

Abstract
Six aspartic proteinase precursors, a pro-cathepsin E (ProCatE) and five pepsinogens (Pgs), were purified from the stomach of adult newts (Cynops pyrrhogaster). On sodium dodecylsulfate-polyacrylamide gel electrophoresis, the molecular weights of the Pgs and active enzymes were 37–38 kDa and 31–34 kDa, respectively. The purified ProCatE was a dimer whose subunits were connected by a disulphide bond. cDNA cloning by polymerase chain reaction and subsequent phylogenetic analysis revealed that three of the purified Pgs were classified as PgA and the remaining two were classified as PgBC belonging to C-type Pg. Our results suggest that PgBC is one of the major constituents of acid protease in the urodele stomach. We hypothesize that PgBC is an amphibian-specific Pg that diverged during its evolutional lineage. PgBC was purified and characterized for the first time. The purified urodele pepsin A was completely inhibited by equal molar units of pepstatin A. Conversely, the urodele pepsin BC had low sensitivity to pepstatin A. In acidic condition, the activation rates of newt pepsin A and BC were similar to those of mammalian pepsin A and C1, respectively. Our results suggest that the enzymological characters that distinguish A- and C-type pepsins appear to be conserved in mammals and amphibians.
Keywords: newt, pepsinogen, pro-cathepsin E, purification, stomach
Aspartic proteinases are proteinases that have two aspartyl residues at the centre of active site include vertebrate pepsin, cathepsin D, cathepsin E and renin. In vertebrates, pepsin plays a role in the digestion of food proteins in the digestive tract, and its precursor is localized in the mucosa. The precursors of pepsins, pepsinogens (Pgs), are secreted into the lumen from the mucosal epithelium. The conversion to pepsins occurs by release of the N-terminal propeptide, a process that occurs autocatalytically in the acidic conditions of the lumen (1). The molecular evolution of Pgs has been well documented based on comparisons of the primary structures (2–4) obtained from many vertebrates. Pgs are classified into two types, A and C, and the two types are further classified into several groups. A-type Pgs are divided into PgA, PgF, prochymosin and fish PgA. C-type Pgs (progastricsin) include PgB, PgBC, PgC1, PgC2 and fish PgC. PgBC was found in Xenopus tropicalis genome sequences and is thought to be an amphibian-specific Pg (3).
Unlike with Pgs, cathepsin D (CatD) and E (CatE) are not secreted into the lumen of the digestive tract (5). It is thought that CatD, which functions as a cell-signaling molecule, plays a physiological role in intracellular digestion. Additionally, pro-cathepsin D/CatD also plays a role in pathological processes such as cancer progression (6). CatE is localized in other cell organelles, such as the endoplasmic reticulum, endosomes, the Golgi body and the plasma membrane (7, 8). CatE plays a physiological role in antigen processing and in defence against microorganism infection, and it has a pathological role in cancer (8, 9). Despite these reports, the functions of cathepsin E are not fully understood.
It is thought that the aspartic proteinase genes were derived from a common ancestor (2, 3, 10). In amphibians, the enzymatic properties and primary structures of Pgs have been documented in anurans, such as the bullfrog (Rana catesbeiana; 11–16) and African clawed frog (Xenopus laevis; 16, 17). Information regarding urodele aspartic proteinases is limited to the histochemical localization of Pg (18). We recently obtained three cDNA Pg clones (PgC1, PgC2 and PgA) from the Japanese fire belly newt (Cynops pyrrhogaster; 19); however, these Pgs have not yet been purified. To improve our knowledge of urodele aspartic proteinases, there is a need to understand the molecular evolution of aspartic proteinases.
We purified and cloned the aspartic proteinases from the stomach of C. pyrrhogaster and performed a gene phylogenetic analysis. The enzymatic properties of PgBC, purified in this study, were investigated and compared with those of other vertebrates. To our knowledge, this is the first report on the purification and cloning of PgBC.
Materials and Methods
Ethics statement
The experiments were performed in accordance with the Law for the Humane Treatment and Management of Animals in Japan (20). The law was enacted in 1973, amended in 2013 and coordinated with Institutional Animal Care and Use Committee (IACUC) protocols (21).
Preparation of crude extract
Adult newts (C. pyrrhogaster) were obtained from a commercial supplier. Thirty adult newts were anesthetized on ice and their spinal cords were cut. The stomach was removed from each individual with scissors and subsequently washed with 0.65% NaCl saline solution, then minced and homogenized in 20 mM Tris-HCl buffer (pH 7.5) (19 volumes [v/w] of stomach weight). The homogenate was centrifuged at 10,000 × g for 30 min at 4°C. The supernatant (the crude extract) was used as the starting material for the purification of aspartic proteinases.
Electrophoresis
Polyacrylamide gel electrophoresis (native-PAGE) was carried out using a CompactPAGE AE7300 (ATTO, Tokyo, Japan) following the manufacturer’s instructions with a slight modification. We used a c-PAGEL (ATTO) 15% or 10% acrylamide gel. The sample (20 μl) was mixed with 2 μl of 50% glycerol and 2 μl of 1% bromophenol blue then subjected to PAGE. Electrophoresis was performed at room temperature.
Zymography
The bands that had acid proteinase activity were visualized using a previously published method (12) with a slight modification. After native-PAGE, the gel was incubated with 2% bovine haemoglobin in 0.1 M sodium formate buffer (pH 3.0) at 37°C for 30 min and then incubated in the buffer without haemoglobin for 30 min at 37°C. The gel was then incubated in Coomassie Brilliant Blue (CBB) solution, EzStainAQua (ATTO), warmed in a microwave oven, then incubated for 30 min at room temperature. To decolorize extra pigment, the gel was immersed in distilled water, warmed in a microwave oven, then incubated for 30 min at room temperature.
Sodium dodecylsulfate PAGE
Sodium dodecylsulfate (SDS)-PAGE was carried out following the method of Laemmli (22) using a 15% gel. The molecular weights of aspartic proteinases and their pro-enzymes were calculated using Molecular Weight Marker II (Daiichi Pure Chemicals Co., Tokyo, Japan) or Ez Standard (ATTO). Staining with CBB for protein bands was conducted using EzStainAQua following the manufacturer’s instructions. Staining with silver for protein bands was conducted using a Silver Stain II Kit (Wako, Osaka, Japan) following the manufacturer’s instructions.
Determination of protein concentration
The amount of protein was estimated using a BCA Protein Assay Kit (Pierce, Rockford, IL) following the manufacturer’s instructions with bovine serum albumin as a standard.
Standard assay for aspartic proteinase
The proteolytic activity of acid proteinases was measured using a previously published method (12) with slight modification. The reaction mixture (0.45 ml) containing 2.5% bovine haemoglobin, 0.25 M sodium formate buffer (pH 3.0) and enzyme sample was incubated at 37°C for 30 min. The reaction was stopped by adding 2.5 ml of 3.0% [w/v] trichloroacetic acid. After the mixture was centrifuged at 440 × g for 10 min, the absorbance of the supernatant was measured at 280 nm. One unit of activity was defined as a change of absorbance of 1.0 during an incubation time of 30 min.
Purification of aspartic proteinase precursors
Chromatography was performed at 4°C, except the Mono Q column (GE Healthcare, Buckinghamshiere, UK) chromatography, which was performed at room temperature.
Q-Sepharose anion exchange column chromatography
The crude extract was applied to a Q-Sepharose fast flow column (2.2 × 11 cm; GE Healthcare) equilibrated with 20 mM Tris-HCl buffer (pH 7.5). After washing the column with the same buffer containing 0.1 M NaCl, elution was performed with a liner gradient of relevant molar NaCl in 20 mM Tris-HCl buffer (pH 7.5).
Sephacryl S-200 gel filtration column chromatography
The fractions obtained from Q-Sepharose chromatography that exhibited proteolytic activity were concentrated using an Amicon U-15 ultrafiltration centrifugal unit (Millipore, Bedford, MA), then applied to a column (2.6 × 60 cm) of Sephacryl S-200 (GE Healthcare) equilibrated with 0.15 M NaCl in 20 mM Tris-HCl buffer (pH 7.5) and fractionated with the same buffer.
Con-A Sepharose affinity column chromatography
The pro-enzyme fraction containing pro-cathepsin E (ProCatE) was applied to a Con-A Sepharose column (1.2 × 8.5 cm; GE Healthcare) equilibrated with 0.5 M NaCl in 20 mM Tris-HCl buffer (pH 7.5). After washing them with the same buffer, the bound proteins were eluted with 0.3 M methyl-α-D-mannopyranoside in the same buffer.
Mono Q column chromatography
The pro-enzyme fraction obtained during the former step was diluted with 20 mM Tris-HCl buffer (pH7.5) and applied to a Mono Q column (5 × 50 mm) equilibrated with the same buffer. The column was washed with 20 mM Tris-HCl buffer (pH 7.5), containing adequate molar NaCl and then eluted with a liner gradient of NaCl.
Time course of Pg activation under acidic conditions
The pH of the purified Pg solution was adjusted to 2.0 by adding 1 N HCl. After incubation at 22°C, aliquots of the solution were withdrawn at appropriate intervals. To stop the activation reaction, the samples were neutralized by adding 1 N NaOH and then subjected to SDS-PAGE.
The inhibitory effect of pepstatin A
The purified Pgs were activated by adding HCl at pH 2.0 and the activated sample was added into a standard assay reaction mixture containing relevant molar pepstatin A. The mixture was incubated at 37°C for 30 min and the remaining activity was determined using the method outlined above (standard assay for aspartic proteinase).
pH dependency of pepsin activity
The pH dependency of acid-treated Pg activity was examined using the standard assay for aspartic proteinases and using sodium acetate-HCl buffer instead of formate buffer over a range of pH 1.0–4.0.
Determination of amino acid sequences of N-terminal regions of pro-enzyme and activated enzyme
The amino acid sequences of the pro-enzymes and activated enzymes were determined by Edman degradation using an automatic amino acid sequencer (Procise 491HT: Applied Biosystems, Foster City, CA). The purified pro-enzyme or activated sample was subjected to SDS-PAGE and protein bands were blotted electrically to PVDF membrane. The membrane was stained by 0.1% CBB (Sigma Aldrich St. Louis, MO). The stained bands were cut out, decolorized in methanol and applied to the sequencer.
Cloning of cDNAs for C. pyrrhogaster aspartic proteinases
Total RNA was extracted from the stomach of adult newts. The cDNA was synthesized using a SMART RACE cDNA Amplification kit (Clontech, Palo Alto, CA). The fragment was amplified by polymerase chain reaction (PCR) using the degenerate primers based on the N-terminal amino acid sequence of purified proteins or the highly conservative sequence at the active site of aspartic proteinases in other vertebrates. The degenerate primers are summarized in Table I. The fragments thus obtained were inserted into a pMD20 T-vector (Takara, Shiga, Japan). Nucleotide sequences of the fragments were determined using a 3130 DNA sequencer (Applied Biosystems) with a Big Dye Cycle Sequencing Kit. The full-length sequence was obtained from 3′-RACE and 5′-RACE using the gene specific primers designed from the cloned fragments.
Table I.
Sequences of degenerate primers used for RT-PCR
| Gene and primer name | Primer sequence |
|---|---|
| Pro-enzyme I | |
| F 1st | CAKAAYYTRGAYATGGTYCARTACGARGA |
| F nest | GGTBCCHTCTGTGTACTGCAYHGCCMAGC |
| R nest | ATCCAGBKSWATYTGCCAGTAGSCTTG |
| R 1st | AARACATCYCCYAGRATCCASARDGGTCC |
| Pro-enzyme II | |
| F | TAYGARCCNATGTAYATGGA |
| R* | AANACRTCNCCNARDATCCA |
| Pro-enzyme III-1 | |
| F | GTNMGNGTNCCNYTNCARAARTA |
| R* | AANACRTCNCCNARDATCCA |
| Pro-enzyme III-2 | |
| F | WSNGARCCNATGACNAAYTA |
| R* | AANACRTCNCCNARDATCCA |
| Pro-enzyme IV-2 | |
| F | YTNGTNCCNYTNATHAARGGNAA |
| R* | AANACRTCNCCNARDATCCA |
Phylogenetic analysis
A multiple sequence alignment was performed using the CLUSTAL X program. A tree was constructed using the amino acid sequences of mature enzymes according to the maximum-likelihood method in the program RAxML with PROTGAMMAIJTT. The reliability of the tree was assessed by a bootstrap analysis with 1000 replicates.
Results
Purification of the pro-enzymes
The stomach of adult newts had several acidic proteinases. These were separated primarily into four bands on zymography using a 15% acrylamide gel (Fig. 1, Lane 1). The four proteinase bands were classified as aspartic proteinases because they were strongly inhibited by pepstatin A (Fig. 1, Lane 2). We designated acidic proteinases corresponding to the four bands as pro-enzymes I, II, III and IV and attempted to purify all of them. First, the crude extract was applied to a Q-Sepharose column and eluted by a linear gradient of NaCl (Fig. 2). The activity was eluted as two broad peaks. Zymographic analysis revealed that the former peak primarily contained pro-enzymes II and III (fraction II+III) and the later contained I and IV (fractions IV+I). The two fractions were fractionated separately using column chromatography. In fraction II+III, pro-enzymes II and III were separated by Q-Sepharose column chromatography with a gentle NaCl slope (Fig. 3A). The two resulting fractions, II and III, were further fractionated by Mono Q column chromatography using a NaCl slope that was gentler than that used in the previous round of Q-Sepharose column chromatography (Fig. 3B and C). A single peak of pro-enzyme II was obtained from ‘Fraction II’ (Fig. 3B). Pro-enzyme III was separated into three peaks, named pro-enzymes III-1, III-2 and III-3 (Fig. 3C). The three peaks (pro-enzymes II, III-1 and III-2) were purified separately by sequential column chromatography using a Mono Q column and a Sephacryl S-200 column.
Fig. 1.

Zymography of the acid proteinases in crude extract of adult newt stomach. Lane 1. Crude extract (15 μl) was subjected to native PAGE and the acid proteinases were visualized by zymography. Lane 2. Same conditions as for lane 1, with the addition of 4 μg/ml of pepstatin A in substrate (haemoglobin) solution.
Fig. 2.

The elution pattern of the crude extract on Q-Sepharose fast flow chromatography. The crude extract obtained from stomachs of 30 adult newts was applied to a Q-Sepharose column and eluted by liner gradient using 0.1–0.6 M NaCl in a total volume of 320 ml. The horizontal dotted bars indicate the eluting position of the pro-enzymes I–IV as judged by zymography. The two fractions (horizontal bars, II+III and IV+I) were pooled separately and used for further purification.
Fig. 3.

Purification of pro-enzymes II, III-1 and III-2. (A) Separation of pro-enzymes II and III on Q-sepharose column chromatography. The fraction II+III obtained from the previous step was applied to a Q-Sepharose column, equilibrated with 0.1 M NaCl in Tris-buffer. After washing the column with the same buffer, the binding proteins were eluted by liner gradient using 0.1–0.4 M NaCl in a total volume of 160 ml. Two fractions (horizontal bars, fraction of pro-enzymes II and III) were pooled separately and used for further purification each other. (B) The elution pattern of pro-enzyme II on Mono Q column chromatography. The fraction containing pro-enzyme II (Fig. 3A) was subjected to a Sephacryl S-200 column, and the active fraction was applied to a Mono Q column, equilibrated with Tris-buffer. After washing the column with the same buffer, the binding proteins were eluted by liner gradient using 0–0.2 M NaCl in a total volume of 80 ml. Fraction II (the horizontal bar) was subjected to further purification. (C) Separation of pro-enzymes III-1 and III-2 on Mono Q column chromatography. The fraction containing pro-enzyme III was subjected to a Sephacryl S-200 column, and the active fraction was applied to a Mono Q column equilibrated with Tris-buffer containing 0.05 M NaCl. After washing with the same buffer, the proteins bound to the column were eluted by liner gradient using 0.05–0.4 M NaCl in a total volume of 80 ml. Fractions III-1 and III-2 (the horizontal bars) were collected separately and then subjected to further purification steps.
Fraction IV+I was separated into the two broad peaks by gel filtration using a Sephacryl S-200 column (Fig. 4A). The former minor peak contained pro-enzyme I and the latter major peak contained pro-enzyme IV based on zymographic analysis. Using information from our previous study, pro-enzyme I was expected to be ProCatE based on the elution position during gel filtration, as CatE is known to form a dimer (10, 13, 23). Pro-enzyme I in the minor peak was further purified by Con-A Sepharose affinity column chromatography (data not shown). This column is suitable for the purification of CatE which has a high concentration of mannose sugar chains (13, 23). Pro-enzyme I was bound to the column and eluted with 0.3 M mannose as a single peak (data not shown). The fraction containing pro-enzyme I was then subjected to Mono Q column chromatography, and the resulting fraction represented the purified sample. The latter major peak (Fig. 4A) containing pro-enzyme IV was applied to a Mono Q column and eluted in two close peaks. The pro-enzymes contained in the two peaks were named pro-enzymes IV-1 and IV-2 (Fig. 4B). To remove the contaminated monomeric form of ProCatE or ProCatD, which can potentially co-elute with pro-enzyme IVs during anion-exchange column chromatography and gel filtration column chromatography, the fractions containing pro-enzymes IV-1 and IV-2 were applied to a Con-A Sepharose column and the pass-through fraction was collected for further purification (data not shown). Pro-enzymes IV-1 and IV-2 were then purified by sequential chromatography using mono Q and Sephacryl S-200 gel-filtration columns.
Fig. 4.

The purification of pro-enzymes I, IV-1 and IV-2 on chromatography. (A) Separation of pro-enzymes I and IV by Sephacryl S-200 column chromatography. The IV+I fraction (Fig. 2) was concentrated and applied to a Sephacryl S-200 column, The proteolytic activity was separated into two peaks, the first minor peak corresponding to pro-enzyme I and the second major peak corresponding to pro-enzyme IV (horizontal bars). (B) Separation of pro-enzymes IV-1 and IV-2 using Mono Q column chromatography. The fraction IV obtained from the previous step (Fig. 4A) was applied to a Mono Q column equilibrated with 0.05 M NaCl in Tris buffer. After washing with the same buffer, the bound proteins were eluted using 0.05–0.4 M NaCl in a total volume of 80 ml. Fractions IV-1 and IV-2 (the horizontal bars) were subjected to sequential column chromatography.
Pro-enzymes I, II, III-1, III-2, III-3, IV-1 and IV-2 were obtained during each of the three purification procedures using fresh crude extract (data not shown). Thus, these seven enzymes were assumed to be synthesized in the stomach of adult newts under normal conditions. In this study, six pro-enzymes, including pro-enzymes I, II, III-1, III-2, IV-1 and IV-2, were purified.
In SDS-PAGE of the purified pro-enzyme I, two close bands were observed at approximately 82 kDa with several faint bands in the non-reduced state. In the reduced state, the molecular mass of the two bands decreased by ∼43 kDa (Fig. 5A). This suggests that pro-enzyme I is a dimer linked with an intra-molecular disulphide bridge. The micro-heterogeneity was likely caused by partial degradation during the purification procedure. Zymography analysis confirmed that the purified pro-enzyme I corresponded to band I, and there was no contamination from other acid proteinases (Fig. 5B). Thus, pro-enzyme I is homo-dimeric and has characteristics similar to those of other vertebrate ProCatEs (10, 13, 23). Additionally, our results suggest that pro-enzyme I has high mannose type oligosaccharide chain(s); therefore, we classified pro-enzyme I as newt ProCatE.
Fig. 5.

SDS-PAGE (A) and zymography (B) of the purified pro-enzyme I. (A) Purified pro-enzyme I was subjected to SDS-PAGE on a 10% gel and stained with silver. M: molecular weight marker, (+) presence and (−) absence of reducing agent. (B) Zymography of the purified pro-enzyme I (lane 1) and the supernatant of the crude extract (lane 2).
The purified pro-enzymes II, III-1, III-2, IV-1 and IV-2 were homogeneous on SDS-PAGE and the molecular weights were 38 kDa for pro-enzymes II, III-1 and III-2 and 37 kDa for IV-1 and IV-2 (Fig. 6), which is similar to those for frog Pgs (40 kDa for bullfrog and African clawed frog A-type and C-type Pgs). Native-PAGE also yielded a single protein band in each purified sample (Fig. 7A). The zymographic analysis confirmed that each pro-enzyme band had activity for acid proteinase (Fig. 7B). The potential specific activities of these purified pro-enzymes, pro-enzymes II, III-1, III-2, IV-1 and IV-2, were 62, 127, 248, 394 and 172 units/mg, respectively.
Fig. 6.
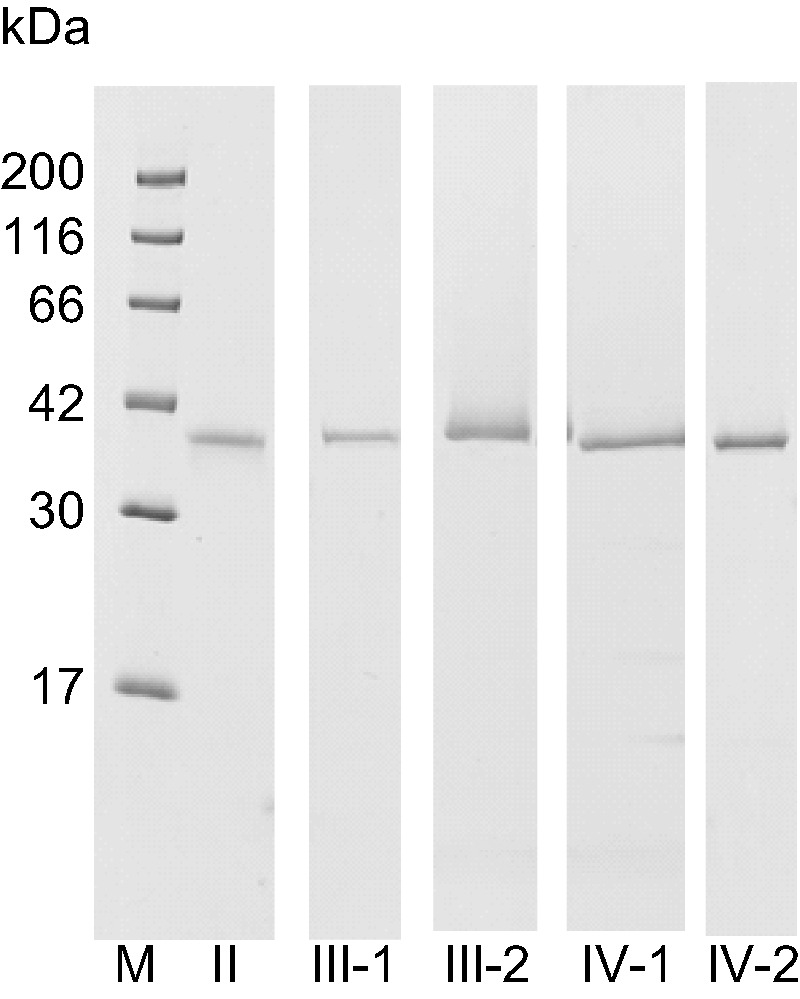
SDS-PAGE of purified pro-enzymes II, III-1, III-1, III-2, IV-1 and IV-2. Purified pro-enzymes (2 μg) were subjected to SDS-PAGE using a 15% gel and stained with CBB. M: molecular weight marker, II: pro-enzymes II, III-1: pro-enzymes III-1, III-2: pro-enzymes III-2, IV-1: pro-enzymes IV-1, IV-2: pro-enzyme IV-2.
Fig. 7.

Native-PAGE and zymography of the purified pro-enzymes II, III-1, III-2, IV-1 and IV-2. (A) Two micrograms of the purified pro-enzymes or 5 μl of the crude extract were subjected to Native-PAGE and stained with CBB. (B) The proteolytic activity of 0.3 μg of the purified pro-enzymes and 10 μl of the supernatant of the crude extract (sup) were visualized by zymography.
Thus, we purified the six aspartic proteinase precursors from the stomach of adult newts. Pro-enzyme I was ProCatE and the other five pro-enzymes, II, III-1, III-2, IV-1 and IV-2, were Pgs.
cDNA cloning of aspartic proteinases and molecular phylogenetic analysis
To carry out cDNA cloning, we first determined the N-terminal amino acid sequences of the pro-enzymes and active enzymes. Then degenerate primers were generated based on their N-terminal amino acid sequences (Table I). For upstream primers, the two degenerate primers were designed from the N-terminal amino acid sequences of the Pgs and pepsins, respectively. The two conserved regions at the Pgs active sites were used to design the two downstream primers. Because the N-terminal amino acid sequences were almost identical between the pro-enzymes and active enzymes of IV-1 and IV-2, we only cloned pro-enzyme IV-2 cDNA. By using the combination of two upstream primers and two downstream primers, the cDNA fragments of pro-enzymes II, III-1, III-2 and IV-2 were successfully amplified by reverse transcription polymerase chain reaction (RT-PCR) followed by nested PCR. Next, the full-length cDNAs were cloned by rapid amplification of cDNA ends PCR (RACE-PCR). The alignment of the deduced amino acid sequences of newt Pg cDNAs together with newt PgA, PgC1 and C2 previously reported (19) are shown in Fig. 8A. The sequences identical to the N-terminal amino acid sequences of pro-enzymes and active enzymes determined from the purified enzymes were found in all the cloned Pg cDNAs. The two active site consensus sequences, DTG or DSG, including a reactive aspartic acid residue, and six cysteins that formed an intra-molecular disulphide bridge were conserved in all the Pgs. Interestingly, four Pg cDNAs that were cloned in our study did not correspond to the newt PgA, PgC1 and C2 that have been reported previously (19).
Fig. 8.

Multiple alignments of the amino acid sequences of the aspartic proteinases. (A) Multiple alignments of amino acid sequences from newt Pg cDNAs. The solid lines and the dotted lines indicate the amino acid sequences identical to the N-terminal sequences of the purified Pgs and pepsins, respectively. Six cysteine residues thought to contribute to intermolecular disulphide bond are indicated by asterisks. Two aspartic acid residues positioned at two active sites are indicated by closed circles. (B) Multiple alignments of the amino acid sequences from human, frog and newt ProCatE cDNAs. The broken line indicates the amino acid sequence identical to that of the purified ProCatE. A triangle indicates the putative cleavage site of the signal peptidase. Asterisks and a diamond indicate the cysteine residues contributing to the intra-molecular disulphide bonds and a disulphide bond forming homo-dimeric form, respectively. The open and closed circles show the putative N-glycosylation site and aspartic acid residues positioned at two active sites, respectively.
To clone newt ProCatE cDNA, the N-terminal amino acid sequence of pro-enzyme I was determined and used to design the degenerate upstream primers. The two conserved active site sequences were used to design downstream primers for RT-PCR. The full-length cDNA was cloned by RT-PCR and RACE-PCR. The alignment of the amino acid sequence deduced from newt ProCatE cDNA and those of human and frog ProCatEs are shown in Fig. 8B. Although the N-terminal sequence of purified pro-enzyme I was identical to the predicted sequence from newt ProCatE, its N-terminus did not correspond to the predicted signal peptide cleavage site. Our results suggest that autolysis of the propeptide region occurs during the purification procedure. Similar to the Pgs, the two active site consensus sequences, DTG or DSG, and the six cysteins that formed the intra-molecular disulphide bridge were conserved in all the ProCatEs. Additionally, a cysteine residue that is reported to form inter-molecular disulphide bridge (5, 24) was also conserved in newt ProCatE, which is consistent with the results of our biochemical analysis suggesting that pro-enzyme I is dimer.
A phylogenetic tree was constructed based on the amino acid sequences of the active enzyme sections of vertebrate acid proteases using the ML method (Fig. 9). The tree shows that acid proteases were divided into four groups, CatE, CatD, A-type Pg and C-type Pg with high bootstrap node values. Newt ProCatE/pro-enzyme I was included in the CatE group. A- and C-type Pgs groups were further divided into several subgroups. Pro-enzymes III-2 and IV-2 formed a group with the previously cloned newt PgA and were included in the tetrapod PgA subgroup. Thus, pro-enzymes III-2 and IV-2 were named newt PgA1 and A3, respectively. Pro-enzymes II and III-1 were located in the PgBC subgroup in the C-type Pgs. Pro-enzymes II and III-1 were sisters of X. tropicalis PgBC2 and BC1, respectively; therefore, we named pro-enzymes II and III-1 as PgBC2 and BC1, as orthologues of anuran PgBC, respectively. Conversely, the previously cloned newt PgC1 and C2 belong to the PgC2 subgroup, which is a different subgroup from PgBC. Our results demonstrate that newts have two subgroups of C-type Pg, PgBC and PgC2.
Fig. 9.

Phylogenetic tree of vertebrate aspartic proteinases. The phylogenetic tree was constructed using the amino acid sequences of proteinase domains of aspartic proteinases. General English animal names and the enzyme names are shown with the accession number in parentheses. Procathepsin Es were used as the out group. The branch length of the tree represents the evolutionary distance. The numbers on each node show bootstrap values obtained from 1000 replications.
In summary, we cloned two PgAs (pro-enzymes III-2/PgA1 and IV-2/PgA3) and two PgBCs (pro-enzymes II/PgBC2 and III-1/PgBC1). Because the N-terminal amino acid sequences of pro-enzyme and active enzyme of IV-1 were almost identical to those of pro-enzyme IV-2, pro-enzyme IV-2 is considered to be a PgA and was therefore named PgA2.
Characterization of PgAs and PgBCs
Mammalian A-type Pg exhibits different enzymological characteristics from mammalian C-type Pg, including differences in the activation profile of pro-enzymes, the inhibitory effect of pepstatin A and the pH optimum; therefore, we examined the enzymological properties of PgAs and PgBCs.
Activation profile of pro-enzymes
The time course for pro-enzyme conversion to active enzyme was analysed by SDS-PAGE (Fig. 10). Pro-enzyme II/PgBC2 was converted to active enzyme (molecular mass was 34 kDa) within 5 min. During activation of pro-enzyme III-1/PgBC1, a major 34-kDa band and two minor bands at molecular weights of 36 and 33 kDa were observed after acid treatment, and the three bands were stable for >120 min. Sequence analysis revealed that the N-terminal sequence of the 34-kDa major band corresponded to that of the active form of pro-enzyme III-1/PgBC1. However, the N-terminal sequence of the 33-kDa band was AVGYEPLSNY, which is identical to the predicted N-terminal sequence of the active enzyme of PgC1 cloned previously, suggesting there was contamination of PgC1 in the purified Pro-enzyme III-1/PgBC1 sample. The sequence of the 36-kDa band could not be determined. Given that the 38-kDa band of pro-enzyme III-1/PgBC1 disappeared within 5 min of incubation, the activation profile of pro-enzyme III-1/PgBC1 was similar to that of pro-enzyme II/PgBC2. Conversely, the activation of pro-enzyme III-2/PgA1 was slower than those of the PgBCs, because a 38-kDa band of the pro-enzyme was observed with the 31-kDa band of the active form up to 20 min during incubation. The pro-enzyme IV-1/PgA2 and IV-2/PgA3 went through the intermediate form (34 kDa) and took more than 30 min for complete conversion to the active form (31 kDa). Pro-enzymes IV-1/PgA2 and IV-2/PgA3 were activated via intermediate forms, whereas pro-enzyme III-2/PgA1 was directly activated, and both PgAs required more than 20 min for complete activation. Our results suggest that the activation rates of newt PgBCs were faster than the PgAs in acidic conditions.
Fig. 10.

Time course of activation of the purified Pgs. Activation was performed at 22°C at pH 2.0. After incubation for a defined time (min), aliquots were withdrawn and neutralized by NaOH. Then, the samples were subjected to SDS-PAGE and stained with CBB. The proteins identical to 2 μg of the purified Pgs were loaded on each lane.
Inhibitory effect of pepstatin A
The inhibitory effect of pepstatin A on pepsin activity was measured after treatment of the purified pro-enzymes in acidic conditions (Fig. 11). We excluded pro-enzyme III-1/BC1 from further experiments because of contamination of PgC1 in the purified sample. At equimolar amounts of pepstatin A and enzyme, the activities of enzyme III-2/A1, IV-1/A2 and IV-2/A3 were completely inhibited. In contrast, enzyme II/BC2 required a 500-fold excess molar ratio for complete inhibition. Our results suggest that the inhibitory effect of pepstatin A on pepsin As activity is much higher than that of pepsin BCs.
Fig. 11.

Inhibitory effect of pepstatin A on proteolytic activity of the purified pepsins. After the purified Pgs were activated, the activities of pepsins were determined in reaction mixtures using the ‘Standard assay method for aspartic proteinase’ containing various concentrations of pepstatin A. Two micrograms (53 pmol) of pro-enzyme II/PgBC2 (•), III-2/PgA1 (▪) and 1.95 μg (53 pmol) of pro-enzyme IV-1/PgA2 (♦) and pro-enzyme IV-2/PgA3 (×) were used for each reaction.
The effect of pH on proteolytic activity
Pgs were converted to pepsins by acid treatment, then the optimum pH of the pepsins was determined using haemoglobin as substrate (Fig. 12). The optimum pH of pepsin II/BC2 and III-2/A1 was 1.5, while that of IV-1/A2 and IV-2/A3 was 2.0. The pH optimum curve of pepsin II/BC2 was broader than that for pepsin As and yielded 85% activity at pH 1.
Fig. 12.

pH dependency curve of acid protease activity. After activation of the purified Pgs, the activity of the pepsins was measured using the ‘Standard assay for aspartic proteinase’ using 0.25 M sodium acetate-HCl buffer over a range of pH. Two micrograms (53 pmol) of pro-enzyme II/PgBC (•) and III-2/PgA (▪), and 1.95 μg (53 pmol) of pro-enzyme IV-1/PgA (♦) and pro-enzyme IV-2/PgA (×) were used for each reaction. The activity is expressed as percentages of the highest activities of the respective pepsins.
Discussion
Five Pgs and ProCatE were purified from the stomach of adult newts. On the basis of the amino acid sequences determined from the purified proteins, we cloned four Pg cDNAs and ProCatE cDNA. The enzymological characters and structures of newt ProCatE, including the formation of a homodimer, were similar to those of mammalian and frog ProCatE. Phylogenetic analysis of the Pg cDNAs revealed that two cDNAs were included in the PgA group, and the remaining two were members of the PgBC group. Recently, Castro et al. (3) proposed a new classification of C-type Pg, which includes PgC1, PgC2, PgB, fish PgC and PgBC. They showed that PgBC genes were only found in the genome sequence of X. tropicalis; however, PgBCs were not characterized to the protein level. We demonstrated that PgBC genes producing functional proteinases are one of the major constituents of aspartic proteinases in the newt stomach. Given that PgBCs are found in urodeles and anurans, but not other vertebrates, PgBCs appear to be amphibian-specific Pgs that have diverged during the evolution of amphibians. The PgBC group is the sister of PgC1/PgB. PgBC, PgC1 and PgB formed a single group in the phylogenetic analysis. A number of tetrapods possess two C-type Pgs in their genomes; for example, amphibians have PgBC and PgC2 and other tetrapods (amniotes) have PgC1/PgB and PgC2. Interestingly, mammalians have lost PgC2 and PgB and avians have lost PgC1in their evolutional lineage. A single copy of PgA was found in the X. tropicalis genome (25). Additionally, a single PgA was purified from the stomach of adult bullfrogs and X. laevis (14, 16). Taken together, these observations suggest that anuran PgA is a single copy gene. However, three PgA cDNAs (two from present study and one from Inokuchi et al. (19)) have been cloned from the newt stomach, with amino acid identity of 73–80%. Thus, multicopy PgA genes are present in the newt genome and are thought to be a result of a duplication event during the evolution of urodeles.
We compared the enzymological properties of newt PgAs and PgBCs with those from other vertebrates (except birds and reptiles). Avian and reptilian Pgs have been purified (26–28), but the corresponding cDNAs have not yet been cloned. In mammals, bullfrog and X. laevis, C-type Pgs are rapidly converted to pepsins, whereas the conversion of A-type Pgs is slow under acidic conditions (14, 16). Two PgBCs, PgII/BC2 and PgIII-1/BC1, were converted to pepsins within 5 min, which was faster than the conversion rate for the three PgAs, PgIII-2/A1, PgIV-1/A2 and PgIV-2/A3. Thus, the activation profiles of newt PgAs and PgBCs are similar to those of other tetrapod A-type and C-type Pgs, respectively.
Moreover, the difference in sensitivity to pepstatin A between newt PgAs and PgBC2 was consistent with the difference between mammalian A-type and C-type Pg reported previously. The activity of newt PgAs, PgIII-2/A1, PgIV-1/A2 and PgIV-2/A3 was completely inhibited by the presence of equimolar pepstatin. This is consistent with results obtained from a number of tetrapod pepsin As that exhibit equimolar inhibition (14, 16, 29–31). In contrast, newt PgII/BC2 required a 500-fold molar excess for complete inhibition. Mammalian pepsin C1s are known to be less sensitive to the inhibitor than pepsin A. For example, mammalian pepsin C1s, such as those from the house musk shrew (29), goat (30) and new world monkey (32), require a 100-molar excess of pepstatin for complete inhibition. Thus, the inhibitory properties of newt PgBC are similar to those of pepsin C1s in mammals. Conversely, the PgC2s from bullfrog and X. laevis require a 10-molar excess of pepstatin for complete inhibition (14, 16). Therefore, the sensitivity of frog PgC2 to pepstatin appears to be higher than that of PgC1 and PgBC. According to the phylogenetic analysis, PgBC and PgC1 groups are sisters, and the PgC2 group diverged at an earlier date. It is interesting that the inhibitory properties of PgBC are more similar to that of PgC1 than PgC2, consistent with their phylogenetic relationship. Although the inhibitory properties of pepsin B have not yet been evaluated, it is generally thought that tetrapod C-type pepsins are less sensitive than tetrapod A-type pepsins.
In mammals, the pH optima of A-type pepsins is usually lower than those of C-type pepsins (32, 33). However, the pH optima for newt pepsin BC (enzyme II/BC2, around pH 1.5) was lower than for newt pepsin As (enzyme IV-1/A2 and IV-2/A3, pH 2.0) and similar to those of another newt pepsin A (enzyme III-2/A1, pH 1.5). Furthermore, the optimum pH for frog pepsin C2s are slightly lower than for frog pepsin As (14, 16); therefore, the pH optimum cannot be used to distinguish the two types of tetrapod pepsin.
The specific activity of newt pepsin As was 172–394 units/mg, which was similar to that of Xenopus pepsin A, 227 units/mg (16). In contrast, the specific activity of newt pepsin BC, 62 and 127, was about four times lower than those of Xenopus pepsin C2, 457 units/mg (16), suggesting the some heterogeneity in the enzyme characters in C type pepsin.
Taken together, the observations above suggest that C-type Pgs of tetrapods, including newt PgBC, can be distinguished from A-type Pgs based on their enzyme characteristics, such as the activation rate of Pg and the inhibitory effects of pepstatin, but not by the pH optimum. Conversely, fish Pgs cannot be distinguished using the enzyme characteristics of tetrapod Pgs. For example, two PgAs from Japanese seabass are rapidly converted to pepsin when compared with their own PgC (34). Similarly, the activation rate, pH dependency and inhibitory effect curve of PgA and PgC (pepsin A and pepsin C) are similar in coelacanths (35). In largemouth bass, one PgA is rapidly converted to pepsin, whereas the conversion of another PgA is slow (36). These observations suggest that the distinguishing characters between tetrapod A- and C-type Pgs were established during evolution of the tetrapod lineage.
The stomach environment is slightly changed by an individual’s diet. Having two types of pepsin, A and C, that exhibit different enzyme characters allows for flexibility. It is interesting to learn which amino acid residues are responsible for such distinguishing properties among the two types of Pgs and how those residues are conserved in each evolutional lineage.
Conflict of Interest
None declared.
Glossary
Abbreviations
- CatD
cathepsin D
- CBB
Coomassie Brilliant Blue
- PAGE
polyacrylamide gel electrophoresis
- PgA
pepsinogen A
- PgC, pepsinogen C; ProCatE
pro-cathepsin E
- SDS
sodium dodecylsulfate
Nucleotide sequences The nucleotide sequences data reported in this article will appear in the DDBJ/EMBL/GenBank nucleotide sequence database with accession number LOC060623, LOC060624, LOC060625, LOC060626, LOC060627.
References
- 1.Richter C., Tanaka T., Yada R.Y. (1998) Mechanism of activation of the gastric aspartic proteinases: pepsinogen, progastricsin and prochymosin. Biochem. J. 335, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanji M., Yakabe E., Kubota K., Kageyama T., Ichinose M., Miki K., Ito H., Takahashi K. (2009) Structural and phylogenetic comparison of three pepsinogens from pacific Bluefin tuna: molecular evolution of fish pepsinogens. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 152, 9–19 [DOI] [PubMed] [Google Scholar]
- 3.Castro L.F.C.C., Lopes-Marques M., Goncalves O., Wilson J.M. (2012) The evolution of pepsinogen C genes in vertebrates: duplication, loss and functional diversification. PLoS One 7, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kageyama T. (2002) Pepsinogens, progastricsins, and prochymosins: structure, function, evolution, and development. Cell. Mol. Life Sci. 59, 288–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chlabicz M., Gacko M., Worowska A., Lapinski R. (2011) Cathepsin E (EC 3.4.23.34)—a review. Folia Histochem. Cytobiol. 49, 547–557 [DOI] [PubMed] [Google Scholar]
- 6.Benes P., Vetvicka V., Fusek M. (2008) Cathepsin D—many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sastradipura D.F., Nakanishi H., Tsukuba T., Nishishita K., Sakai H., Kato Y., Gotow T., Uchiyama Y., Yamamoto K. (1998) Identification of cellular compartments involved in processing of cathepsin E in primary cultures of rat microglia. J. Neurochem. 70, 2045–2056 [DOI] [PubMed] [Google Scholar]
- 8.Zaidi N., Kalbacher H. (2013). Cathepsin E. In: Salvesen N. D., Rawlings G. (Eds.), Handbook of Proteolytic Enzymes. 3rd edn, Academic Press, Salt Lake City, UT, pp. 42–49 [Google Scholar]
- 9.Matsuo K., Kobayashi I., Tsukuba T., Kiyoshima T., Ishibashi Y., Miyoshi A., Yamamoto K., Sakai H. (1996) Imunohistochemical localization of cathepsin D and E in human gastric cancer: a possible correlation with local invasive and metastatic activities of carcinoma cells. Hum. Pathol. 27, 184–190 [DOI] [PubMed] [Google Scholar]
- 10.Kageyama T., Ichinose M., Tsukuda S., Miki K., Kurokawa K., Koiwai O., Tanji M., Yakabe E., Athauda S.B.P., Takahashi K. (1992) Gastric pro-cathepsin E and progastricsin from guinea pig purification, molecular cloning of cDNAs, and characterization of enzymatic properties, with special reference to pro-cathepsin E. J. Biol. Chem. 267, 16450–16459 [PubMed] [Google Scholar]
- 11.Yakabe E., Tanji M., Ichinose M., Goto S., Miki K., Kurokawa K., Ito H., Kageyama T., Takahashi K. (1991) Purification, characterization, and amino acid sequences of pepsinogens and pepsins from the esophageal mucosa of bullfrog (Rana catesbeiana). J. Biol. Chem. 266, 22436–22443 [PubMed] [Google Scholar]
- 12.Inokuchi T., Kobayashi K., Horiuchi S. (1991) Acid proteinases of the fore-gut in metamorphosing tadpoles of Rana catesbeiana. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 111, 111–117 [DOI] [PubMed] [Google Scholar]
- 13.Inokuchi T., Kobayashi K., Horiuchi S. (1994) Purification and characterization of cathepsin E type acid proteinase from gastric mucosa of bullfrog, Rana catesbeiana. J. Biochem. 115, 78–81 [DOI] [PubMed] [Google Scholar]
- 14.Inokuchi T., Kobayashi K., Horiuchi S. (1995) Isolation of pepsinogen A from gastric mucosa of bullfrog, Rana catesbeiana. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 111, 111–117 [DOI] [PubMed] [Google Scholar]
- 15.Inokuchi T., Kobayashi K., Horiuchi S. (2003) Molecular cloning of prepro-cathepsin E cDNA from the stomach of bullfrog Rana catesbeiana. J. Biochem. 115, 76–81 [DOI] [PubMed] [Google Scholar]
- 16.Ikuzawa M., Inokuchi T., Kobayashi K., Yasumasu S. (2001) Amphibian pepsinogens: purification and characterization of Xenopus pepsinogens, and molecular cloning of Xenopus and bullfrog pepsinogens. J. Biochem. 129, 147–153 [DOI] [PubMed] [Google Scholar]
- 17.Ikuzawa M., Yasumasu S., Inokuchi T., Kobayashi K., Nomura K., Iuchi I. (2003) Differential expression of two cathepsin Es during metamorphosis-associated remodeling of the larval to adult type epithelium in Xenopus stomach. J. Biochem. 134, 385–394 [DOI] [PubMed] [Google Scholar]
- 18.Liquori G.E., Zizza S., Mastrodonato M., Scillitani G., Calamita G., Ferri D. (2005) Pepsinogen and H, K-ATPase mediate acid secretion in gastric glands of Triturus carnifex (amphibian, caudata). Acta Histochem. 107, 133–141 [DOI] [PubMed] [Google Scholar]
- 19.Inokuchi T., Ikuzawa M., Yamazaki S., Watanabe Y., Shiota K., Katoh T., Kobayashi K. (2013) Molecular cloning of pepsinogens A and C from adult newt (Cynops pyrrhogaster) stomach. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 165, 226–235 [DOI] [PubMed] [Google Scholar]
- 20.Law for the Humane Treatment and Management of Animals in Japan. (2013) https://www.env.go.jp/nature/doubutsu/aigo/2_data\laws/nt_h25_84.pdf [Google Scholar]
- 21.Institutional Animal Care and Use Committee. http://iacuc.usc.edu/
- 22.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto K., Katsuda N., Kato K. (1978) Affinity purification and properties of cathepsin-E-like acid proteinase from rat spleen. Eur. J. Biochem. 92, 499–508 [DOI] [PubMed] [Google Scholar]
- 24.Fowler S.D., Kay J., Dunn B.M., Tatnell P.J. (1995) Monomeric human cathepsin E. FEBS Lett. 366, 72–74 [DOI] [PubMed] [Google Scholar]
- 25.Castro L.F., Goncalves O., Mazan S., Tay B.H., Venkatesh B., Wilson J.M. (2013) Recurrent gene loss correlates with the evolution of stomach phenotypes in gnathostome history. Proc. Biol. Sci. 281, 20132669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pletschke R.J., Naude R.J., Oelofsen W., Muramoto K., Yamauchi F. (1995) Ostrich pepsinogens I and II: purification, activation and chemical and immunochemical characterization of the enzymes from the proventriculus. Int. J. Biochem. Cell Biol. 27, 613–624 [DOI] [PubMed] [Google Scholar]
- 27.Hirasawa A., Athauda S.B.P., Takahashi K. (1996) Purification and characterization of turtle pepsinogen and pepsin. J. Biochem. 120, 407–414 [DOI] [PubMed] [Google Scholar]
- 28.Yonezawa H., Nonaka T., Uchikoba T., Hattori S., Ohno M., Kaneda M. (2000) Isolation and characterization of pepsinogen from Trimeresurus flavoviridis (Habu snake). J. Biochem. 127, 755–760 [DOI] [PubMed] [Google Scholar]
- 29.Narita Y., Oda S., Moriyama A., Tanaka O., Kageyama T. (1997) Pepsinogens and pepsins from house musk shrew, Sucus murinus: purification, characterization, determination of the amino-acid sequences of the activation segments, and analysis of proteolytic specificities. J. Biochem. 121, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 30.Suzuki M., Narita Y., Oda S., Moriyama A., Takenaka O., Kageyama T. (1999) Purification and characterization of goat pepsinogens and pepsins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 122, 453–460 [DOI] [PubMed] [Google Scholar]
- 31.Kageyama T. (2000) New world monkey pepsinogens A and C, and prochymosins. Purification, analysis of the conversion process to pepsin and determination of the NH2-terminal amino-acid sequences. Eur. J. Biochem. 141, 261–269 [DOI] [PubMed] [Google Scholar]
- 32.Richmond V., Tang J., Wolf S., Trucco R.E., Caputto R. (1958) Chromatographic isolation of gastricsin, the proteolytic enzyme from gastric juice with pH optimum 3.2. Biochim. Biophys. Acta 29, 453–454 [DOI] [PubMed] [Google Scholar]
- 33.Tang J., Wolf S., Caputto R., Trucco R.E. (1959) Isolation and crystallization of gastricsin from human gastric juice. J. Biol. Chem. 234, 1174–1178 [PubMed] [Google Scholar]
- 34.Cao M., Chen W., Du C., Yoshida A., Lan W., Liu G., Su W. (2011) Pepsinogens and pepsins from Japanese seabass (Lateolabrax japonicus). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 158, 259–265 [DOI] [PubMed] [Google Scholar]
- 35.Tanji M., Yakabe E., Kageyama T., Yokobori S., Ichinose M., Miki K., Ito H., Takahashi K. (2007) Purification and characterization of pepsinogens from the gastric mucosa of African coelacanth, Latimeria chalumnae, and properties of the pepsins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 146, 412–420 [DOI] [PubMed] [Google Scholar]
- 36.Miura Y., Kageyama T., Moriyama A. (2015) Pepsinogens and pepsins from largemouth bass, Micropterus salmoides: purification and characterization with special reference to high proteolytic activities of bass enzymes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 183, 42–48 [DOI] [PubMed] [Google Scholar]