

Abstract
Major intrinsic protein (MIP) is a functional water-channel (AQP0) that also plays a key role in establishing lens fiber cell architecture. Genetic variants of MIP have been associated with inherited and age-related forms of cataract; however, the underlying pathogenic mechanisms are unclear. Here we have used lens transcriptome profiling by microarray-hybridization and qPCR to identify pathogenic changes during cataract development in Mip-mutant (Lop/+) mice. In postnatal Lop/+ lenses (P7) 99 genes were up-regulated and 75 were down-regulated (>2-fold, p=<0.05) when compared with wild-type. A pathway analysis of up-regulated genes in the Lop/+ lens (P7) was consistent with endoplasmic reticulum (ER)-stress and activation of the unfolded protein response (UPR). The most up-regulated UPR genes (>4-fold) in the Lop/+ lens included Chac1 > Ddit3 > Atf3 > Trib3 > Xbp1 and the most down-regulated genes (>5-fold) included two anti-oxidant genes, Hspb1 and Hmox1. Lop/+ lenses were further characterized by abundant TUNEL-positive nuclei within central degenerating fiber cells, glutathione depletion, free-radical overproduction, and calpain hyper-activation. These data suggest that Lop/+ lenses undergo proteotoxic ER-stress induced cell-death resulting from prolonged activation of the Eif2ak3/Perk-Atf4-Ddit3-Chac1 branch of the UPR coupled with severe oxidative-stress.
Keywords: Lens, cataract, unfolded protein response, oxidative stress, mouse
1. Introduction
The major intrinsic protein (MIP) of the vertebrate eye lens is a structurally conserved member of the aquaporin family of water channels that is also referred to as aquaporin-0 (AQP0) (1). MIP constitutes over 40% of the mouse lens membrane proteome (2) and has been shown to function as a relatively weak water-channel in the tightly packed, concentric layers of specialized fiber cells that form the crystalline mass of the lens (3-6). Beyond water transport, ultrastructural and immuno-localization studies have implicated MIP in the formation of junctional micro-domains between lens fiber cells and MIP is particularly enriched at interlocking protrusions that line the edges of fiber cell membranes - consistent with a specialized function in fiber cell adhesion (7-9). The combined water-channel and cell-cell junctional properties of MIP have been proposed to play a synergistic functional role in establishing the refractive index gradient and biomechanical properties of the lens (10,11).
Genetic variations in MIP have been associated with lens opacities or cataract in both humans and rodents (12) [http://cat-map.wustl.edu/]. So far, at least 16 mutations in the human MIP gene (MIP), mostly resulting in missense substitutions, have been linked with autosomal dominant forms of cataract that present during infancy or childhood exhibiting wide variability in size, shape, and density of lens opacities. Further, several common single nucleotide variants have been tentatively associated with age-related cataract. In mice, at least four mutant strains harboring mutations in the MIP gene (Mip) have been reported with semi-dominant, highly-penetrant cataract (13-16). Whereas three of these mouse strains (Fr, Hfi, Tohm) inherit different deletion mutations in Mip, the lens opacity (Lop) mutant inherits a missense mutation (p.Ala51Pro) similar to those typically found in humans. Mice functionally lacking (null) or deficient in MIP also develop fully penetrant cataract (6) and a 5-bp insertion mutation in the rat MIP gene that results in nonsense mediated decay of MIP transcripts causes autosomal recessive cataract in the kfrs rat strain (17).
The pathogenic mechanisms underlying MIP-related cataract are only partially understood. While MIP loss-of-function at the lens fiber cell membrane, due to absence or deficiency of the protein itself, is sufficient to cause ‘recessive’ forms of cataract, it is unclear how mutant forms of MIP elicit ‘dominant’ forms of cataract. A common feature of all four strains of Mip-mutant mice is that the resulting mutant form of MIP accumulates in intracellular endoplasmic reticulum (ER)-like membranes of lens fiber cells (6, 14-16). Similarly, ectopic expression of several human MIP mutants in Xenopus oocytes or cultured cells also results in abnormal intracellular accumulation and impaired water transport across the cell membrane (18-21). Such intracellular accumulation of mutant MIP in lens fiber cells is likely to result in ER-stress and activation of an evolutionarily conserved, adaptive, intracellular signaling mechanism known as the unfolded protein response (UPR) (22). The classical mammalian UPR is comprised of two key components and three signaling branches. Three ER-resident transmembrane sensors - eukaryotic translation initiation factor 2-alpha kinase 3 or PRKR-like ER kinase (EIF2AK3/PERK), ER to nucleus signaling 1 or inositol-requiring 1 alpha (ERN1/IRE1a), and activating transcription factor 6 (ATF6) -detect protein mis-folding stress and subsequently activate three downstream nuclear transcription factors - ATF4, X-box binding protein 1 (XBP1), and cleaved ATF6 (ATF6f), respectively, - to re-program gene expression in distressed cells. Initially, the UPR triggers pro-survival (proteostasis) mechanisms by 1) molecular chaperone expression (e.g. HSPA5) and ER-expansion to increase protein folding capacity, 2) global attenuation of protein translation (via phosphorylation of EIF2A) to reduce biosynthetic demand on the ER, and 3) increased ER-associated degradation (ERAD) to facilitate clearance of unfolded proteins. However, under conditions of severe or chronic unmitigated ER-stress a ‘terminal’ UPR triggers pro-apoptotic mechanisms that lead to cell death to protect surrounding tissue (23-25).
Early signs that the UPR played an important role in lens development was demonstrated when mice lacking the Eif2ak3/Perk activated transcription factor Atf4 were found to exhibit microphthalmia as a result of lens fiber cell degeneration (26, 27). Subsequently, variable activation of several UPR components (e.g. HSPA5, XBP1) has been reported during normal mouse lens development and in lenses of mice that ectopically express certain transgenes or inherit specific mutations in the genes for collagen-4α1, connexin-50, α-crystallin, or β-crystallin (28-34). In vitro studies of transformed human lens epithelial cells or intact rat lenses treated with chemicals that induce ER-stress (e.g. selenite, galactose) have also implicated UPR activation in cataract formation (35-37) and ex vivo lens epithelia derived from aged human cadavers or patients undergoing cataract surgery have been reported to exhibit UPR activation (38, 39). In order to investigate the role of the UPR and other potential pathogenic stress mechanisms in MIP-related cataract we have used a global microarray expression profiling approach to compare lens transcriptome changes in Lop-mutant mice.
2. Materials and methods
2.1. Mice and lenses
Lens opacity (Lop) mutant mice were backcrossed (>10-generations) with C57BL/6J (B6J, Jackson Lab, Bar Harbor ME; stock # 000664) to produce a congenic strain that retains the same semi-dominant, fully-penetrant, bilateral, congenital cataract phenotype of the original mutant (40). PCR-genotyping confirmed that the Lop-mutant strain was ‘rescued’ for a deletion mutation in the phakinin (CP49) gene carried by the 129 and other strains (41). Heterozygous Lop (Lop/+) mice were bred to generate Mendelian ratios of heterozygous and wild-type (+/+) littermates for transcript profiling on embryonic day 14 (E14), postnatal day 1 (P1) and P7. Embryo ages were timed from the appearance of the vaginal plug (E0). For genotyping, genomic DNA was prepared from toe or tail snips using the hot NaOH and Tris (HotShot) procedure (42) then PCR-amplified using allele-specific primers located in exon-1 of Mip (Supplemental Table 1). Mice were humanely killed by CO2 asphyxiation followed by cervical dislocation or by decapitation. Eyes were enucleated into either fixative (4% paraformaldehyde/PBS) for microscopy sections or into pre-warmed PBS (37°C) for lens dissection. After removal of visible remnants of ciliary-body, iris and vasculature (TVL) lenses were stored (-20°C) in RNAlater stabilization solution (ThermoFisher Scientific, Waltham, MA). All mouse studies were approved by the Washington University Animal Studies Committee in compliance with the guidelines published by the Institute for Laboratory Animal Research (ILAR) and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
2.2. Lens RNA preparation
Lens total RNA was extracted from age-matched mouse lenses (E14, P1, P7) of each genotype (Lop/+, +/+) using the RNeasy Kit (Qiagen, Valencia, CA). Triplicate pools of RNA each isolated from 10-20 lenses were quantified by spectrophotometry (ND-2000, NanoDrop, Wilmington NJ) then sized by electrophoresis (Agilent 2100 Bioanalyzer, RNA 6000 kit) according to the manufacturer's protocol (Agilent Technologies, Santa Clara, CA). Samples with RNA integrity number (RIN) values >8.0 were selected for microarray analysis.
2.3. Microarray profiling and statistical analysis
Lens mRNA profiling was performed by the DirectHyb Assay using the MouseWG-6 v2.0 and MouseRef-8 v2.0 (1883 genes) Expression BeadChip Kit according to the manufacturers' instructions (Illumina, San Diego, CA). Both microarrays contain probes (>25,000) to the mouse RefSeq database (Build 36, Release 22; NCBI) and the WG-6 array is further supplemented with probes (>45,000) that target the Mouse Exonic Evidence Based Oligonucliotide (MEEBO) set and the Exemplar protein-coding sequences in the RIKEN FANTOM2 database. Briefly, RNA was reverse transcribed into cDNA (T7-promoter/oligo-dTprimer) then amplified by linear in vitro transcription (IVT) with T7 RNA polymerase in the presence of biotin-UTP using the Illumina TotalPrep RNA Amplification Kit. Following purification and quantitation, biotin-labeled cRNA (∼1.5 μg) was hybridized to the BeadChips, stringency washed and stained (streptavadin-Cy3) using the Illumina Direct Hybridization Array Kit. Following washing and scanning (Illumina BeadArray Reader), microarray data was extracted, normalized, and summarized using the GenomeStudio Gene Expression (GX) Module (Illumina). The raw data were quality assessed by box-plot analysis (Supplemental Fig. 1). The quantile normalized log2 signal data were subject to statistical analysis using the R package ‘limma’, to detect differential expression and p-values were adjusted for false discovery rate (FDR) using Bonferroni correction. Differentially expressed genes (p<0.05 and fold-change minimum cut-off >2.0) were subject to pathway enrichment analysis using the R package ‘gage’ available at http://www.bioconductor.org (43).
2.4. Reverse transcript PCR (RT-PCR)
Mouse lens total RNA was analyzed by SYBR Green-based real-time quantitative PCR (qPCR) using the Mouse UPR RT2 Profiler PCR-array according to the manufacturer's instructions (Qiagen). This 96-well-array-format profiles 84 pathway-related genes and includes house-keeping/reference genes as normalization controls and controls for genomic DNA contamination, RNA quality and PCR performance. Briefly, cDNA was prepared using the RT2 First Strand Kit then combined with the SYBR Green/Fluorescein qPCR master mix and amplified with optimized primer-sets (amplicon size range 100-250 bp) using an iQ5 Real-time PCR system (Bio-Rad, Hercules, CA). Primer specificity was confirmed using melt-curve analysis (iQ5 software v.2.1 Standard Edition). For quantitation of transcripts, threshold cycle (Ct) values were calculated (iQ5 software v.2.1), and pair-wise fold-changes in gene expression calculated by the ΔΔCt method via the RT2 Profiler PCR Array Data Analysis Portal (http://www.sabiosciences.com/pcr/arrayanalysis.php). For end-point RT-PCR, lens RNA was reverse transcribed using the iScript kit (Bio-Rad) and cDNA amplified using gene-specific primers (Supplemental Table 1) and TopTaq polymerase (Qiagen) followed by agarose gel-electrophoresis.
2.5. Immunoblot analysis
Lenses were re-suspended (50 μl, 10 min) in detergent lysis buffer (1% IGEPAL, 50 mM Tris-HCL, 150 mM NaCl, pH 7.8) containing Halt Protease Inhibitor (ThermoFisher) then centrifuged (10,000 × g, 10 min) to pellet cell nuclei. Post-nuclear lysate was removed and soluble protein concentration was determined using the Non-interfering assay (G-Bioscience, St. Louis, MO). Soluble proteins (20-40 μg) and molecular weight markers (10-250 kDa, Li-Cor, Lincoln, NE) were separated on SDS-PAGE gels (4-12% gradient mini-gels, ThermoFisher) then transferred onto nitrocellulose and serially incubated with primary antibody followed by species-appropriate IRDye 680LT and/or IRDye 800CW secondary antibody (1:30,000 dilution, Li-Cor). Protein bands were visualized using an Odyssey Infrared Imaging System (Li-Cor) running Image Studio (v 5.2) software. Primary antibodies used were, anti-caspase-12 (#2202, Cell Signaling Technology, CST, Danvers, MA), anti-spectrin (MAB1622, EMD Millipore, Billerica, MA), and anti-p62 (GP62-C, Progen, Heidelberg, Germany), and anti-β-actin (#3700, CST) to control for sample loading.
2.6. Immunofluorescence microscopy
Eyes were fixed (∼16 hr, 4°C) in 4% paraformaldehyde/PBS and processed using standard paraffin-section or cryo-section techniques. Sections were permeabilized (0.1% Triton/PBS 10 min), blocked (1 hr, 20°C) in Image-iT FX Signal Enhancer (ThermoFisher) then serially incubated with primary antibody (∼16 hr, 4°C) followed by species-appropriate secondary antibody (1 hr, 20°C) conjugated with different Alexa Fluors (ThermoFisher). Images were captured using confocal microscopy (Fluoview1000 MPE; Olympus, Center Valley, PA) and managed in Photoshop (Adobe Systems, San Jose, CA). Primary antibodies used were anti-aquaporin 0 (AB3071, EMD Millipore) and anti-GADD153 (sc-573, Santa Cruz Biotechnology, Dallas, TX).
2.7. Terminal deoxynucleotidal transferase [TdT]-mediated dUTP nick-end labeling (TUNEL) assay
DNA double-strand breaks (3′-OH ends) were detected in mouse lenses (P7) using the Click-iT Plus TUNEL assay according to the manufacturer's protocol (ThermoFisher). Briefly, eye-sections were de-waxed, digested with proteinase K, serially incubated in TdT;EdUTP reaction mix (1 hr, 37°C), followed by Alexa Fluor 488;Cu detection mix (30 min, 37°C protected from light). Cell nuclei were stained with DAPI (15 min, 20°C) and imaged with a confocal microscope (FV1000, Olympus).
2.8. Glutathione assay
Free glutathione (GSH) concentration was measured in mouse lenses using Ellman's sulfhydryl assay reagent (5,5′-dithio-bis-[2-nitrobenzoic acid], DTNB, Sigma-Aldrich, St.Louis, MO) essentially as described (44). Briefly, eyes (P7) were removed into PBS immediately after sacrifice and lens pairs dissected into cold 10% trichloroacetic acid (100 μl 10% TCA, Sigma), homogenized (2 min, Bullet Blender, Next Advance, Averill Park, NY) and centrifuged (16,000×g for 10 min at 4 °C). De-proteinized supernatant (∼100 μl) was mixed with Tris-EDTA pH 8 (890 μl), DTNB (10 μl stock solution - 4 mg/ml in ethanol), incubated for 20 min (20°C) protected from light and absorbance measured at 412 nm (Beckman DU640 Spectrophotometer) against a standard curve for GSH (2-10 μg/ml, Sigma-Aldrich). Protein concentration in the TCA pellet was determined using the Non-Interfering (NI) Potein Assay according to the manufacturer's instructions (G-Biosciences).
2.9. Reactive oxygen species (ROS) assay
Intracellular ROS levels in live mouse lenses were measured using the cell-permeant, free-radical sensor Dihydrorhodamine 123 (DHR123, Sigma) essentially as described (45). Briefly, lenses (P7) were carefully dissected (6 lenses per genotype) into Hanks balanced salt solution with Ca2+ and Mg2+ (HBSS/Ca/Mg, ThermoFisher) then stained by incubating (20°C, 45 min) in DHR123 (7.5 μM) protected from light. Lens cell nuclei were counterstained by adding a drop of NucBlue live cell stain (ThermoFisher) during the final 15 min of incubation with DHR123. Vitally double-stained lenses were rinsed twice in HBSS (2 × 5 min, 20°C) then imaged using a confocal microscope (FV1000, Olympus). Anterior lens epithelial cells were scanned from the surface to a depth of 20 μm with a 2 μm step Z-scan (25× water-immersion objective lens) at wavelengths of 405nm/450nm (NucBlue) and 488nm/550nm (Rhodamine 123). The Z-scan images were projected with Fluoview software and managed in Photoshop (Adobe).
3. Results
3.1. Lens phenotype in the Lop/+ mouse
Dark-field microscopy revealed that the Lop/+ lens at P7 was considerably smaller (microphakia) than wild-type and manifest a dense white central opacity surrounded by a relatively transparent cortical region (Fig. 1). Histochemical staining of eye sections (P7) confirmed severe degeneration of central fiber cells in the Lop/+ lens along the anterior-posterior polar (optical) axis flanked by many displaced cell nuclei derived from the grossly disturbed ‘bow regions’ at the lens equator (Fig. 1). Immuno-fluorescent staining revealed abnormal distribution of MIP in the Lop/+ lens compared with wild-type (Fig. 2). In wild-type lenses MIP was exclusively localized to fiber cell membranes commencing in the penultimate layer of equatorial fiber cells (Fig. 2). By contrast in Lop/+ lenses, while some MIP staining was localized to fiber cell membranes much appeared concentrated in intracellular compartments particularly surrounding cell nuclei and at the anterior and posterior tips of fiber cells (Fig. 2). Overall, these observations were consistent with impaired targeting of mutant MIP to the fiber cell membrane and progressive central fiber cell death in the Lop/+ lens.
Fig. 1.
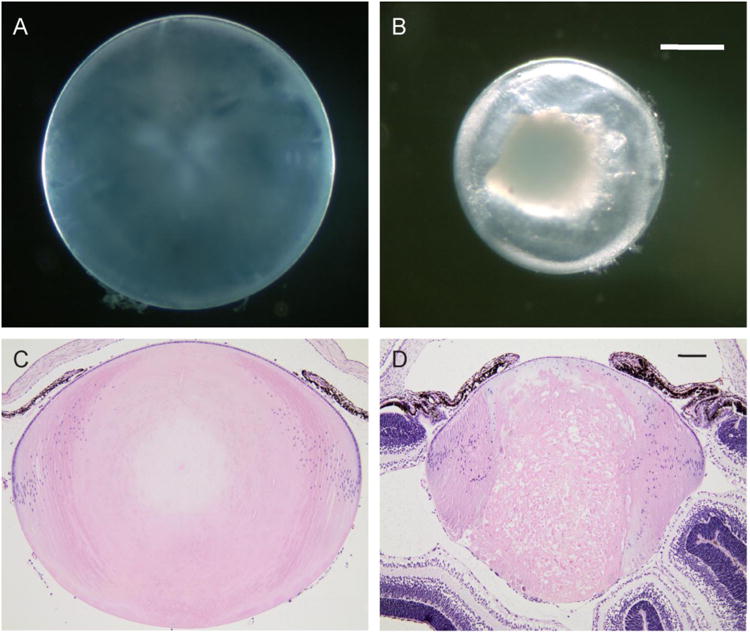
Lens phenotype of the Lop/+ mouse. (A-B) Dark-field images of the wild-type (A) and Lop/+ (B) lenses (P7) showing dense central opacification in the Lop/+ lens. (C-D) Saggital sections of eyes from wild-type (C) and Lop/+ (D) mice stained with H&E showing degeneration of central lens fiber cells and disturbed nuclei close to the equator of the latter. Scale bar: 300 μm (A, B) and 100 μm (C, D).
Fig. 2.

Immuno-fluorescent localization of MIP in the Lop/+ lens (P7). (A-F) Saggital sections of the wild-type lens (A - C) and Lop/+ lens (D - F) showing membrane-specific localization MIP in wild-type (B, C) and intracellular accumulation of MIP around fiber cell nuclei and at the tips of fiber cells in the Lop/+ lens (E, F). Scale bar: 200 μm (A, D) and 30 μm (B, C, E, F).
3.2. Microarray analysis of the Lop/+ lens transcriptome
Initially, we undertook a pilot experiment using the Mouse-6 BeadChip to detect differential transcript expression in the embryonic Lop/+ lens. At E14 no genes were differentially regulated >2-fold in the Lop/+ lens compared with wild-type. By reducing the cut-off threshold to ≥1.3-fold we detected 131 differentially regulated genes with 95 genes up-regulated and 36 genes down-regulated (Supplemental Table 2). However, when adjusted for false discovery rate (FDR) none of these genes attained a significant p-value of 0.05. Because differential regulation of Lop/+ lens gene expression at E14 was not significant we focused subsequent studies on the early postnatal ages P1 and P7 by interrogating the Mouse Ref-8 BeadChip.
At P7 in the Lop/+ lens 174 genes were significantly, differentially regulated (>2-fold change, p=0.05) with 99 genes up-regulated and 75 genes down-regulated (Supplemental Table 3). The most up-regulated genes (>10-fold) at P7 in descending order included Chac1 > Cox6a2 > Retnla > Ddit3 > Emp1 > Fos, whereas, the most down-regulated genes (>5-fold) included Hspb1 > Rgs4 > 1190002N15Rik > Trex1 > Rsad2 > Hmox1. At P1 in the Lop/+ lens 24 genes were up-regulated (>2-fold, p=0.05) and 8 genes down-regulated (Supplemental Table 4). Of the 24 genes up-regulated at P1, 16 remained significantly up-regulated (>4-fold) at P7 including; Arg1 > Retnla > Il4 > Plk3 > Tnfrsf12α > Fos > Cox6a. Similarly, six of the eight genes down-regulated at P1 were also significantly down-regulated at P7 including; Hspb1 > 1190002N15Rik > Trex1 > Hmox1.
Pathway analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway database (http://www.genome.jp/kegg/) indicated that the most up-regulated genes in the Lop/+ lens at P1 were associated with the lysosome pathway (KEGG_ID map04142, p=0.0003) and at P7 with protein processing in the ER (KEGG_ID map04141, p=0.0004) (Supplemental Tables 5 and 6). Similarly, the Gene Ontology (GO) Biological Process database (http://geneontology.org/) indicated that response to ER-stress (GO:0034976, p=0.0025) was the most strongly up-regulated process in the Lop/+ lens at P7 (Supplemental Table 7). The most up-regulated gene (>80-fold) in the Lop/+ lens at P7 was Chac1 (Table 1) – a previously identified pro-apoptotic component of the UPR that acts downstream of the Eif2ak3/Perk-Atf4-Atf3-Ddit3 branch of the UPR (46, 47). In addition, several of the other most up-regulated genes (>2-fold) in the Lop/+ lens coded for well-characterized transcription-factor components of the UPR including, Ddit3, Atf3, Cebpb, and Xbp1 along with at least three of their downstream target genes including, Dnajb9 > Trib3 > Herpud1 (Table 1). By contrast, in the Lop/+ lens at P1 only three UPR genes (Insig1, Atf3, Chac1) were up-regulated >2-fold and in two cases (Insig1, Chac1) the increase was not significant (Table 1). Taken overall, microarray profiling suggested that progressive ER-stress during Lop/+ lens development triggers strong transcriptional activation of several UPR genes around birth.
Table 1.
Most up-regulated UPR genes (>2-fold) in the Lop/+ lens (P1 and P7) detected by microarray and validated by qPCR.
| Gene symbol | P7 fold-change | P7 adjusted p-value | P7 qPCR fold-change | P1 fold-change | P1 adjusted p-value | P1 qPCR fold-change | Mus musculus mRNA transcript | Gene ID | Alias |
|---|---|---|---|---|---|---|---|---|---|
| Chac1 | 84.19 | 6.73E-09 | nd | 2.03 | 9.09E-01 | nd | ChaC, cation transport regulator-like 1 (E. coli) | 69065 | Botch |
| Ddit3 | 11.81 | 1.38E-05 | 12.25 | 1.28 | 8.20E-01 | 1.66 | DNA-damage inducible transcript 3 | 13198 | Chop, Gadd153 |
| Atf3 | 5.18 | 1.59E-04 | nd | 2.90 | 2.09E-02 | nd | Activating transcription factor 3 | 11910 | |
| Trib3 | 4.70 | 1.23E-04 | nd | 1.10 | 9.62E-01 | nd | Tribbles homolog 3 (Drosophila) | 228775 | |
| Xbp1 | 4.15 | 9.09E-07 | 3.76 | 1.55 | 2.12E-01 | 1.37 | X-box binding protein 1 | 22433 | |
| Dnajb9 | 3.02 | 2.17E-04 | 5.83 | 1.42 | 6.37E-01 | 2.03 | Dnaj (Hsp40) homolog, subfamily B, member 9 | 27362 | |
| Cebpb | 2.50 | 8.75E-03 | 5.14 | 1.50 | 2.19E-02 | 1.44 | CCAAT/enhancer binding protein (C/EBP), beta | 12608 | |
| Herpud1 | 2.08 | 5.05E-04 | 2.03 | 1.06 | 9.83E-01 | 1.27 | Homocysteine-inducible, ER stress-inducible, ubiquitin-like domain member 1 | 64209 | |
| Insig1 | 1.47 | 8.70E-03 | 2.26 | 4.79 | 8.64E-01 | -1.19 | Insulin induced gene 1 | 231070 | |
| Hspa5 | nd | nd | 2.55 | nd | nd | 1.13 | Heat shock protein 5 | 14828 | Grp78, Bip |
3.3. Validation of UPR and other differentially regulated genes in the Lop/+ lens
Based on microarray and pathway analyses above we first selected the mouse UPR RT-qPCR array to validate transcript levels in the Lop/+ lens. At P7, 11 of 84 UPR genes profiled on the qPCR-array were differentially regulated >2-fold (Supplemental Table 8). Seven genes were up-regulated in the descending order Ddit3 > Dnajb9 > Cebpb > Xbp1 > Hspa5 > Insig1 > Herpud1; however Cebpb and Herpud1 fell outside the preferred PCR-cycle threshold range. Five of these genes were similarly up-regulated on the microarray platform; however, Insig1 up-regulation on the microarray was < 2-fold and the microarray probe-set for Hspa5 failed QC criteria preventing further analysis. In order to confirm Hspa5 up-regulation on the qPCR-array we performed an independent qPCR assay and since Chac1 was not represented on the qPCR-array we used end-point PCR to confirm its up-regulation on the microarray (Supplemental Fig. 2). Further, using end-point PCR we also confirmed that the Xbp1 transcript underwent unconventional cytoplasmic splicing of a 26 bp exon to generate an Xbp1s transcript that encodes the active form of this transcription factor (48) (Supplemental Fig. 2). Of the four UPR-genes that were down-regulated >2-fold in the Lop/+ lens at P7, only Dnajb2 fell within the preferred cycle threshold range (Supplemental Table 8). At P1 in the Lop/+ lens only one UPR gene (Dnajb9) was up-regulated >2-fold within the preferred cycle threshold range. However, several other UPR genes were marginally up-regulated (>1.3-fold) including, Ddit3 > Cebpb > Xbp1 (Supplemental Table 9). Overall, combined profiling with the microarray and qPCR-array platforms detected up-regulation (>2-fold) of at least nine UPR genes in the Lop/+ lens at P7 (Table 1).
Beyond UPR associated genes, microarray analysis of the Lop/+ lens detected many other functionally diverse genes that were either highly up-regulated (>5-fold) including; Cox6a2 > Retnla > Sqstm1 or strongly down-regulated including, Hspb1 > Hmox1 (Supplemental Table 3). End-point PCR confirmed up-regulation of Retnla and Cox6a2 transcripts and immuno-blotting detected increased Sqstm1 protein levels in Lop/+ lenses at P7 (Supplemental Fig. 2). Together, these observations suggest that in addition to ER-stress many other stress-response mechanisms likely operate during cataractogenesis in Mip-mutant lenses.
3.4. Oxidative stress in the Lop/+ lens
Recently, Chac1 was reported to function as a γ-glutamyl cyclo-transferase (GGCT) that catalyzes conversion of glutathione (GSH), a tripeptide thiol antioxidant (γ-Glu-Cys-Gly), to 5-oxyProline and Cys-Gly (49, 50). Since Chac1 was highly up-regulated in Lop/+ lenses we predicted that GSH levels may be depleted as a consequence of GGCT activity. Therefore, we measured GSH levels in lens lysates (P7) using the sulfhydryl assay reagent DTNB that reacts with free –SH groups (derived from GSH and proteins) to yield a mixed disulfide and a yellow-colored anion (TNB2-). Free -SH levels in the Lop/+ lens were decreased about 50% when compared to wild-type (Fig. 3). While we cannot exclude the possibility that some GSH loss occurred due to fiber cell membrane damage, these data suggest that GSH was depleted in the Lop/+ lens. Since GSH is a key anti-oxidant molecule in the lens (51, 52) we assessed the overall red-ox status in the live Lop/+ lens (P7) by vital staining with a cell-permeant, non-fluorescent indicator (DHR123) that upon oxidation by reactive oxygen species (ROS) is converted to fluorescent rhodamine 123, which accumulates within mitochondria (45). Fluorescent imaging revealed that ROS production was greatly increased in the Lop/+ lens compared with wild-type (Fig. 4). Collectively, these data suggest that Chac1 up-regulation in the Lop/+ lens results in GSH depletion and ROS overproduction consistent with oxidative stress.
Fig. 3.
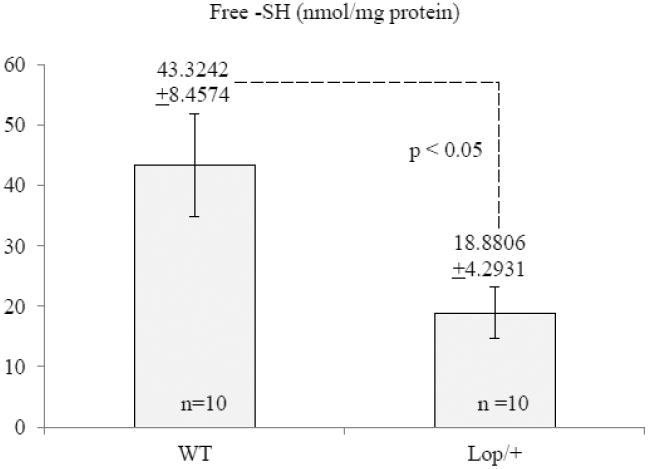
Free GSH levels in the Lop/+ lens (P7) estimated with Ellman's sulfhydryl assay reagent (DTNB).
Fig. 4.
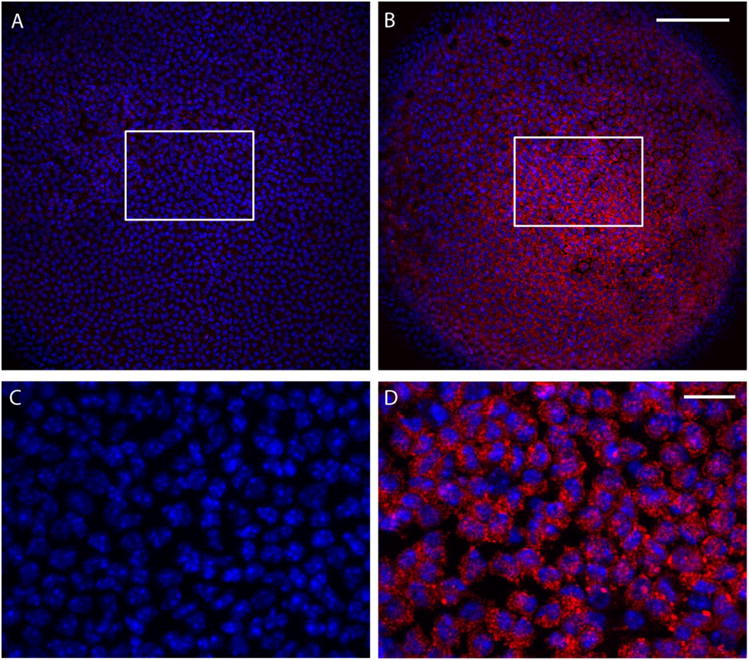
Fluorescence imaging of ROS production in Lop/+ lens epithelial cells (P7) vitally stained with DHR123. The Lop/+ lens (B, D) displayed much greater ROS production (red) when compared to wild-type (A, C) lenses. Cell nuclei were stained with DAPI (blue).
3.5. DNA damage in Lop/+ lens
In order to support up-regulation of Ddit3 expression at the post-translational level we compared in situ DNA fragmentation in Lop/+ and wild-type lenses (P7). In the Lop/+ lens, but not wild-type, Ddit3 expression was immuno-localized to nuclei of degenerating fiber cells within the core but was absent from equatorial epithelial cells and nascent fiber cells (Supplemental Fig. 3). Similarly, TUNEL positive nuclei were also clearly detected throughout the degenerating fiber cells in the center of Lop/+ lens consistent with arrest of the normal de-nucleation process observed in the wild-type lens (Fig. 5). However, TUNEL-positive nuclei were absent from equatorial epithelial cells and nascent fiber cells lying outside the central degeneration zone. These observations suggest that while apoptotic cell-death is not activated in equatorial lens epithelial cells and nascent fiber cells of the Lop/+ lens, apoptosis may be active deeper into these lenses in mature fiber cells that have accumulated high levels of mutant MIP and endured prolonged ER-stress. However, as TUNEL is specific for the 3′-OH ends of fragmented DNA rather than apoptosis per se, it is also possible that the observed DNA-fragmentation may result from cell-death other than classical apoptosis.
Fig. 5.
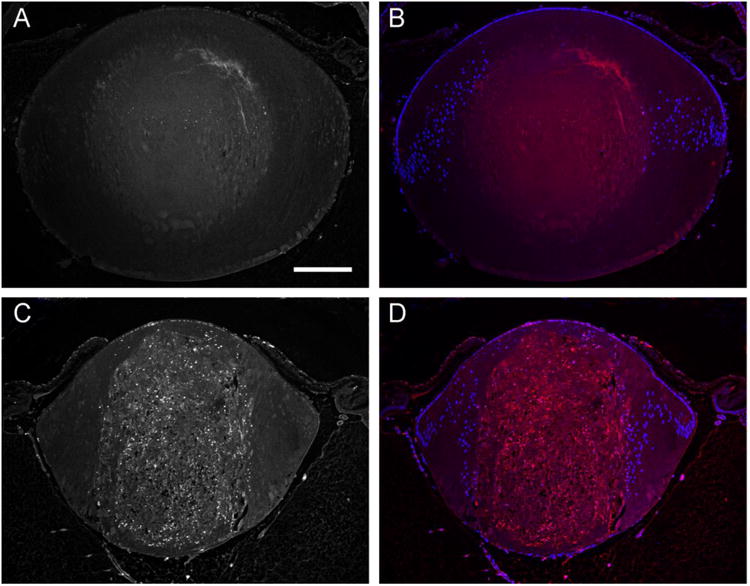
TUNEL analysis of DNA-fragmentation in Lop/+ lens. Saggital sections of wild-type (A, B) and Lop/+ (C, D) lenses (P7) showing many TUNEL positive nuclei throughout the central zone of fiber cell degeneration in the Lop/+ lens (C) with only trace levels in the wild-type lens (A). TUNEL positive nuclei were also detected in the anterior epithelial cell layer that overlies severe fiber cell degeneration in the Lop/+ lens (B, D) Merged images of TUNEL positive nuclei (red) and DAPI stained nuclei (blue) in wild-type (B) and Lop/+ (D) lenses. Scale bar: 200 μm.
3.6. Caspase and calpain activation in the Lop/+ lens
Since up-regulation of Chac1 and Ddit3 are widely regarded as pro-apoptotic responses to ER-stress we sought to find evidence of increased proteolytic enzyme activity associated with cell-death mechanisms in the Lop/+ lens that would not otherwise be detected by transcript profiling. Pro-caspase 12 is an ER-resident cysteine-aspartate protease in mice that undergoes cleavage to an active form by caspase-3/-7 and calpain in order to initiate ER-stress induced apoptosis (53-55). Calpains are cytosolic, calcium-activated cysteine proteases and at least four active isoforms (1-3 and 7) are abundantly expressed in the mouse lens (56). In addition to their role in remodeling of the membrane cytoskeleton during lens fiber cell differentiation, calpain-mediated proteolysis of lens proteins, including αII-spectrin (α-fodrin), has also been associated with cataract formation in rodents (57-59). Immunoblot analysis revealed that the level of pro-caspase 12 cleavage products in the Lop/+ lens was mildly elevated compared to that detected in wild-type (Fig. 6a). By contrast, αll-spectrin cleavage to several specific breakdown products, including C-terminal fragments of 150 kDa and 145 kDa, was greatly increased consistent with robust calpain activation in the Lop/+ lens (Fig. 6b).
Fig. 6.

Immunoblot analysis of cysteine protease cleavage activity in the Lop/+ lens (P7). A) Pro-caspase 12 cleavage products were mildly elevated in Lop/+ lenses compared with wild-type. ** denotes suspected partial cleavage products (∼ 48 kDa), * denotes fully cleaved product (42 kDa). The signal intensity of fully cleaved caspase 12 in the Lop/+ lens, normalized to β-actin, was approximately 1.5-fold greater than that of wild-type. B) αII-spectrin cleavage was greatly increased in the Lop/+ lens compared with wild-type consistent with calpain activation. * denotes cleavage products of 150 kDa and 145 kDa. The normalized (β-actin) signal intensities of these αII-spectrin cleavage products in the Lop/+ lens was over 30-fold greater than those of wild-type. M, molecular weight markers.
4. Discussion
In this study we have used microarray-based transcriptome profiling to identify pathogenic changes in lens gene expression underlying inherited cataract in Mip-mutant (Lop/+) mice. While global gene expression changes in the Lop/+ lens at E14 were not significantly different to wild-type, we detected 32 differentially regulated genes (>2-fold) around birth (24 up-regulated, 8 down-regulated). One week after birth, we identified 174 differentially expressed genes (>2-fold) in the Lop/+ lens (99 up-regulated, 75 down-regulated) compared with wild-type. A pathway analysis of up-regulated genes in the postnatal Lop/+ lens indicated that the most significant underlying processes involved the lysosome at P1 progressing at P7 to protein processing in the ER and response to ER-stress associated with activation of several pro-apoptotic UPR genes including Chac1 and Ddit3 (Table 1). Constitutive activation of the UPR has been detected during embryonic development of the mouse lens (29). The data presented here support the notion that expression and progressive accumulation of the mutant, presumably, mis-folded form of MIP in the Lop/+ lens triggers robust ER-stress and prolonged UPR activation culminating in fiber cell death and cataract formation (Fig. 7).
Fig. 7.
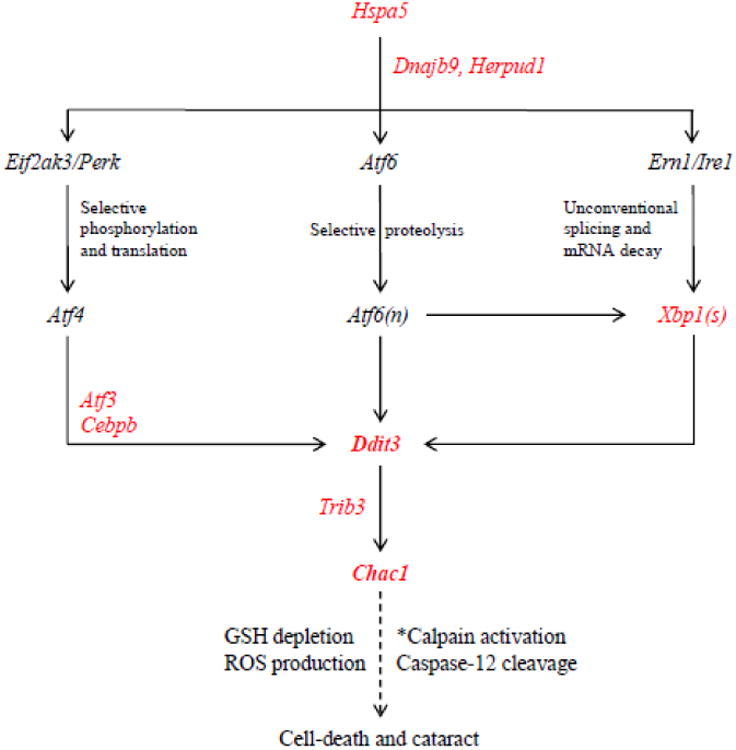
Schematic summarizing terminal activation of the UPR in the Lop/+ lens (P7). Genes in red were up-regulated in this study. *Calpain activation is calcium-dependent.
The most up-regulated gene in the Lop/+ lens - Chac1 - was first identified as a novel pro-apoptotic component of the UPR that acted downstream of the Atf4-Atf3-Ddit3/Chop cascade (45, 46). Subsequently, Chac1 was shown to encode a cytosolic γ-glutamyl cyclo-transferase that catalyzes the degradation of GSH previously attributed to Ddit3 activation (49, 50, 60). Our data suggest that up-regulation of Chac1 in the Lop/+ lens was correlated with an estimated 50% decrease in GSH levels (Fig. 3). Free GSH is a critical red-ox buffer in the lens and its depletion is associated with ROS generation in lens aging and cataractogenesis (51, 52). Vital staining with a red-ox indicator revealed that ROS production was highly elevated in the Lop/+ lens epithelium suggesting that both ER-stress and oxidative stress contribute to cataract development (Fig. 4). To the best of our knowledge, Chac1 has not previously been associated with cataract. However, outside the lens we note that Chac1 activity de-glycinates and antagonizes Notch during neurogenesis (61) and that Chac1 transcription is up-regulated following retinal axon injury (62).
Ddit3 encodes a well known pro-apoptotic, basic region-leucine zipper (bZip) transcription factor -CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) - that under conditions of prolonged ER-stress becomes an important transcriptional target of the Eif2ak3/Perk-Atf4 branch, and to a lesser extent the Ern1/Ire1-Xbp1 and Atf6 branches, of the UPR - thereby providing a pivotal link between ER-stress and apoptosis (24). In the Lop/+ lens, Ddit3 expression was dramatically (∼12-fold) up-regulated (Table 1) and immuno-localized to nuclei of degenerating fiber cells within the lens core (Supplemental Fig. 3). Many such fiber cell nuclei were also TUNEL positive consistent with DNA fragmentation at 3′ -OH strand-breaks that are often associated with programmed cell-death via apoptosis.
Although we did not detect strong up-regulation of Atf4 or its upstream ER-sensor (Eif2ak3/Perk) in the Lop/+ lens, at least three other pro-apoptotic target genes of Atf4 (Atf3 > Cebpb > Trib3) were up-regulated along with Chac1 and Ddit3 (Table 1). Atf3 encodes a bZip transcription factor that acts as an integration hub for various stress responses including ER-stress downstream of the Eif2ak3/Perk-Atf4 branch of the UPR (63). Similarly, Cebpb encodes a bZip transcription factor that is responsive to ER-stress and along with Atf4 and Atf3 is involved in regulation of Chac1 transcription (50). Trib3 encodes the mammalian homolog of Drosophila tribbles, a putative protein kinase that is induced by the Atf4-Ddit3/Chop pathway and sensitizes cells to Ddit3-dependent apoptosis (64). Taken overall, our transcript data in the Lop/+ lens are consistent with terminal activation of the Eif2ak3/Perk-Atf4-Ddit3-Chac1 branch of the UPR leading to fiber cell death.
Despite up-regulation of at least five pro-apopotic UPR genes (Chac1 > Ddit3 >Atf3 > Cebpb > Trib3) we detected relatively mild activation of the ER-stress associated caspase-12 in the Lop/+ lens. By contrast, calpain-mediated cleavage of αII-spectrin was dramatically increased in the Lop/+ lens (Fig. 6). While we cannot exclude caspase-dependent apoptosis, especially prior to P7, our data suggest that calpain-mediated cell-death, associated with impaired de-nucleation of fiber cells, predominated in the Lop/+ lens. Calcium-dependent activation of calpains, especially calpain-2 and calpain-3 (Lp82), in the rodent lens is an established cause of crystallin and cytoskeleton proteolysis resulting in cataract formation (57-59). In the Lop/+ lens there are at least two possible sources of calcium ion (Ca2+) flux necessary for calpain activation. First, prolonged ER-stress as a result of mutant MIP accumulation particularly within the Lop/+ lens cortex may result in uncontrolled Ca2+ leakage from ER-stores. The adverse effects of such efflux may be compounded by the programmed loss of organelles including the ER that is characteristic of lens fiber cell maturation (65). Second, fiber cell membrane damage arising from MIP dysfunction, particularly within the Lop/+ lens nucleus, may permit uncontrolled Ca2+ influx. Such cell membrane damage likely disrupts the lens microcirculation system that allows ions and water to flow in at the anterior and posterior poles and out at the equator [66]. Once activated, calpain-mediated proteolysis of crystallins leading to aggregate formation may also contribute to the UPR in the Lop/+ lens. For example, activation of the UPR has been detected in lenses of mice that accumulate mutant forms of alpha- or beta-crystallins (30, 33, 34). It is noteworthy, however, that in contrast to rodent lenses, primate lenses are resistant to calpain activation since they lack calpain-3 activity and human lens epithelial cells contain high constitutive levels of the calpain inhibitor, calpastatin (67). Such variability in calpain activation between rodents and primates may, in part, explain the increased severity of lens opacities in the Lop/+ mouse when compared with those observed in human families segregating MIP mutations.
In addition to the Eif2ak3/Perk-Atf4 branch of the UPR, several other UPR genes were also up-regulated in the Lop/+ lens. First, Xbp1 and its splicing product (Xbp1s) were up-regulated (Table 1, Supplemental Fig. 2). Xbp1 encodes a bZIP transcription factor that in response to ER-stress – downstream of Ern1/Ire1a - undergoes unconventional cytoplasmic splicing to generate an activated form, which translocates to the nucleus and regulates UPR gene expression (48). Neither Ern1/Ire1a nor Atf6 were strongly up-regulated in the Lop/+ lens; however, the Atf6 cleavage product is known to up-regulate Xbp1 expression and Ern1/Ire1a facilitates the splicing of Xbp1 necessary for its transcription factor function. Generally, Xbp1 splicing signals pro-survival responses, including ER-chaperone up-regulation. However, Xbp1s can also up-regulate Ddit3 expression (24). Second, while the Lop/+ dataset did not directly detect strong up-regulation of the genes coding for ER-stress/UPR sensors (Eif2ak3/Perk, Ern1/Ire1, and Atf6), three other genes encoding ER-resident components of the UPR (Hspa5, Dnajb9, and Herpud1) were also up-regulated (Table 1). Notably, Hspa5 encodes a member of the heat-shock protein 70 (HSP70) family of molecular chaperones - alias 78 kDa glucose-regulated protein (GRP78) or immunoglobulin heavy chain-binding protein (BIP) - that constitutively binds and inactivates the ER-stress/UPR sensors. Accumulation of unfolded or misfolded proteins within the ER triggers release of HSPA5 from the sensors and simultaneously activates UPR pro-survival signaling including up-regulation of ER-resident chaperones (25). Dnajb9 encodes a member of the J-domain heat-shock protein 40 (HSP40) family that acts as a molecular co-chaperone for HSPA5 (68). Herpud1 encodes a ubiquitin-like domain protein that is believed to function in the ER-associated degradation of misfolded proteins (69). When combined with Xbp1 splicing above, these data by extension suggest that both the Ern1/Ire1a-Xbp1 and Atf6 branches of the UPR may also be activated in the Lop/+ lens, possibly, prior to the Eif2ak3/Perk-Atf4 branch.
Beyond UPR genes, many other functionally divergent genes were also differentially regulated (>2-fold) in the Lop/+ lens and may contribute to the cataract phenotype (Supplemental Tables 3 and 4). Noteworthy non-UPR genes that were up-regulated included Cox6a2, Retnla, and Sqstm1. Cox6a2 is a nuclear gene that encodes one of 13 subunits of the mitochondrial cytochrome c oxidase complex that if deficient in mice results in ROS overproduction in skeletal muscle (70). Retnla encodes an adipokine with cholesterol catabolism properties (71) raising the possibility of an anti-inflammatory-like response during cataract development in the Lop/+ lens. Sqstm1 encodes a stress-induced scaffold protein (p62) with multiple domains that act as a signaling hub for diverse cellular events including, NF-kB activation, mTORC1 activation, selective autophagy, and Nrf2 signaling (72). Recently, p62 up-regulation has been linked with impaired autophagy in lenses from α-crystallin mutant mice (33, 73) and with an oxidative-stress response via the Nrf2-Keap1 pathway (74). Several non-UPR genes were also strongly down-regulated in the Lop/+ lens including Hspb1, 1190002N15Rik, Trex1, and Hmox1. Interestingly, both Hspb1 and Hmox1 have been directly associated with red-ox status. Hspb1 encodes a member of the small heat-shock proteins (27 kDa) that in addition to its chaperone properties has been implicated in glutathione status, actin re-modeling, and anti-apoptosis (75). Hmox1 encodes heme oxygenase 1 (alias HSP37), an essential antioxidant heme-catabolism enzyme found to be cytoprotective against H2O2- induced oxidative-stress in lens epithelial cells (76). Trex1 encodes an ER-associated 3′-exonuclease implicated in suppressing innate immune responses (77) and 1190002N15Rik codes for a secreted Golgi protein (GoPro49) with cytoprotective properties (78). Taken together, the differential regulation of many genes unrelated to the UPR suggests that additional stress-response mechanisms may operate during Mip-related cataractogenesis.
In summary, our data suggest that prolonged activation the Eif2ak3/Perk-Atf4-Ddit3-Chac1 branch of the UPR in the Lop/+ mouse lens contributes to fiber cell death and cataract development associated with GSH-depletion, ROS-overproduction, and calpain hyper-activation. Additionally, many other non-UPR related genes were differentially regulated suggesting that therapeutic targeting of cataract via the lens UPR will require consideration of multiple stress response pathways especially oxidative stress.
Supplementary Material
Highlights.
Microarray and qPCR analyses detected 174 transcript changes in lenses of Lop/+ mice
Pathway analysis indicated ER-stress and activation of five pro-apoptotic UPR genes
Chac1 up-regulation was correlated with GSH depletion and ROS overproduction
Ddit3 up-regulation was correlated with TUNEL-positive DNA fragmentation
Calpain hyper-activation was associated with lens fiber cell death and cataract
Acknowledgments
We thank the Genome Technology Access Center (GTAC) at Washington University School of Medicine for help with transcriptome analysis, Dr. J. Yu for help with statistical analysis, B. McMahan and G. Ling for histology support, and J. King for technical assistance. This work was supported by NIH/NEI grants EY012284 and EY023549 (to A.S.) and P30 EY02687 (Core Grant for Vision Research), and an unrestricted grant to the Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness (RPB). The genome Technology Access Center (GTAC) at Washington University School of Medicine is partially supported by NIH grants P30 CA91842 and UL1 TR000448.
Footnotes
Appendix A. Supplementary data
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chepelinsky AB. Structural function of MIP/aquaporin 0 in the eye lens; genetic defects lead to congenital inherited cataracts. Handbook Expt Pharmacol. 2009:265–297. doi: 10.1007/978-3-540-79885-9_14. [DOI] [PubMed] [Google Scholar]
- 2.Bassnett S, Wilmarth PA, David LL. The membrane proteome of the mouse lens fiber cell. Mol Vis. 2009;15:2448–2463. [PMC free article] [PubMed] [Google Scholar]
- 3.Varadaraj K, Kushmerick C, Baldo GJ, Bassnett S, Shiels A, Mathias RT. The role of MIP in lens fiber cell membrane transport. J Memb Biol. 1999;170:191–203. doi: 10.1007/s002329900549. [DOI] [PubMed] [Google Scholar]
- 4.Varadaraj K, Kumari S, Shiels A, Mathias RT. Regulation of aquaporin water permeability in the lens. Invest Ophthalmol Vis Sci. 2005;46:1393–1402. doi: 10.1167/iovs.04-1217. [DOI] [PubMed] [Google Scholar]
- 5.Varadaraj K, Kumari SS, Mathias RT. Functional expression of aquaporins in embryonic, postnatal, and adult mouse lenses. Dev Dyn. 2007;236:1319–1328. doi: 10.1002/dvdy.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiels A, Bassnett S, Varadaraj K, Mathias R, Al-Ghoul K, Kuszak J, Donoviel D, Lilleberg S, Friedrich G, Zambrowicz B. Optical dysfunction of the crystalline lens in aquaporin-0-deficient mice. Physiol Genomics. 2001;7:179–186. doi: 10.1152/physiolgenomics.00078.2001. [DOI] [PubMed] [Google Scholar]
- 7.Zampighi GA, Eskandari S, Hall JE, Zampighi L, Kreman M. Micro-domains of AQP0 in lens equatorial fibers. Exp Eye Res. 2002;75:505–519. doi: 10.1006/exer.2002.2041. [DOI] [PubMed] [Google Scholar]
- 8.Mangenot S, Buzhynskyy N, Girmens JF, Scheuring S. Malformation of junctional microdomains in cataract lens membranes from a type II diabetes patient. Pflugers Arch. 2009;457:1265–1274. doi: 10.1007/s00424-008-0604-4. [DOI] [PubMed] [Google Scholar]
- 9.Lo WK, Biswas SK, Brako L, Shiels A, Gu S, Jiang JX. Aquaporin-0 targets interlocking domains to control the integrity and transparency of the eye lens. Invest Ophthalmol Vis Sci. 2014;55:1202–1212. doi: 10.1167/iovs.13-13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumari SS, Varadaraj K. Aquaporin 0 plays a pivotal role in refractive index gradient development in mammalian eye lens to prevent spherical aberration. Biochemical and biophysical research communications. 2014;452:986–991. doi: 10.1016/j.bbrc.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sindhu Kumari S, Gupta N, Shiels A, FitzGerald PG, Menon AG, Mathias RT, Varadaraj K. Role of Aquaporin 0 in lens biomechanics. Biochem Biophys Res Commun. 2015;462:339–345. doi: 10.1016/j.bbrc.2015.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–2015. [PMC free article] [PubMed] [Google Scholar]
- 13.Shiels A, Bassnett S. Mutations in the founder of the MIP gene family underlie cataract development in the mouse. Nat Genet. 1996;12:212–215. doi: 10.1038/ng0296-212. [DOI] [PubMed] [Google Scholar]
- 14.Shiels A, Mackay D, Bassnett S, Al-Ghoul K, Kuszak J. Disruption of lens fiber cell architecture in mice expressing a chimeric AQP0-LTR protein. Faseb J. 2000;14:2207–2212. doi: 10.1096/fj.99-1071com. [DOI] [PubMed] [Google Scholar]
- 15.Sidjanin DJ, Parker-Wilson DM, Neuhauser-Klaus A, Pretsch W, Favor J, Deen PM, Ohtaka-Maruyama C, Lu Y, Bragin A, Skach WR, Chepelinsky AB, Grimes PA, Stambolian DE. A 76-bp deletion in the Mip gene causes autosomal dominant cataract in Hfi mice. Genomics. 2001;74:313–319. doi: 10.1006/geno.2001.6509. [DOI] [PubMed] [Google Scholar]
- 16.Okamura T, Miyoshi I, Takahashi K, Mototani Y, Ishigaki S, Kon Y, Kasai N. Bilateral congenital cataracts result from a gain-of-function mutation in the gene for aquaporin-0 in mice. Genomics. 2003;81:361–368. doi: 10.1016/s0888-7543(03)00029-6. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe K, Wada K, Ohashi T, Okubo S, Takekuma K, Hashizume R, Hayashi J, Serikawa T, Kuramoto T, Kikkawa Y. A 5-bp insertion in Mip causes recessive congenital cataract in KFRS4/Kyo rats. PloS one. 2012;7:e50737. doi: 10.1371/journal.pone.0050737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis P, Chung JJ, Yasui M, Berry V, Moore A, Wyatt MK, Wistow G, Bhattacharya SS, Agre P. Functional impairment of lens aquaporin in two families with dominantly inherited cataracts. Hum Mol Genet. 2000;9:2329–2334. doi: 10.1093/oxfordjournals.hmg.a018925. [DOI] [PubMed] [Google Scholar]
- 19.Varadaraj K, Kumari SS, Patil R, Wax MB, Mathias RT. Functional characterization of a human aquaporin 0 mutation that leads to a congenital dominant lens cataract. Exp Eye Res. 2008;87:9–21. doi: 10.1016/j.exer.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumari SS, Gandhi J, Mustehsan MH, Eren S, Varadaraj K. Functional characterization of an AQP0 missense mutation, R33C, that causes dominant congenital lens cataract, reveals impaired cell-to-cell adhesion. Exp Eye Res. 2013;116:371–385. doi: 10.1016/j.exer.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shentu X, Miao Q, Tang X, Yin H, Zhao Y. Identification and Functional Analysis of a Novel MIP Gene Mutation Associated with Congenital Cataract in a Chinese Family. PloS one. 2015;10:e0126679. doi: 10.1371/journal.pone.0126679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 24.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells. 1998;3:801–810. doi: 10.1046/j.1365-2443.1998.00230.x. [DOI] [PubMed] [Google Scholar]
- 27.Hettmann T, Barton K, Leiden JM. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev Biol. 2000;222:110–123. doi: 10.1006/dbio.2000.9699. [DOI] [PubMed] [Google Scholar]
- 28.Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J Biol Chem. 2009;284:35872–35884. doi: 10.1074/jbc.M109.060384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Firtina Z, Duncan MK. Unfolded Protein Response (UPR) is activated during normal lens development. GEP. 2011;11:135–143. doi: 10.1016/j.gep.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson GW, Andley UP. Activation of the unfolded protein response by a cataract-associated alphaA-crystallin mutation. Biochem Biophys Res Commun. 2010;401:192–196. doi: 10.1016/j.bbrc.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reneker LW, Chen H, Overbeek PA. Activation of unfolded protein response in transgenic mouse lenses. Invest Ophthalmol Vis Sci. 2011;52:2100–2108. doi: 10.1167/iovs.10-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alapure BV, Stull JK, Firtina Z, Duncan MK. The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp Eye Res. 2012;102:28–37. doi: 10.1016/j.exer.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andley UP, Goldman JW. Autophagy and UPR in alpha-crystallin mutant knock-in mouse models of hereditary cataracts. Biochim Biophys Acta. 2016;1860:234–239. doi: 10.1016/j.bbagen.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Z, Yao W, Chan CC, Kannabiran C, Wawrousek E, Hejtmancik JF. Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells. Biochim Biophys Acta. 2016;1862:1214–1227. doi: 10.1016/j.bbadis.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikesugi K, Yamamoto R, Mulhern ML, Shinohara T. Role of the unfolded protein response (UPR) in cataract formation. Expt Eye Res. 2006;83:508–516. doi: 10.1016/j.exer.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 36.Mulhern ML, Madson CJ, Danford A, Ikesugi K, Kador PF, Shinohara T. The unfolded protein response in lens epithelial cells from galactosemic rat lenses. Invest Ophthalmol Vis Sci. 2006;47:3951–3959. doi: 10.1167/iovs.06-0193. [DOI] [PubMed] [Google Scholar]
- 37.Palsamy P, Bidasee KR, Shinohara T. Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp Eye Res. 2014;121:26–34. doi: 10.1016/j.exer.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Zhou S, Gu J, Wang Y, Guo M, Liu Y. Differences in Unfolded Protein Response Pathway Activation in the Lenses of Three Types of Cataracts. PloS one. 2015;10:e0130705. doi: 10.1371/journal.pone.0130705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang HZ, Yang LM. Activation of the unfolded protein response in aged human lenses. Mol Med Rep. 2015;12:389–393. doi: 10.3892/mmr.2015.3417. [DOI] [PubMed] [Google Scholar]
- 40.Lyon MF, Jarvis SE, Sayers I, Holmes RS. Lens opacity: a new gene for congenital cataract on chromosome 10 of the mouse. Genet Res. 1981;38:337–341. doi: 10.1017/s0016672300020668. [DOI] [PubMed] [Google Scholar]
- 41.Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, FitzGerald PG. Characterization of a mutation in the lens-specific CP49 in the 129 strain of mouse. Invest Ophthalmol Vis Sci. 2004;45:884–891. doi: 10.1167/iovs.03-0677. [DOI] [PubMed] [Google Scholar]
- 42.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) BioTechniques. 2000;29:52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 43.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry RA, Lawrence M, Love MI, MacDonald J, Obenchain V, Oles AK, Pages H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, Morgan M. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lou MF, Dickerson JE, Jr, Garadi R, York BM., Jr Glutathione depletion in the lens of galactosemic and diabetic rats. Expt Eye Res. 1988;46:517–530. doi: 10.1016/s0014-4835(88)80009-5. [DOI] [PubMed] [Google Scholar]
- 45.Pendergrass W, Penn P, Possin D, Wolf N. Accumulation of DNA, nuclear and mitochondrial debris, and ROS at sites of age-related cortical cataract in mice. Invest Ophthalmol Vis Sci. 2005;46:4661–4670. doi: 10.1167/iovs.05-0808. [DOI] [PubMed] [Google Scholar]
- 46.Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol. 2009;182:466–476. doi: 10.4049/jimmunol.182.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh-Hashi K, Nomura Y, Shimada K, Koga H, Hirata Y, Kiuchi K. Transcriptional and post-translational regulation of mouse cation transport regulator homolog 1. Mol Cell Biochem. 2013;380:97–106. doi: 10.1007/s11010-013-1663-1. [DOI] [PubMed] [Google Scholar]
- 48.Glimcher LH. XBP1: the last two decades. Ann Rheum Dis. 2010;69 Suppl 1:i67–71. doi: 10.1136/ard.2009.119388. [DOI] [PubMed] [Google Scholar]
- 49.Kumar A, Tikoo S, Maity S, Sengupta S, Sengupta S, Kaur A, Bachhawat AK. Mammalian proapoptotic factor ChaC1 and its homologues function as gamma-glutamyl cyclotransferases acting specifically on glutathione. EMBO Reports. 2012;13:1095–1101. doi: 10.1038/embor.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford RR, Prescott ET, Sylvester CF, Higdon AN, Shan J, Kilberg MS, Mungrue IN. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J Biol Chem. 2015;290:15878–15891. doi: 10.1074/jbc.M114.635144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giblin FJ. Glutathione: a vital lens antioxidant. J Ocul Pharmacol Ther. 2000;16:121–135. doi: 10.1089/jop.2000.16.121. [DOI] [PubMed] [Google Scholar]
- 52.Lou MF. Redox regulation in the lens. Prog Ret Eye Res. 2003;22:657–682. doi: 10.1016/s1350-9462(03)00050-8. [DOI] [PubMed] [Google Scholar]
- 53.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–1493. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- 56.De Maria A, Shi Y, Kumar NM, Bassnett S. Calpain expression and activity during lens fiber cell differentiation. J Biol Chem. 2009;284:13542–13550. doi: 10.1074/jbc.M900561200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Azuma M, Fukiage C, David LL, Shearer TR. Activation of calpain in lens: a review and proposed mechanism. Exp Eye Res. 1997;64:529–538. doi: 10.1006/exer.1996.0234. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura Y, Fukiage C, Shih M, Ma H, David LL, Azuma M, Shearer TR. Contribution of calpain Lp82-induced proteolysis to experimental cataractogenesis in mice. Invest Ophthalmol Vis Sci. 2000;41:1460–1466. [PubMed] [Google Scholar]
- 59.Sakamoto-Mizutani K, Fukiage C, Tamada Y, Azuma M, Shearer TR. Contribution of ubiquitous calpains to cataractogenesis in the spontaneous diabetic WBN/Kob rat. Exp Eye Res. 2002;75:611–617. doi: 10.1006/exer.2002.2065. [DOI] [PubMed] [Google Scholar]
- 60.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chi Z, Byrne ST, Dolinko A, Harraz MM, Kim MS, Umanah G, Zhong J, Chen R, Zhang J, Xu J, Chen L, Pandey A, Dawson TM, Dawson VL. Botch is a gamma-glutamyl cyclotransferase that deglycinates and antagonizes Notch. Cell Reports. 2014;7:681–688. doi: 10.1016/j.celrep.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yasuda M, Tanaka Y, Nishiguchi KM, Ryu M, Tsuda S, Maruyama K, Nakazawa T. Retinal transcriptome profiling at transcription start sites: a cap analysis of gene expression early after axonal injury. BMC Genomics. 2014;15:982. doi: 10.1186/1471-2164-15-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunt D, Raivich G, Anderson PN. Activating transcription factor 3 and the nervous system. Front Mol Neurosci. 2012;5:7. doi: 10.3389/fnmol.2012.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cunard R. Mammalian tribbles homologs at the crossroads of endoplasmic reticulum stress and mammalian target of rapamycin pathways. Scientifica. 2013;2013:750871. doi: 10.1155/2013/750871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res. 2009;88:133–139. doi: 10.1016/j.exer.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Candia OA, Mathias R, Gerometta R. Fluid circulation determined in the isolatedbovine lens. Invest Ophthalmol Vis Sci. 2012;53:7087–7096. doi: 10.1167/iovs.12-10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakajima T, Shearer TR, Azuma M. Loss of calpastatin leads to activation of calpain in human lens epithelial cells. Invest Ophthalmol Vis Sci. 2014;55:5278–5283. doi: 10.1167/iovs.14-14778. [DOI] [PubMed] [Google Scholar]
- 68.Shen Y, Meunier L, Hendershot LM. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J Biol Chem. 2002;277:15947–15956. doi: 10.1074/jbc.M112214200. [DOI] [PubMed] [Google Scholar]
- 69.Ho DV, Chan JY. Induction of Herpud1 expression by ER stress is regulated by Nrf1. FEBS Lett. 2015;589:615–620. doi: 10.1016/j.febslet.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quintens R, Singh S, Lemaire K, De Bock K, Granvik M, Schraenen A, Vroegrijk IO, Costa V, Van Noten P, Lambrechts D, Lehnert S, Van Lommel L, Thorrez L, De Faudeur G, Romijn JA, Shelton JM, Scorrano L, Lijnen HR, Voshol PJ, Carmeliet P, Mammen PP, Schuit F. Mice deficient in the respiratory chain gene Cox6a2 are protected against high-fat diet-induced obesity and insulin resistance. PloS one. 2013;8:e56719. doi: 10.1371/journal.pone.0056719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee MR, Lim CJ, Lee YH, Park JG, Sonn SK, Lee MN, Jung IH, Jeong SJ, Jeon S, Lee M, Oh KS, Yang Y, Kim JB, Choi HS, Jeong W, Jeong TS, Yoon WK, Kim HC, Choi JH, Oh GT. The adipokine Retnla modulates cholesterol homeostasis in hyperlipidemic mice. Nat Commun. 2014;5:4410. doi: 10.1038/ncomms5410. [DOI] [PubMed] [Google Scholar]
- 72.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 73.Wignes JA, Goldman JW, Weihl CC, Bartley MG, Andley UP. p62 expression and autophagy in alphaB-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp Eye Res. 2013;115:263–273. doi: 10.1016/j.exer.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Rad Biol Med. 2015;88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kampinga HH, Garrido C. HSPBs: small proteins with big implications in human disease. Int J Biochem Cell Biol. 2012;44:1706–1710. doi: 10.1016/j.biocel.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 76.Zheng Y, Liu Y, Ge J, Wang X, Liu L, Bu Z, Liu P. Resveratrol protects human lens epithelial cells against H2O2-induced oxidative stress by increasing catalase, SOD-1, and HO-1 expression. Mol Vis. 2010;16:1467–1474. [PMC free article] [PubMed] [Google Scholar]
- 77.Barchet W, Zillinger T. Exonuclease TREX1 also has a sweet tooth. Immunity. 2015;43:411–413. doi: 10.1016/j.immuni.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 78.Huang J, Guo J, Beigi F, Hodgkinson CP, Facundo HT, Zhang Z, Espinoza-Derout J, Zhou X, Pratt RE, Mirotsou M, Dzau VJ. HASF is a stem cell paracrine factor that activates PKC epsilon mediated cytoprotection. J Mol Cell Cardiol. 2014;66:157–164. doi: 10.1016/j.yjmcc.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.