

Abstract
Mitochondria with decreased membrane potential are characterized by defects in protein import into the matrix and impairments in high-efficiency synthesis of ATP. These low-quality mitochondria are marked with ubiquitin for selective degradation. Key factors in this mechanism are PTEN-induced putative kinase 1 (PINK1, a mitochondrial kinase) and Parkin (a ubiquitin ligase), disruption of which has been implicated in predisposition to Parkinson’s disease. Previously, the clearance of damaged mitochondria had been thought to be the end result of a simple cascading reaction of PINK1–Parkin–ubiquitin. However, in the past year, several research groups including ours unexpectedly revealed that Parkin regulation is mediated by PINK1-dependent phosphorylation of ubiquitin. These results overturned the simple hierarchy that posited PINK1 and ubiquitin as the upstream and downstream factors of Parkin, respectively. Although ubiquitylation is well-known as a post-translational modification, it has recently become clear that ubiquitin itself can be modified, and that this modification unexpectedly converts ubiquitin to a factor that functions in retrograde signalling.
Keywords: mitophagy, Parkin, phosphorylation, PINK1, ubiquitin
Various Factors Involved in Mitophagy
ATP, an intracellular energy molecule, is essential for the survival of all cells. Although cells can produce ATP via the glycolysis pathway, the inner mitochondrial membrane (IMM)–localized ATP synthase and associated proton gradient across the IMM [hereafter, described as mitochondrial membrane potential (ΔΨm)] play critical roles in high-efficiency production of ATP. In addition to ATP, mitochondria concomitantly generate reactive oxygen species (ROS) as an inevitable toxic byproduct. Low-quality mitochondria that produce less ATP and more ROS are targeted for degradation by a mitochondrial quality control pathway. Mitophagy, an autophagic degradation mechanism specific to mitochondria, has recently been identified as an essential step in the mitochondrial quality control pathway. Atg32 in yeast and Bcl2-L-13 (Bcl-2-like protein 13), PTEN-induced putative kinase 1 (PINK1), Parkin, NIX/BNIP3L and FUNDC1 in mammalian cells are factors involved in mitophagy. PINK1 and Parkin degrade damaged mitochondria in response to a reduction in ΔΨm (1), whereas NIX/BNIP3L promotes mitochondrial degradation through erythrocyte differentiation, and FUNDC1 in response to hypoxia (2, 3). Atg32, Bcl2-L-13, NIX/BNIP3L and FUNDC1 localize on the outer mitochondrial membrane (OMM), and are thought to promote mitophagy by directly binding to components of the autophagy machinery such as Atg8 (in the case of yeast Atg32) or LC3 (in the cases of mammalian Bcl2-L-13, NIX/BNIP3L and FUNDC1) (2–6). In contrast, PINK1 (a Ser/Thr kinase) and Parkin (a ubiquitin ligase) do not directly interact with components of the autophagy machinery, instead they generate ubiquitylated OMM proteins on depolarized mitochondria, which then functions as an autophagy machinery recognition signal (1, 5, 7).
How to Monitor Mitophagy
Ubiquitin is well-known as a signal for proteasome-dependent degradation; however, it also functions in autophagic degradation. Increasing evidence indicates that selective autophagy functions in intracellular quality control by using ubiquitin tags to delineate aggregated proteins, damaged organelles and invading bacteria for degradation (8). In this process, the predominant mechanism involves simultaneous binding of ubiquitin-tagged cargo and components of the autophagy machinery such as LC3 by autophagy receptors (e.g. p62/SQSTM1, NBR1, NDP52, TAX1BP1 and Optineurin) to promote autophagic degradation (9, 10); however, another mechanism for this pathway (i.e. a direct interaction between ubiquitin and Atg16L1) has also been reported (11).
Around 2010, we believed a simple cascading model in the clearance of damaged mitochondria, namely PINK1→Parkin→ubiquitin→autophagy receptor(s) such as p62/SQSTM1. However, this model was later challenged on multiple fronts. First, although OMM proteins such as TOMM20/Tom20 were conventionally used to monitor mitophagy in early studies (12), later papers reported that some OMM proteins ubiquitylated by Parkin are subjected to proteasomal degradation rather than autophagic degradation (13–15). Second, although Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) had been used routinely to induce mitochondrial stress to trigger mitophagy, CCCP also inhibits lysosomal acidification/function, thus potentially confounding interpretations of the data (16). Moreover, the contribution of p62/SQSTM1 itself to PINK1/Parkin-mediated mitophagy has been controversial (17–19). These findings challenged the validity of early studies and the deduced hypothesis.
To address these issues, the research community established a more reliable system for monitoring mitophagy. Recently, the measurement of mitochondrial DNA as a degradation indicator has been touted as a more robust means of following mitophagy rather than relying on the degradation of OMM proteins (20). In addition, some researchers have implemented a pH-based spectral shift in a mitochondrial fluorescent protein (mt-mKeima) following lysosomal engulfment (20, 21). To depolarize mitochondria, respiratory complex inhibitors oligomycin and antimycin A have been recently utilized as they target mitochondria more specifically than pan-depolarizing agents such as CCCP (20). We believe these methodological changes in experimental design will facilitate mitophagy studies and generate a more accurate picture of the mechanisms driving this cellular process.
Parkin: An RBR-Type Ubiquitin Ligase
Ubiquitylation is a reaction that transfers ubiquitin to a substrate via the functions of three enzymes, E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme) and E3 (ubiquitin ligase). This cascading reaction guarantees a vast variety and specificity of substrates. The PARKIN gene was first identified as a causal gene for autosomal recessive hereditary Parkinsonism (22), and its E3 activity was reported later (23, 24). Parkin possesses an N-terminal ubiquitin-like domain (Ubl domain) and four zinc finger domains (RING0/UPD-RING1-IBR-RING2) in the C terminus (25). Parkin is categorized in RING-IBR-RING (RBR)-type E3 based on the presence of two domains (RING1 and RING2) with amino acid sequence similarity to a canonical RING finger motif and a zinc-binding IBR domain between the RING1 and RING2 domains (IBR means in-between-RING fingers). Subsequently, another zinc finger domain was found closer to the N-terminal end of RING1 and was named the RING0 domain (also referred to as unique PARKIN domain/UPD) (26, 27). However, subsequent structural analysis in 2013 showed that RING1 is the only domain that is structurally similar to a classical RING finger motif, whereas the structures of the other zinc-binding domains (e.g. RING0, IBR and RING2) are completely different from that of a typical RING finger motif (27–30).
The unique enzymatic properties of Parkin as an E3 enzyme were revealed in studies published from 2011 to 2013. Rachel Klevit’s group initially suggested that RBR-type E3s such as human homolog of Drosophila ariadne (HHARI) and Parkin form a thioester bond with ubiquitin at a catalytic cysteine in the RING2 domain, and that this ubiquitin–thioester bond serves as a reaction intermediate for catalyzing ubiquitylation (31). We now postulate that when Parkin catalyzes ubiquitylation, the thioester-bonded ubiquitin on E2 is transferred to Cys431 (the active centre) in the RING2 domain as a transthiolation reaction. This is then followed by an acyl transfer reaction in which the Cys431 ubiquitin is transferred to the lysine residue on the substrate to yield a stable isopeptide bond (29, 32–34).
Mechanism for How PINK1 Recognizes Mitochondrial Damage and How It Is Activated on Damaged Mitochondria
In 2004, PINK1 was reported as a causal gene for autosomal recessive hereditary Parkinsonism (35). The PINK1 protein has a mitochondrial targeting signal (MTS), an N-terminal transmembrane region, and C-terminal kinase domain (35, 36) (Fig. 1A). Subsequent to its identification, genetic interactions between PINK1 and PARKIN were elucidated (37).
Fig. 1.
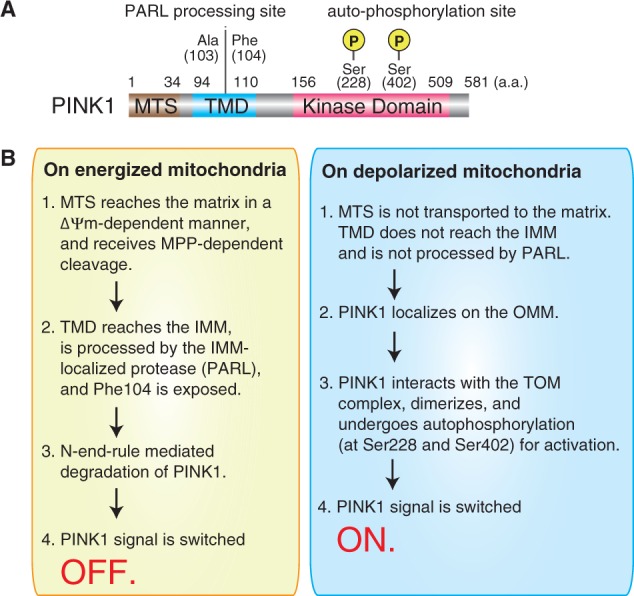
Schematic depiction of PINK1. (A) Domain structure and important residues of PINK1. (B) Molecular mechanism underlying PINK1 accumulation and activation on depolarized mitochondria (see text for details).
An unique feature of PINK1 is its stringent and specific localization on damaged mitochondria (7) (Fig. 1B). Under steady-state (normal) conditions, PINK1 is targeted to mitochondria in a ΔΨm-dependent manner with its N-terminal MTS localized to the mitochondrial matrix, and then receives mitochondrial processing peptidase–dependent cleavage of MTS. During this process, the neighbouring transmembrane region becomes embedded in the IMM and is cleaved by an intramembrane cleaving protease referred to as presenilins-associated rhomboid-like (PARL) that localizes in the IMM. After this cleavage, a 104th phenylalanine residue exposed at the N terminus of the processed PINK1 serves as a signal for N-end rule pathway-dependent degradation (38) (Fig. 1A). Therefore, under physiological conditions with normal ΔΨm, PINK1 is rapidly degraded and barely detectable (39, 40). In contrast, a decrease in ΔΨm inhibits both the matrix targeting of MTS and the IMM localization of the transmembrane domain, and as a consequence, PINK1 escapes both PARL-dependent cleavage and N-end rule pathway-dependent degradation, and accumulates on the OMM of damaged mitochondria (41) (Fig. 1B). In other words, the presence or absence of ΔΨm determines which mitochondrial compartment PINK1 is targeted to, and is the key for identifying damaged mitochondria.
To activate PINK1 on damaged mitochondria, interaction with the translocase of the outer membrane (TOM) complex and autophosphorylation of PINK1 are important. When ΔΨm decreases, PINK1 forms a super-molecular complex (∼850 kDa) comprised of components of the TOM complex and dimeric PINK1 that facilitates intermolecular autophosphorylation between two PINK1 molecules (42–44) (Fig. 1A). This active form of PINK1 phosphorylates both Parkin and ubiquitin, an event that turns on the switch for mitochondrial degradation.
PINK1 Phosphorylates Parkin
In 2013, Parkin auto-inhibition was clarified and the molecular mechanism that governs this state was revealed. Parkin ubiquitylation of either pseudo-substrates fused to Parkin or genuine substrates on the OMM is dependent on a reduction in ΔΨm. In 2008, Parkin-mediated degradation of damaged mitochondria following its translocation was first reported (12). Two years later, several research groups reported that PINK1 is essential for both Parkin recruitment to damaged mitochondria and Parkin E3 activity (17, 39, 40, 45, 46). In 2013, structural studies of Parkin revealed that the basis for the auto-inhibition mechanism is occlusion of the catalytically active centre (Cys431) in the RING2 domain by the RING0 (UPD) domain (27–29).
PINK1-dependent phosphorylation of Parkin is a required step in Parkin activation. Ser65 in the Ubl domain of Parkin is phosphorylated by PINK1 in a ΔΨm-dependent process (32, 47, 48) (Fig. 2A). However, the E3 activity of a Ser65 phosphomimetic Parkin mutant is barely detectable, and its activation requires both a decrease in ΔΨm and PINK1 involvement, suggesting that another PINK1-catalyzed phosphorylation event is critical.
Fig. 2.
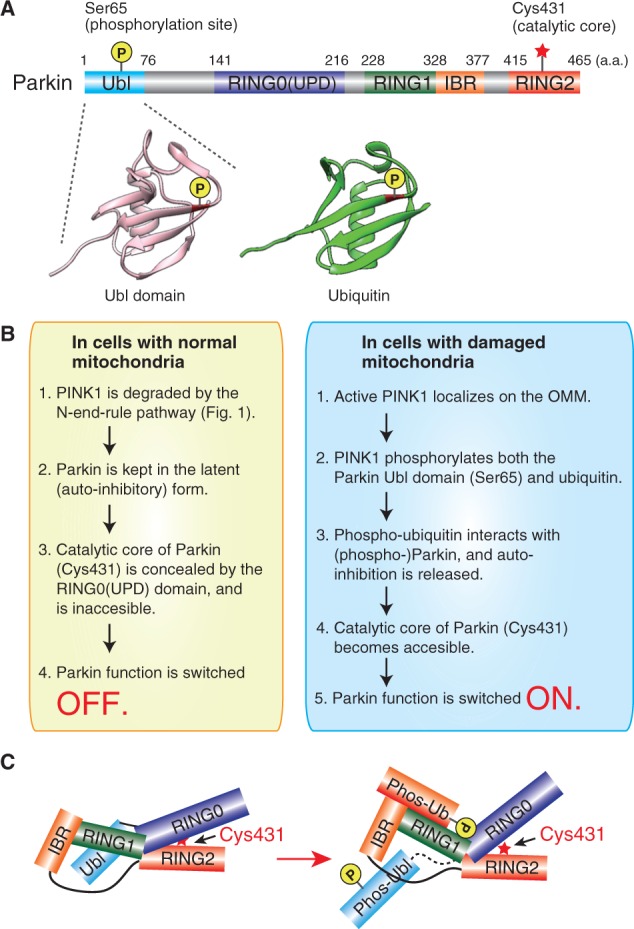
Schematic depiction of Parkin. (A) Domain structure and important residues of Parkin. The structural similarities between Ubl domain and ubiquitin are also illustrated. (B) Current model for the molecular mechanism underlying Parkin activation on depolarized mitochondria. (C) Very recently, structural and biochemical analyses revealed how phosphorylated ubiquitin interacts with Parkin and changes its structure as illustrated. However, because of space and time limitations, these reports have regrettably not been covered in this review.
Phosphorylated Ubiquitin Activates Parkin and Changes Its Localization
Based on structural similarities between the Ubl domain of Parkin and ubiquitin, and determination that the Ser65 phosphorylation site is conserved between Parkin Ubl and ubiquitin (Fig. 2A), we hypothesized that ubiquitin may be phosphorylated by PINK1. Multiple reports have since verified that ubiquitin is indeed phosphorylated by PINK1, and to our surprise, this phosphorylation event is the missing step needed to convert Parkin to its fully active form (49–51). When phosphomimetic mutations were introduced in both the Parkin Ubl domain and ubiquitin in cells, Parkin E3 activity was observed even in the absence of PINK1 and in the presence of normal ΔΨm, and phosphorylated ubiquitin itself can activate phosphorylated Parkin in vitro (49–51). Physical interactions between Parkin and ubiquitin have been observed in cells only when both proteins possess phosphomimetic mutations (51). These results suggest that phosphorylated ubiquitin functions as a Parkin activator by liberating the auto-inhibition mechanism (Fig. 2B and C). Ubiquitin-like modifiers (Ubls) that possess ubiquitin-like structures (e.g. SUMO1 and ISG15) are not phosphorylated by PINK1 (50, 51), suggesting that PINK1 specifically phosphorylates ubiquitin and the Ubl domain of Parkin, and does not phosphorylate other proteins despite the presence of a ubiquitin-like structure.
After this discovery, the mechanism driving Parkin localization to damaged mitochondria became the prime focus of study. PINK1 can phosphorylate not only monoubiquitin but also ubiquitin chains, and the importance of a phosphorylated ubiquitin chain for Parkin localization to damaged mitochondria was recently revealed (52–54).
Translocation of Parkin to damaged mitochondria was inhibited when its E3 activity was attenuated by RING2 domain mutations such as C431S/F/N. Intriguingly, co-expression with wild-type Parkin, an E3-competent R275W mutant, or a tandem ubiquitin chain containing a mitochondrial localization signal (Tom70-4xUbiG76V) efficiently restored mitochondrial localization of the Parkin mutants lacking E3 activity (e.g. C431S and C431N) (33, 34). In these cases, a decrease in ΔΨm and the presence of PINK1 were required, suggesting a cooperative product of Parkin-catalyzed ubiquitylation and PINK1 recruits E3-imcompetent Parkin mutants to damaged mitochondria. Importantly, introduction of a phospho-mimetic mutation into both Parkin and a mitochondria-localized tandem ubiquitin chain (Mt-4xUb) enabled Parkin to localize on mitochondria irrespective of ΔΨm and PINK1 (54). Furthermore, Parkin preferentially binds phosphorylated polyubiquitin chains in vitro (52–54). These results strongly suggest that a PINK1 phosphorylated ubiquitin chain is the most probable receptor for Parkin.
Model for the Tangled Relationship between PINK1, Parkin and Ubiquitin
An important issue in PINK1-mediated ubiquitin phosphorylation is that it overturns the previous hierarchical hypothesis of PINK1, Parkin and ubiquitin. As indicated earlier, the predominant model in this field proposed that PINK1 accumulation on mitochondria following dissipation of ΔΨm recruits Parkin, which then conjugates ubiquitin to OMM substrates, and that this ubiquitylation links depolarized mitochondria to the proteasomal and autophagic degradation pathways. In this context, a simple cascading reaction of PINK1→Parkin→ubiquitin leads to the clearance of damaged mitochondria. However, in 2014–2015, several research groups including ours unexpectedly revealed that PINK1 phosphorylates ubiquitin (49–51). These results overturned the simple hierarchy that posited PINK1 and ubiquitin as the upstream and downstream factors of Parkin, respectively. Instead, the new findings prompted us to consider that already-existing ubiquitin could be upstream of Parkin because it functions as a Parkin receptor/activator when phosphorylated by PINK1, and that PINK1 might be downstream of Parkin because in the subsequent round Parkin-catalyzed ubiquitin becomes a PINK1 substrate. In fact, although Parkin is activated and recruited to depolarized mitochondria by phosphorylated ubiquitin, localization of phosphorylated ubiquitin on damaged mitochondria depends on Parkin, revealing the interdependent relationship in mitochondrial localization of both Parkin and phosphorylated ubiquitin (54). To explain these results, a positive feedback model is conceivable. Namely, PINK1 phosphorylates a basal-level mitochondrial ubiquitin chain and Parkin is subsequently recruited to the mitochondria to ubiquitylate OMM substrates. The initiating ubiquitin chain functions as a PINK1 substrate again, thereby causing a local accumulation of phosphorylated ubiquitin (49, 52–54). Collectively, PINK1, Parkin and ubiquitin do not make a linear cascading reaction but rather they form tripartite interdependent reactions (Fig. 3).
Fig. 3.
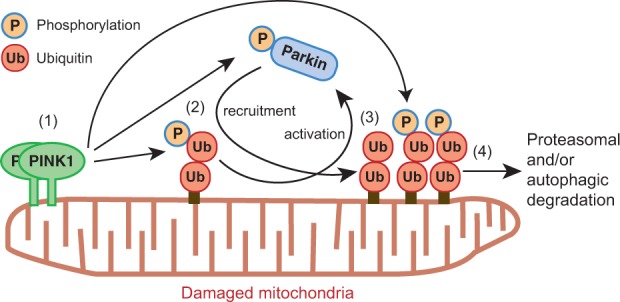
Positive feedback model for PINK1- and Parkin-catalyzed ubiquitylation. Previously, it was thought that PINK1 accumulation on damaged mitochondria recruited Parkin, and that Parkin-catalyzed ubiquitylation was the key signal for degradation. A new model suggests that accumulated PINK1 on damaged mitochondria (1) phosphorylates Parkin and ubiquitin, which (2) induces Parkin activation and its recruitment to the phosphorylated ubiquitin chain. Activated Parkin produces more ubiquitin chain (3), and the resultant ubiquitin is phosphorylated by PINK1 in a positive feedback cycle. Parkin thus functions as an amplifier of the (phospho-)ubiquitin chain on depolarized mitochondria (4) for degradation.
Autophagy Receptors Interact with Phosphorylated Ubiquitin
The mechanisms of how Parkin is transported to the damaged mitochondria and how Parkin is activated by PINK1 have been clarified considerably. In contrast, the molecular mechanisms of the subsequent process of how damaged mitochondria are degraded remains obscure and sometimes controversial. Very recently, Richard Youle’s group proposed a novel hypothesis in which phospho-ubiquitin again plays a main part in the process. They measured mitophagy activity following knockout of five autophagy receptors (p62/SQSTM1, NBR1, NDP52, TAX1BP1 and Optineurin) individually or in combination and found that p62/SQSTM1 and NBR1 are dispensable, whereas NDP52 and Optineurin are essential for PINK1/Parkin-mediated mitophagy (20). This finding is partly consistent with previous studies (18, 19, 55). Interestingly, NDP52 and Optinurin are also known as autophagy receptors for Xenophagy, a specialized process to eliminate the infecting pathogens via autophagy (8). The ubiquitin-binding domain of either NDP52 or Optineurin is essential for their translocation to depolarized mitochondria and mitophagy activity. Intriguingly, coupling exogenous PINK1 to normal mitochondria was sufficient to recruit Optineurin and NDP52 even in the absence of Parkin and mitochondrial damage, suggesting that Parkin-catalyzed ubiquitylation is dispensable for their mitochondrial recruitment. Nevertheless, ubiquitin-binding mutants do not translocate to mitochondria under this experimental condition. These results seemingly contradict each other; however, phosphorylated ubiquitin can disentangle this complex process.
Endogenous Optineurin and NDP52 are preferentially co-immunoprecipitated with phosphomimetic ubiquitin from cells, and are even more readily pulled down with phosphorylated ubiquitin in vitro. Given that Optineurin and NDP52 induce mitophagy in response to recognizing phosphorylated ubiquitin, the discrepancy in results described above can be clarified if PINK1 phosphorylates already-existing ubiquitin. The resulting phospho-ubiquitin then functions as the key signal in recruiting Optineurin and NDP52, which engage components of the autophagy pathway (e.g. ULK1 and LC3) to initiate mitophagy (Fig. 4). This model is also consistent with confounding results which showed that the ubiquitin-binding activities of Optineurin and NDP52 are required even for Parkin-independent mitochondrial recruitment.
Fig. 4.
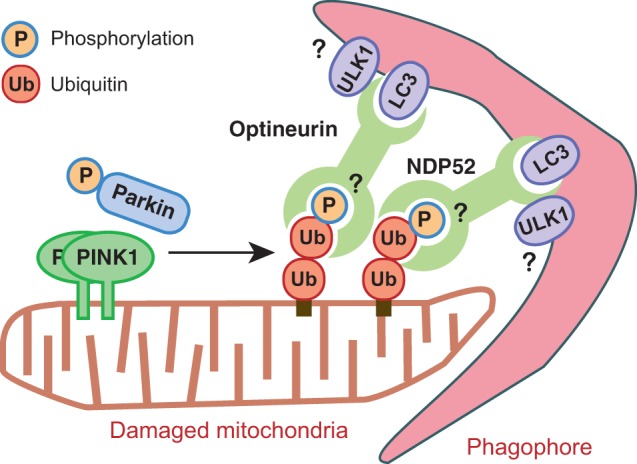
Recently proposed new model for ubiquitin-mediated mitophagy. Although further validation studies remain to be demonstrated, this new model suggests that PINK1 and Parkin cooperate to generate phosphorylated ubiquitin chains on depolarized mitochondria, which then induce Optineurin and NDP52 recruitment to initiate mitophagy. In this context, PINK1 generates a mitophagy signal per se, and Parkin serves as an amplifier of the signal.
This new model, however, does not discount the role of Parkin in mitophagy. Parkin expression dramatically increased Optineurin-dependent mitophagy, presumably because Parkin assists the process as an amplifier of phospho-ubiquitin by setting up the aforementioned positive feedback cycle (Fig. 3). Strong genetic interactions between PINK1 and PARKIN in model systems have been reported (37), and recessive familial Parkinsonism with similar symptoms is caused by mutations of either PINK1 or PARKIN in humans (56), clearly indicating the essential role of Parkin under physiological conditions.
Although further biochemical and structural studies should help to understand this novel model, the phospho-ubiquitin–binding activity of Optineurin and NDP52 (20) provides a significant clue to understand how phosphorylated ubiquitin assists degradation of damaged mitochondria.
Conclusion Remarks
Phosphorylation of ubiquitin Ser65, which we and other researchers discovered, can be used as an indicator of active PINK1/Parkin and damaged mitochondria. This knowledge could potentially be developed as a pathological or clinical molecular marker for Parkinson’s disease in the future. However, I think more important aspects of this finding are that it overturns the classical hierarchy between the upstream ubiquitin ligase and the downstream ubiquitin, and can open new avenues for further exploration in ubiquitin and autophagy studies. To date, ubiquitylation has been a well-known post-translational modification; however, it is becoming increasingly clear that modified ubiquitin itself plays a critical cellular function (57). This work can establish new principles of how a simple ubiquitin tag plays more varied roles than expected.
Note Added in Proof
After this manuscript was accepted, Heo et al. reported (58) that Parkin-catalyzed ubiquitin chains on mitochondria trigger autophagy adaptor recruitment concomitantly with activation and recruitment of TBK1 kinase. Importantly, the TBK1 kinase activated by mitochondrial depolarization results in phosphorylation of S473 in OPTN and a dramatic increase in the binding of OPTN to poly-ubiquitin chains in vitro. Under their experimental conditions, phosphorylation of S65 in the poly-ubiquitin chains inhibited OPTN interactions, however, the S473 phosphorylated OPTN retained the ability to interact with the S65 phosphorylated poly-ubiquitin chains, albeit less efficiently than with unphosphorylated poly-ubiquitin chains. These results suggest that OPTN S473 phosphorylation, rather than ubiquitin S65 phosphorylation, is a key step for efficient mitophagy. Thus to develop more accurate models for the role phospho-ubiquitin plays in ubiquitin-mediated mitophagy further validation studies are required.
Funding
This work was supported by JSPS PRESTO, KAKENHI Grant Number 26650042, MEXT KAKENHI Grant Numbers 26111729 and 15H01196, the Tomizawa Jun-ichi and Keiko Fund for Young Scientist and by the Takeda Science Foundation to N.M.
Conflict of Interest
None declared.
References
- 1.Eiyama A., Okamoto K. (2015) PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101 [DOI] [PubMed] [Google Scholar]
- 2.Youle R.J., Narendra D.P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu L., Sakakibara K., Chen Q., Okamoto K. (2014) Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murakawa T., Yamaguchi O., Hashimoto A., Hikoso S., Takeda T., Oka T., Yasui H., Ueda H., Akazawa Y., Nakayama H., Taneike M., Misaka T., Omiya S., Shah A.M., Yamamoto A., Nishida K., Ohsumi Y., Okamoto K., Sakata Y., Otsu K. (2015) Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6, 7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okamoto K. (2014) Organellophagy: eliminating cellular building blocks via selective autophagy. J. Cell Biol. 205, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanki T., Furukawa K., Yamashita S.I. (2015) Mitophagy in yeast: molecular mechanisms and physiological role. Biochim. Biophys. Acta. 1853, 2756–2765 [DOI] [PubMed] [Google Scholar]
- 7.Pickrell A.M., Youle R.J. (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N., Komatsu M. (2011) Autophagy: renovation of cells and tissues. Cell 147, 728–741 [DOI] [PubMed] [Google Scholar]
- 9.Birgisdottir A.B., Lamark T., Johansen T. (2013) The LIR motif—crucial for selective autophagy. J. Cell Sci. 126, 3237–3247 [DOI] [PubMed] [Google Scholar]
- 10.Ichimura Y., Kumanomidou T., Sou Y.S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 283, 22847–22857 [DOI] [PubMed] [Google Scholar]
- 11.Fujita N., Morita E., Itoh T., Tanaka A., Nakaoka M., Osada Y., Umemoto T., Saitoh T., Nakatogawa H., Kobayashi S., Haraguchi T., Guan J.L., Iwai K., Tokunaga F., Saito K., Ishibashi K., Akira S., Fukuda M., Noda T., Yoshimori T. (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J. Biol. 203, 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narendra D., Tanaka A., Suen D.F., Youle R.J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka A., Cleland M.M., Xu S., Narendra D.P., Suen D.F., Karbowski M., Youle R.J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshii S.R., Kishi C., Ishihara N., Mizushima N. (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan N.C., Salazar A.M., Pham A.H., Sweredoski M.J., Kolawa N.J., Graham R.L., Hess S., Chan D.C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padman B.S., Bach M., Lucarelli G., Prescott M., Ramm G. (2013) The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 9, 1862–1875 [DOI] [PubMed] [Google Scholar]
- 17.Geisler S., Holmstrom K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 18.Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y.S., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narendra D., Kane L.A., Hauser D.N., Fearnley I.M., Youle R.J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarou M., Sliter D.A., Kane L.A., Sarraf S.A., Wang C., Burman J.L., Sideris D.P., Fogel A.I., Youle R.J. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katayama H., Kogure T., Mizushima N., Yoshimori T., Miyawaki A. (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18, 1042–1052 [DOI] [PubMed] [Google Scholar]
- 22.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 23.Shimura H., Hattori N., Kubo S., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., Suzuki T. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 [DOI] [PubMed] [Google Scholar]
- 24.Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., Tanaka K. (2006) Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281, 3204–3209 [DOI] [PubMed] [Google Scholar]
- 25.Koyano F., Matsuda N. (2015) Molecular mechanisms underlying PINK1 and Parkin catalyzed ubiquitylation of substrates on damaged mitochondria. Biochim. Biophys. Acta 1853, 2791–2796 [DOI] [PubMed] [Google Scholar]
- 26.Hristova V.A., Beasley S.A., Rylett R.J., Shaw G.S. (2009) Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 284, 14978–14986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wauer T., Komander D. (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 32, 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trempe J.F., Sauve V., Grenier K., Seirafi M., Tang M.Y., Menade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G., Nagar B., Fon E.A., Gehring K. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 29.Riley B.E., Lougheed J.C., Callaway K., Velasquez M., Brecht E., Nguyen L., Shaler T., Walker D., Yang Y., Regnstrom K., Diep L., Zhang Z., Chiou S., Bova M., Artis D.R., Yao N., Baker J., Yednock T., Johnston J.A. (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spratt D.E., Julio Martinez-Torres R., Noh Y.J., Mercier P., Manczyk N., Barber K.R., Aguirre J.D., Burchell L., Purkiss A., Walden H., Shaw G.S. (2013) A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat. Commun. 4, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenzel D.M., Lissounov A., Brzovic P.S., Klevit R.E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iguchi M., Kujuro Y., Okatsu K., Koyano F., Kosako H., Kimura M., Suzuki N., Uchiyama S., Tanaka K., Matsuda N. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng X., Hunter T. (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res. 23, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazarou M., Narendra D.P., Jin S.M., Tekle E., Banerjee S., Youle R.J. (2013) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol. 200, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G., Albanese A., Nussbaum R., Gonzalez-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W.P., Latchman D.S., Harvey R.J., Dallapiccola B., Auburger G., Wood N.W. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 36.Unoki M., Nakamura Y. (2001) Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 20, 4457–4465 [DOI] [PubMed] [Google Scholar]
- 37.Pallanck L., Greenamyre J.T. (2006) Neurodegenerative disease: pink, parkin and the brain. Nature 441, 1058. [DOI] [PubMed] [Google Scholar]
- 38.Yamano K., Youle R.J. (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., Sou Y.S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okatsu K., Kimura M., Oka T., Tanaka K., Matsuda N. (2015) Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 128, 964–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okatsu K., Uno M., Koyano F., Go E., Kimura M., Oka T., Tanaka K., Matsuda N. (2013) A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 288, 36372–36384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazarou M., Jin S.M., Kane L.A., Youle R.J. (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev. Cell 22, 320–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okatsu K., Oka T., Iguchi M., Imamura K., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., Sato S., Shimizu H., Fukunaga Y., Taniguchi H., Komatsu M., Hattori N., Mihara K., Tanaka K., Matsuda N. (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziviani E., Tao R.N., Whitworth A.J. (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U. S. A. 107, 5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R.L., Kim J., May J., Tocilescu M.A., Liu W., Ko H.S., Magrane J., Moore D.J., Dawson V.L., Grailhe R., Dawson T.M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H.I., Campbell D.G., Gourlay R., Burchell L., Walden H., Macartney T.J., Deak M., Knebel A., Alessi D.R., Muqit M.M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori N. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kane L.A., Lazarou M., Fogel A.I., Li Y., Yamano K., Sarraf S.A., Banerjee S., Youle R.J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D.G., Ritorto M.S., Hofmann K., Alessi D.R., Knebel A., Trost M., Muqit M.M. (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E.A., Trempe J.F., Saeki Y., Tanaka K., Matsuda N. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 [DOI] [PubMed] [Google Scholar]
- 52.Ordureau A., Sarraf S.A., Duda D.M., Heo J.M., Jedrychowski M.P., Sviderskiy V.O., Olszewski J.L., Koerber J.T., Xie T., Beausoleil S.A., Wells J.A., Gygi S.P., Schulman B.A., Harper J.W. (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shiba-Fukushima K., Arano T., Matsumoto G., Inoshita T., Yoshida S., Ishihama Y., Ryu K.Y., Nukina N., Hattori N., Imai Y. (2014) Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 10, e1004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okatsu K., Koyano F., Kimura M., Kosako H., Saeki Y., Tanaka K., Matsuda N. (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong Y.C., Holzbaur E.L. (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U. S. A. 111, E4439–E4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corti O., Lesage S., Brice A. (2011) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 91, 1161–1218 [DOI] [PubMed] [Google Scholar]
- 57.Herhaus L., Dikic I. (2015) Expanding the ubiquitin code through post-translational modification. EMBO Rep. 16, 1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heo J.M., Ordureau A., Paulo J.A., Rinehart J., Harper J.W. (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]