% paraformaldehyde. Whole brains were carefully removed and sliced coronally at 30-μm intervals beginning at −3.6 mm from the Bregma. Sections were prepared for hematoxylin and eosin (H&E) staining, immunohistochemistry for Iba 1 and cleaved-caspase-3 and the TUNEL assay. The hippocampal CA1 regions were separated on wet ice snap-frozen in liquid nitrogen and stored at −80°C until enzyme-linked immunosorbent assay (ELISA) and Western blotting analysis.
H&E Staining
After tissue fixation, 3-μm tissue sections were obtained. The sections were stained with H&E using standard procedures and examined under the light microscope.
TUNEL Assay of Hippocampal Cell Apoptosis
The fixed tissues were embedded in paraffin and 3-μm tissue sections obtained. The sections were processed for the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay according to the manufacturer's protocol (Roche, Basel, Switzerland) using an in situ cell death detection fluorescein kit. In brief, the sections were incubated with the TUNEL reaction mixture in a humidified atmosphere for 60 minutes at 37°C in the dark and then washed 3 times with phosphate-buffered saline.
Each hippocampal section was 1st examined for cell apoptosis (magnification, 100×) and then 3 nonredundant views were selected (magnification, 200×) for the counting of positively staining cells and total cell number; the percentage of positively staining cells was then calculated. The average value for the percentage of positively staining cells was taken to represent the level of cell apoptosis. The apoptotic cells showed volume reduction, chromatin condensation and fragmentation to form apoptotic bodies.
Immunohistochemistry for Cleaved Caspsase-3 and Ionized Calcium-Binding Adapter Molecule 1
The hippocampal sections (3 μm thick) were stained with either rabbit anti-rat ionized calcium-binding adapter molecule 1 (Iba1) polyclonal antibody (Wako Pure Chemicals Industries, Ltd, Osaka, Japan) or rabbit anti-rat cleaved caspase-3 polyclonal antibody (Cell Signaling Technology, Danvers, MA). The sections were then incubated with goat anti-rabbit secondary antibody (BIOSS Company, Beijing, China). The staining of each hippocampal section was 1st examined at 100× magnification and then 3 nonredundant views were selected for further inspection at 200× magnification to count the number of positively staining cells and the total cell number; the percentage of positively staining cells was then calculated. The average value for the percentage of positively staining cells was taken to represent the expression level of cleaved caspase-3 or Iba1.
Immunoblotting
To determine the levels of GSK-3β, phosphorylated GSK-3β (p-GSK-3β), NF-κBp65 and phosphorylated NF-κBp65 (p-NF-κBp65) in the cortex and hippocampus, 50 mg of rat brain tissue was lysed in 500 μL of lysis buffer and incubated with rabbit anti-p-GSK-3β (1:1,000), mouse anti-GSK-3β (1:1,000), rabbit anti-p-NF-κBp65 (1:500) or mouse anti-NF-κBp65 (1:500) (all from Cell Signaling Technology).
ELISA Assay of TNF-α and IL-6 Levels
Each hippocampal tissue sample was accurately weighed, and 9 times the volume (ie, 9 mL per 1 g) of homogenization medium (0.9% saline) was added. Of note, 10% tissue homogenates were prepared mechanically and centrifuged at 10,000 rpm for 15 minutes at 4°C. The protein concentration of the supernatant from each sample was determined using a bicinchoninic acid protein assay kit (Enjing Biotech Ltd, Nanjing, China). The TNF-α levels were assayed using a TNF-α ELISA kit (Shanghai ExCell Bio Inc, Shanghai, China). The IL-6 levels were assayed in hippocampal tissue and rat serum using an IL-6 ELISA kit (Shanghai ExCell Bio Inc).
Statistical Analysis
All data were analyzed using SPSS statistical software version 18.0 (SPSS Inc, Chicago, IL) for Windows. Comparisons of TUNEL staining and immunohistochemical staining for cleaved caspase-3 and Iba1 were made using nonparametric tests; the Kruskal–Wallis H test was used to assess for differences between the experimental groups and the Mann-Whitney's U test for pairwise comparisons. One-way analysis of variance and Bonferroni correction was used for comparisons between the different groups in the results of the immunoblot and ELISA experiments. P < 0.05 was considered to be statistically significant.
RESULTS
Establishment of a Rat Diabetes Model
At the end of the 3-week experimental period, none of the animals had died. A murine model of diabetes was successfully established in all 30 rats in the T1DM group: after the establishment of diabetes, blood glucose levels in the T1DM group were significantly higher than those in the control group (P < 0.05 at 3 days, 1 week, 2 weeks and 3 weeks after I/R surgery; Table 1). Furthermore, within each of the T1DM and control groups, there were no significant differences in the blood glucose levels between weeks 1, 2 and 3 post-I/R surgery. Rats in the control group exhibited a moderate increase in body size (as would be expected for rats of this age), a smooth and glossy coat and no abnormalities in mental state, responsiveness or agility. In contrast, rats in the T1DM group showed signs of emaciation, a distended abdomen, slow responsiveness, bradykinesia, mental fatigue, dulling of their coat, fur loss, increased water and food intake and increased urine volume and odor. There was no significant difference in rat body weight between the T1DM and control groups before the establishment of diabetes; however, after the induction of diabetes, rats in the T1DM group had a significantly lower body weight (P < 0.05) than those in the control group (Table 1).
TABLE 1.
Body weights and blood glucose levels of rats in the T1DM and control groups
Diabetes Exacerbates the Brain Injury Induced by Limb I/R
The results of the H&E staining experiments (Figure 1A) suggested that the brain injury induced by I/R was worse in rats in the T1DM group than in those in the control group. I/R surgery in control rats (control-I/R group) induced swelling of most of the pyramidal cells of the brain cortex with decreases in the cytoplasmic and nuclear densities; some of the neurons showed loss of the nucleolus. Obvious lesions were evident in the hippocampal CA1 region with karyopyknosis, deep staining and the appearance of an edematous fissure around the cell. In the diabetes-sham group, there was shrinkage and deep staining of some of the cortical pyramidal cells and some of the neurons in the hippocampal CA1 region, with the appearance of a surrounding edematous fissure. Compared with the other groups, rats in the T1DM group exposed to I/R surgery showed an exacerbation of the lesions in the brain cortex, a degeneration of the hippocampal CA1 region and an increase in the number of deep-staining neurons (Figure 1A). Interestingly, the DM-I/R + TDZD-8 group (ie, rats treated with the GSK-3β inhibitor, TDZD-8) showed reduced degeneration of the brain cortex and hippocampal CA1 region and less pronounced neuronal staining compared with the DM-I/R group (Figure 1A).
FIGURE 1.

Cerebral injury induced by hindlimb ischemia–reperfusion is aggravated by diabetes. (A) Representative images of brain tissue from the hippocampal CA1 area taken from rats in the control-sham, control-I/R, T1DM-sham, T1DM-I/R and DM-I/R+TDZD-8 groups, showing (A) H&E staining, (B) TUNEL staining, (C) immunohistochemical staining of cleaved caspase-3 and (D) immunohistochemical staining of Iba1 (magnification, 200×). (E–G) Quantitative analysis of the data shown in (B–D), respectively. *P < 0.05 versus the control-sham group; #P < 0.05 versus the DM-I/R group.
The TUNEL assay revealed very few apoptotic cells in the control-sham group; in contrast, the proportion of apoptotic cells was significantly increased in the T1DM-sham, control-I/R and T1DM-I/R groups (P < 0.05) with the T1DM-I/R group exhibiting a significantly larger increase than the other groups (P < 0.05) (Figures 1B and 1E). In addition, the T1DM-I/R + TDZD-8 group had a significantly smaller percentage of apoptotic cells than the T1DM-I/R group (Figures 1B and 1E).
Immunohistochemistry demonstrated that the T1DM-sham, control-I/R and T1DM-I/R groups had significantly more cells positive for cleaved caspase-3 (Figures 1C and 1F) and Iba1 (Figures 1D and 1G) than the control-sham group (P < 0.05); the largest increases (for both cleaved caspase-3 and Iba1) occurred in the T1DM-I/R group (P < 0.05). Furthermore, TDZD-8 treatment significantly reduced the proportion of cells positive for cleaved caspase-3 (Figures 1C and 1F) or Iba1 (Figures 1D and 1G) compared with the T1DM-I/R group (P < 0.05).
GSK-3β/NF-κB/TNF-α Signaling Pathway Is Involved in the Process of Brain Cell Apoptosis
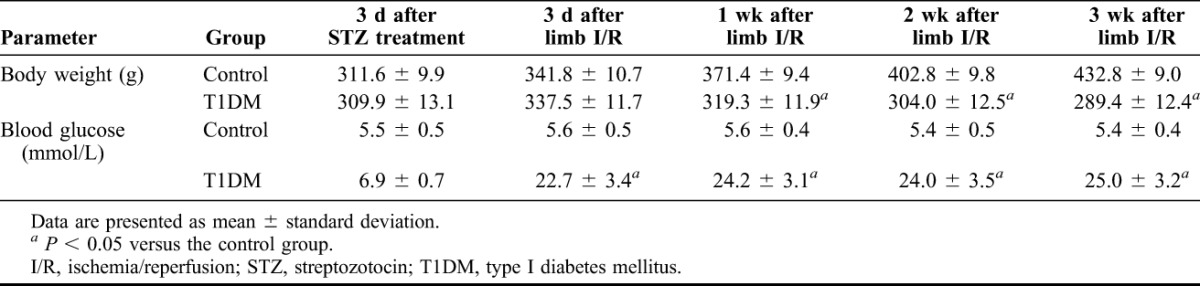
The results of the immunoblotting assay (Figure 2) showed that the expression of p-GSK-3β and p-NF-κBp65 proteins were significantly lower in the control-I/R, T1DM-sham and T1DM-I/R groups than in the control-sham group (P < 0.05); however, the expression of GSK-3β and NF-κBp65 did not differ significantly. The decreased expression of p-GSK-3β and p-NF-κBp65 were most pronounced in the T1DM-I/R group (P < 0.05). Furthermore, treatment with TDZD-8 significantly increased the expression of p-GSK-3β (P < 0.05) but significantly decreased the expression of p-NF-κBp65 (P < 0.05).
FIGURE 2.
Phosphorylation of GSK-3β and NF-κB is associated with the cerebral injury induced by hindlimb ischemia–reperfusion. (A) GSK-3β and p-GSK-3β levels, detected using immunoblotting, in the control-sham (C-S), control-I/R (C-I), T1DM-Sham (D-S), T1DM-I/R (D-I) and T1DM-I/R + TDZD-8 (D-I + T) groups. (B) Quantitative analysis of the GSK-3β and p-GSK-3β levels. (C) NF-κB and p-NF-κB levels, detected using immunoblotting, in the control-sham (C-S), control-I/R (C-I), T1DM-Sham (D-S), T1DM-I/R (D-I) and T1DM-I/R + TDZD-8 (D-I + T) groups. (D) Quantitative analysis of the NF-κB and p-NF-κB levels. *P < 0.05 versus the Control-sham group; #P < 0.05 versus the DM-L I/R group.
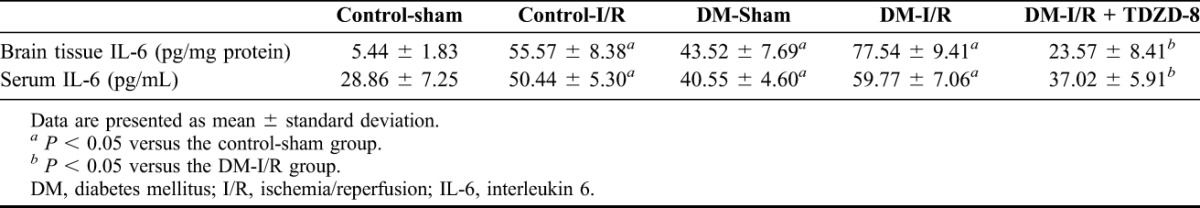
As presented in Table 2, the TNF-α levels in both the serum and brain tissue were significantly higher in the T1DM-sham, control-I/R and T1DM-I/R groups than in the control-sham group (P < 0.05), with the T1DM-I/R group showing the greatest increase (P < 0.05). Moreover, TDZD-8 significantly reduced the production of TNF-α (Table 2). The IL-6 levels as a measure of inflammation showed a similar pattern to the TNF-α with significantly higher levels in the T1DM-sham, control-I/R and T1DM-I/R groups than in the control-sham group (P < 0.05) and the T1DM-I/R group showing the greatest increase (P < 0.05). TDZD-8 also significantly reduced the production of IL-6 (Table 3).
TABLE 2.
TNF-α levels in the brain tissues and sera of rats in the various experimental groups
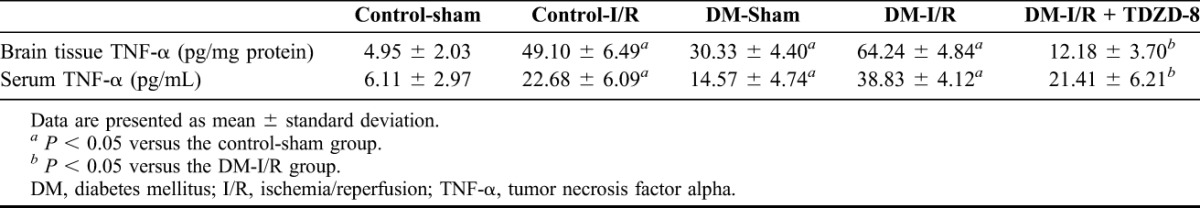
TABLE 3.
IL-6 levels in the brain tissues and sera of rats in the various experimental groups
DISCUSSION
The main findings of this study were that cortical and hippocampal injury, cellular apoptosis, cleaved caspase-3 expression and Iba1 expression were more pronounced in the T1DM-I/R group than in the control-I/R group and that TDZD-8 reduced the extent of the injury/apoptosis and cleaved caspase-3/Iba1 expressions. Moreover, the expression of p-GSK-3β and p-NF-κBp65 were lower in the T1DM-I/R group than in the control-I/R group, whereas TDZD-8 increased p-GSK-3β expression and decreased p-NF-κBp65 expression. Furthermore, serum and brain tissue TNF-α levels were elevated more substantially in the T1DM-I/R group than in the control-I/R group, whereas TDZD-8 reduced TNF-α production. Taken together, data indicate that diabetes can exacerbate the cerebral damage caused by limb I/R and that this may involve a mechanism recruiting GSK-3β, NF-κB and TNF-α.
The results demonstrated that limb I/R was associated with injury and apoptosis in the hippocampal CA1 region and in cortical areas, and that this was aggravated by diabetes. There is now substantial evidence that I/R injury comprises a series of inflammatory responses, induced by hypoxia of the ischemic limb and subsequent reperfusion, which can result in both local and remote damage.22 Although various remote organs, including the heart, brain, liver, lungs, kidneys and gastrointestinal tract, are potentially susceptible to injury,7 it is well known that pyramidal cells in the hippocampal CA1 region are particularly vulnerable to ischemia23 and hence may also be more prone to secondary injury from limb I/R. For this reason, the CA1 region in the hippocampus likely represents an important area of the brain that can be easily injured after I/R.
GSK-3β is an important protein that is modulated by the insulin-signaling transduction pathway. Insulin signaling can inactivate GSK-3β through the PI3K/Akt pathway; hence, the low-insulin levels associated with diabetes can result in a reduction in the level of GSK-3β phosphorylation and enhanced GSK-3β activation.24–26 In this study, the authors found a relationship between GSK-3β signaling and I/R injury and observed notable effects of TDZD-8, a non-ATP competitive GSK-3β inhibitor that increases GSK-3β phosphorylation.20 They found that treatment with TDZD-8 enhanced the phosphorylation of GSK-3β; decreased NF-κB activation, microglial cell activation, TNF-α expression in serum and hippocampal tissue, the number of cleaved caspase-3 positive cells and neuronal cell apoptosis and ameliorated I/R injury. Taken together, these results indicate that phosphorylation of GSK-3β may participate in the mechanisms by which diabetes exacerbates I/R injury.
There are many factors involved in inflammatory responses. NF-κB is an important component of inflammatory pathways and can initiate the production of various inflammatory mediators and function in cascade amplification. For this reason, the NF-κB–related pathway is considered to be a “control center” and the critical initiating step in the inflammatory response.27,28 Lin et al29 reported that I/R injury could phosphorylate NF-κBp65 to activate it. The involvement of NF-κB/TNF-α pathway is well-established in cerebral I/R injury.30,31 NF-κB is one of the most important transcription factors activated after cerebral ischemia. NF-κB is involved in inflammatory responses that potentiate ischemic injury activating many genes involved in the pathogenesis of cerebral ischemia, such as iNOS, IL-1β, TNF-α, ICAM-1, COX-2 and IL-6.32 This study found that limb I/R did not influence the expression of NF-κBp65 protein but increased the hippocampal levels of phosphorylated NF-κBp65 indicating significant activation of NF-κB in this brain region. Sustained NF-κBp65 activation would induce a substantial production of proinflammatory cytokines resulting in an exacerbation of brain injury.
TNF-α can be secreted by vascular endothelial cells, astrocytes and microglial cells in the central nervous system.33 It is a proinflammatory cytokine that can be produced during the early stages of I/R and can exert a variety of biological effects including the activation of many cascade reactions.34,35 The 2 possible sources of TNF-α after limb I/R are the affected limb itself or the microglial cells in the central nervous system. Despite the blood–brain barrier, cytokines can still enter the central nervous system through passive diffusion into the circumventricular organ, which lacks a blood–brain barrier or through active transport.36,37 In addition, microglial cells and infiltrating immune cells in the brain can also secret cytokines.38,39 This study found low TNF-α expression in the serum and brain tissue of control rats but significant increases in TNF-α levels in the serum and brain tissue after I/R indicating substantial activation of proinflammatory cytokine secretion. Furthermore, diabetes enhanced the elevation in TNF-α levels after I/R suggesting that diabetes can exacerbate the inflammatory responses induced by I/R. The authors also found a similar pattern with IL-6 levels supporting the role of inflammation in this study, but the role and expression patterns of other inflammatory indices and the role of treatment on inflammation will require further research.
Iba1 is a calcium-binding protein that can specifically bind to macrophages and microglial cells. An in vivo and ex vivo study has confirmed that only microglial cells express Iba1 in brain tissues.40 Iba1 is often used as a marker of microglial activation because activation of these cells during brain inflammation and injury is associated with enhanced expression of Iba1. In this study, the expression of Iba1 in control rats was low but increased notably after I/R indicating the enhanced activation of microglial cells.
A limitation of this study, shared by other investigations using animal models, is that the existing model cannot be expected to exactly imitate the clinical pathology that would be observed in human patients. There are 3 main methods for modeling I/R injury: the tourniquet model,41 the vascular clipping model42 and the McGivney hemorrhoidal ligator and orthodontic rubber ring model.43 This study used the vascular clipping model with the use of a tension band to block the collateral circulation. Furthermore, this model of streptozotocin-induced diabetes may not be representative of the mechanisms underlying diabetes in patients. There are many different rat models available for investigating the effects of diabetes each of which have different advantages or disadvantages.44 The authors used a traditional STZ-induced diabetes model but made modifications to establish the model in all of the rats without any resulting deaths. They used this as a straightforward model of diabetes establishment but whether this is truly representative of diabetes needs to be established in detail; other more established methods may have been better suited to this study. They also did not separate the cytoplasmic and nuclear cell fractions during the protein expression analysis; so, they cannot say whether transcription factors are binding to promoters; as required by these signaling pathways, the details of these mechanisms will have to be revealed in later studies. Nonetheless, this study provides important novel data implicating GSK-3β signaling in the mechanisms by which diabetes aggravates cerebral injury after lower limb I/R.
CONCLUSIONS
Limb I/R in this rat model can lead to cerebral damage. Furthermore, this brain injury is exacerbated by STZ-induced diabetes mellitus through a mechanism likely to involve activation of GSK-3β.
Footnotes
The authors have no conflicts of interest to disclose.
Supported by the Administration of Traditional Chinese Medicine of Guangdong Province Scientific Research Fund Project (20121055).
H.L. and S.O. contributed equally to this study and share first authorship.
REFERENCES
- 1.Sims-Robinson C, Kim B, Rosko A, et al. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 2010;6:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gold SM, Dziobek I, Sweat V, et al. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 2007;50:711–9. [DOI] [PubMed] [Google Scholar]
- 3.Cox DJ, Kovatchev BP, Gonder-Frederick LA, et al. Relationships between hyperglycemia and cognitive performance among adults with type 1 and type 2 diabetes. Diabetes Care 2005;28:71–7. [DOI] [PubMed] [Google Scholar]
- 4.Wrighten SA, Piroli GG, Grillo CA, et al. A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta 2009;1792:444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biessels GJ, van der Heide LP, Kamal A, et al. Ageing and diabetes: implications for brain function. Eur J Pharmacol 2002;441:1–14. [DOI] [PubMed] [Google Scholar]
- 6.Li PA, He QP, Yi-Bing O, et al. Phosphorylation of extracellular signal-regulated kinase after transient cerebral ischemia in hyperglycemic rats. Neurobiol Dis 2001;8:127–35. [DOI] [PubMed] [Google Scholar]
- 7.Yassin MM, Harkin DW, Barros D'Sa AA, et al. Lower limb ischemia-reperfusion injury triggers a systemic inflammatory response and multiple organ dysfunction. World J Surg 2002;26:115–21. [DOI] [PubMed] [Google Scholar]
- 8.Bianco-Batlles MD, Sosunov A, Polin RA, et al. Systemic inflammation following hind-limb ischemia-reperfusion affects brain in neonatal mice. Dev Neurosci 2008;30:367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi-Yanaga F. Activator or inhibitor? GSK-3 as a new drug target. Biochem Pharmacol 2013;86:191–9. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita K, Nakada M, Shakoori A, et al. An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3 beta. Anticancer Agents Med Chem 2009;9:1114–22. [DOI] [PubMed] [Google Scholar]
- 11.Cole AR, Astell A, Green C, et al. Molecular connexions between dementia and diabetes. Neurosci Biobehav Rev 2007;31:1046–63. [DOI] [PubMed] [Google Scholar]
- 12.Qu Z, Jiao Z, Sun X, et al. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res 2011;1383:300–6. [DOI] [PubMed] [Google Scholar]
- 13.Jolivalt CG, Lee CA, Beiswenger KK, et al. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer's disease and correction by insulin. J Neurosci Res 2008;86:3265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King MR, Anderson NJ, Guernsey LS, et al. Glycogen synthase kinase-3 inhibition prevents learning deficits in diabetic mice. J Neurosci Res 2013;91:506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 2010;31:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Zhang X, Wang C, et al. Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: up-regulated pAkt, pGSK and pCREB, down-regulated NF-kappaB expression, ameliorated BBB permeability. Brain Res 2012;1459:61–70. [DOI] [PubMed] [Google Scholar]
- 17.Wang MJ, Huang HY, Chen WF, et al. Glycogen synthase kinase-3beta inactivation inhibits tumor necrosis factor-alpha production in microglia by modulating nuclear factor kappaB and MLK3/JNK signaling cascades. J Neuroinflammation 2010;7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock 2007;27:113–23. [DOI] [PubMed] [Google Scholar]
- 19.Kim B, Sullivan KA, Backus C, et al. Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid Redox Signal 2011;14:1829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collino M, Aragno M, Castiglia S, et al. Insulin reduces cerebral ischemia/reperfusion injury in the hippocampus of diabetic rats: a role for glycogen synthase kinase-3beta. Diabetes 2009;58:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albadawi H, Oklu R, Cormier NR, et al. Hind limb ischemia-reperfusion injury in diet-induced obese mice. J Surg Res. 2014;190(2):683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szijarto A, Turoczi Z, Szabo J, et al. Rapidly progressing fatal reperfusion syndrome caused by acute critical ischemia of the lower limb. Cardiovasc Pathol 2013;22:493–500. [DOI] [PubMed] [Google Scholar]
- 23.Ordy JM, Wengenack TM, Bialobok P, et al. Selective vulnerability and early progression of hippocampal CA1 pyramidal cell degeneration and GFAP-positive astrocyte reactivity in the rat four-vessel occlusion model of transient global ischemia. Exp Neurol 1993;119:128–39. [DOI] [PubMed] [Google Scholar]
- 24.Draznin B, Miles P, Kruszynska Y, et al. Effects of insulin on prenylation as a mechanism of potentially detrimental influence of hyperinsulinemia. Endocrinology 2000;141:1310–6. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Ma D, Wang Y, et al. Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J Alzheimers Dis 2013;33:329–38. [DOI] [PubMed] [Google Scholar]
- 26.Zhang T, Pan BS, Sun GC, et al. Diabetes synergistically exacerbates poststroke dementia and tau abnormality in brain. Neurochem Int 2010;56:955–61. [DOI] [PubMed] [Google Scholar]
- 27.Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res 2011;21:223–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yenari MA, Han HS. Influence of hypothermia on post-ischemic inflammation: role of nuclear factor kappa B (NFkappaB). Neurochem Int 2006;49:164–9. [DOI] [PubMed] [Google Scholar]
- 29.Lin M, Li L, Li L, et al. The protective effect of baicalin against renal ischemia-reperfusion injury through inhibition of inflammation and apoptosis. BMC Complement Altern Med 2014;14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun BZ, Chen L, Wu Q, et al. Suppression of inflammatory response by flurbiprofen following focal cerebral ischemia involves the NF-κB signaling pathway. Int J Clin Exp Med 2014;7:3087–95. [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed MA, El Morsy EM, Ahmed AA. Pomegranate extract protects against cerebral ischemia/reperfusion injury and preserves brain DNA integrity in rats. Life Sci 2014;110:61–9. [DOI] [PubMed] [Google Scholar]
- 32.Seegers H, Grillon E, Trioullier Y, et al. Nuclear factor-kappa B activation in permanent intraluminal focal cerebral ischemia in the rat. Neurosci Lett 2000;288:241–5. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Kawamura N, Schmelzer JD, et al. Decreased peripheral nerve damage after ischemia-reperfusion injury in mice lacking TNF-alpha. J Neurol Sci 2008;267:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huck V, Niemeyer A, Goerge T, et al. Delay of acute intracellular pH recovery after acidosis decreases endothelial cell activation. J Cell Physiol 2007;211:399–409. [DOI] [PubMed] [Google Scholar]
- 35.Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav 2007;87:179–97. [DOI] [PubMed] [Google Scholar]
- 36.Banks WA. The blood-brain barrier in psychoneuroimmunology. Immunol Allergy Clin North Am 2009;29:223–8. [DOI] [PubMed] [Google Scholar]
- 37.Banks WA, Farr SA, La Scola ME, et al. Intravenous human interleukin-1alpha impairs memory processing in mice: dependence on blood-brain barrier transport into posterior division of the septum. J Pharmacol Exp Ther 2001;299:536–41. [PubMed] [Google Scholar]
- 38.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 2007;8:57–69. [DOI] [PubMed] [Google Scholar]
- 39.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 2012;12:623–35. [DOI] [PubMed] [Google Scholar]
- 40.Pifarre P, Prado J, Giralt M, et al. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J Neurochem 2010;112:807–17. [DOI] [PubMed] [Google Scholar]
- 41.Bonheur JA, Albadawi H, Patton GM, et al. A noninvasive murine model of hind limb ischemia-reperfusion injury. J Surg Res 2004;116:55–63. [DOI] [PubMed] [Google Scholar]
- 42.Wilson JS, Rushing G, Johnson BL, et al. The effects of dichloroacetate in a rabbit model of acute hind-limb ischemia and reperfusion. J Am Coll Surg 2003;197:591–5. [DOI] [PubMed] [Google Scholar]
- 43.Crawford RS, Hashmi FF, Jones JE, et al. A novel model of acute murine hindlimb ischemia. Am J Physiol Heart Circ Physiol 2007;292:H830–7. [DOI] [PubMed] [Google Scholar]
- 44.Islam MS, Loots du T. Experimental rodent models of type 2 diabetes: a review. Methods Find Exp Clin Pharmacol 2009;31:249–61. [DOI] [PubMed] [Google Scholar]